Introduction
Lattice corneal dystrophy (LCD) is an inherited disorder of the eye characterized by the deposition of amyloid resulting in steadily progressive loss of vision. These deposits create linear, “lattice-like” opacities arising primarily in the central cornea, while the peripheral cornea is often spared. They are radially oriented and are accompanied by gradual, superficial opacification of the cornea. Recurrent epithelial erosions are often present, causing ocular irritation and additional vision loss. The erosions may appear before any noticeable stromal deposits. LCD belongs to a broader family of corneal dystrophies and has several subtypes, as described below.[1][2][3][4][5]
Type I LCD (LCD1), also known as classic lattice corneal dystrophy or Biber-Haab-Dimmer dystrophy, is the primary form of LCD. It is autosomal dominant and results from mutations in the transforming human growth factor beta-induced (TGFBI) gene. Although TGFBI and its protein transcript are found throughout the body, there are no known systemic effects outside of the ocular pathology for which it is named. It usually presents in the first or second decade of life.[5][6]
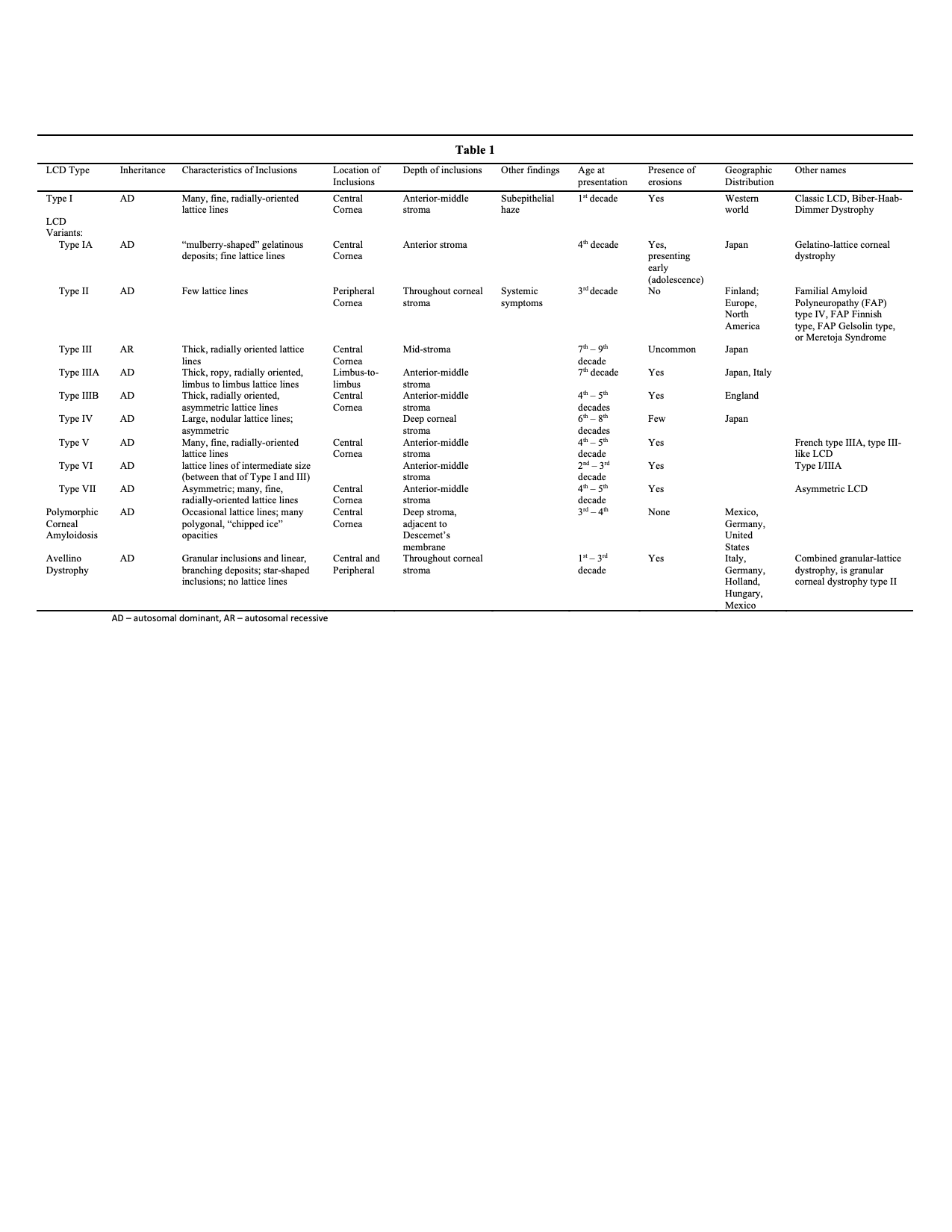
The LCD variants are subtypes of LCD caused by a variety of mutations on the TGFBI gene. The variants were formerly described as types IA, III, IIIA, IIIB, IV, V, VI, VII, and polymorphic corneal amyloidosis. These are variations of the same disease process that causes type I LCD, with minor changes in their phenotypic features. Specific phenotypic patterns are traceable to specific mutations in the TGFBI gene, which resulted in their being initially described as separate diseases. They are now considered to be subtypes of the same disease, in which type I is the classic and most common presentation.[6] The variants are often geographically specific and are occasionally traceable to single founder mutations.[5][6]
LCD type II is no longer included among the corneal dystrophies as it is a primarily systemic disorder with ophthalmologic features. While it was initially included in the LCD family because of the lattice-like ocular deposits, it is now more accurately described as familial amyloid polyneuropathy (FAP) type IV, FAP Finnish type, FAP Gelsolin type, or Meretoja syndrome.[6] Systemic symptoms include neuropathy (due to amyloid infiltration of nerves), facial paralysis, and extreme skin laxity. It tends to present in the twenties.[7]
Another disease process often mistakenly included in the LCD family is granular corneal dystrophy type II (GCD type II). Also called combined granular-lattice dystrophy or Avellino dystrophy, GCD type II was formerly considered a hybrid of granular and lattice dystrophies since it exhibits symptoms of both diseases. It is characterized by both granular and branching linear deposits that make it challenging to distinguish it from the LCDs. Physical exam findings that differentiate it from the lattice dystrophies are further discussed in this article under the name Avellino dystrophy, to minimize confusion with type II LCD.[6]
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
LCD type I is caused by mutations in the transforming human growth factor beta-induced (TGFBI) gene, located on chromosome 5 at locus 5q31.[5][6] This gene is also called BIGH3.[1][8] Multiple different nucleotide substitutions in this gene can cause type I LCD. The phenotypic variation seen in LCD may correlate with specific nucleotide substitutions. Mutations also cause the LCD variants in this same gene locus.[6]
Mutations cause type II LCD in the 9q34 locus of the GSN gene.[6] As in type I, several different nucleotide substitutions have been identified at this location.[9]
Epidemiology
Lattice corneal dystrophy affects males and females equally, with age of onset varying as described in history and physical.[4]
Type I LCD is among the most common corneal dystrophies. It is found worldwide, most commonly in the western world.[3] The LCD variants are most common in Japan and Italy.[6]
Type II LCD is most common in Finland, although a few cases have been found in America, Denmark, the Netherlands, the Czech Republic, and elsewhere.[3][9]
Pathophysiology
Type I LCD is caused by the accumulation of a mutant form of the transforming growth factor beta-induced protein, also called kerato-epithelin. It is the product of the TGFBI gene. While this protein is found throughout the body, its effects are limited to the cornea. These effects are most likely mediated by abnormal protein folding and precipitation as amyloid.[1][5][10] The LCD variants are less common forms of the same disease, also caused by kerato-epithelin-derived amyloid deposits.[5]
Type II LCD is caused by the accumulation of a mutant form of the protein gelsolin, resulting in amyloid deposits throughout the body. It is caused by the substitution of uncharged amino acids for asparagine at residue 187, resulting in a change in conformation. It is suggested that this conformational change causes the protein to form the previously described amyloid deposits.[9]
Histopathology
LCD type I is characterized histopathologically by multiple eosinophilic amyloid inclusions between the epithelium and the Bowman layer, as well as within the corneal stroma. The deposits stain well with Congo red, exhibiting birefringence and dichroism under polarized light. They also show metachromasia when stained with crystal violet, and fluorescence when stained with thioflavin T.[6][8] Epithelial atrophy is seen with degeneration of basal epithelial cells and loss of or damage to the Bowman layer.[4]
Confocal microscopy shows linear, branching structures in the corneal stroma with indistinct margins.[6]
Optical computed tomography shows atrophic epithelium, disruption of the Bowman layer, and inclusions extending from the Bowman layer into the anterior stroma.[4]
Electron microscopy shows deposits of extracellular, electron-dense fibrils that are 8-10 nm diameter, consistent with amyloid protein. Corneal fibroblasts are reduced in the number surrounding these deposits.[6][8]
Type II LCD is characterized by thin lattice lines concentrated in the anterior and middle stroma. These are initially seen peripherally and spread centrally. There are fewer lines as compared to type I LCD, and epithelial erosions are not seen. Similar to type I, there is a disruption of Bowman’s layer by amyloid invading the subepithelial space.[3][6][11][12]
In Avellino dystrophy, histology shows hyaline and amyloid deposits spanning from the basal epithelium to the deep stroma.[4][6]
The location of these inclusions and their relative size varies between the different types and is described below.[6]
History and Physical
Type I LCD patients often present with visual impairment and ocular irritation. These symptoms are progressive, starting as early as the first decade of life and causing visual impairment between the fourth and sixth decade.[5][6] Type I shows an autosomal dominant inheritance pattern, and patients usually have a strong family history of similar symptoms. It is a bilateral process but may affect the eyes asymmetrically.[5] The LCD variants often present later than type I LCD but with similar symptoms. Most are autosomal dominant, with the possible exception of type III.[5][6]
Type II LCD symptoms may begin after the age of 20, with mean diagnosis at 39 years of age.[13] Mild lattice corneal dystrophy is often among the first symptoms to manifest.[7] Vison is often minimally affected until later in life, as late as the seventh decade. Symptom onset varies by the specific mutation, with homozygous patients developing visual symptoms at an earlier age. It also shows an autosomal dominant inheritance pattern.[6][7][14]
Evaluation
Type I diagnosis is made clinically. On slit-lamp examination, subepithelial spots or flecks are seen, starting centrally, sometimes with diffuse haziness and a ground-glass appearance. Retroillumination reveals branching or lattice-like lines in the superficial stroma spreading from the central cornea toward the periphery. (See figures 1 and 2) The deposits result in recurrent erosions. The deep stroma, Descemet’s membrane, and endothelial layers are spared. LCD1 is almost always a bilateral process, but may not be symmetric and has been seen unilaterally. Genetic analysis will also reveal the TGFBI mutation.[6]
LCD variants, now considered offshoots of LCD1, vary from type I only slightly, and subtype diagnosis may not be necessary for appropriate treatment.[6] A brief description of each is below. See table 1 for details.
- Type IA has lattice lines similar to type I, with the addition of “mulberry shaped” gelatinous deposits in the affected corneas. Erosions appear in adolescence. Also called gelatino-lattice corneal dystrophy.[15]
- Type III has thicker lattice lines, radially oriented, which occur after the seventh decade of life.[5][10][16]
- Type IIIA is the most well-defined of the variants and shows thicker, ropy lattice lines extending from limbus to limbus. It also occurs later in life.[5][10]
- Type IIIB is poorly elucidated, has asymmetric corneas, and occurs in middle age as opposed to type I and III, which occur in young and older ages, respectively.[5][10]
- Type IV has deeper deposits than the earlier types and therefore exhibits erosions less often than other types. It has large, nodular lattice lines that progress from posterior to anterior, unlike the other varieties which progress from anterior to posterior. It has a late onset and shows substantial phenotypic variation.[5][17]
- Type V is poorly elucidated and has deposits that are indistinguishable from type I. It is also called French type IIIA and type III-like LCD.[5][18]
- Type VI, also called type I/IIIA, has thinner lattice lines than type III but thicker than type I, and presents in the second to third decade of life.[5][19]
- Type VII is also poorly elucidated, onsets late in life, and is characterized by patients with bilateral symptoms but a greater degree of asymmetry than is seen in other LCDs. It is indistinguishable from type 1 on histology or electron microscopy. Also known as Asymmetric LCD.[5][18]
- Polymorphic corneal amyloidosis has mostly polygonal inclusions in the deep, central stroma with few or no lattice lines. No erosions are seen.[6][20][21][22]
Type II diagnosis is also made clinically by the triad of lattice corneal dystrophy, cutis laxia (loose skin), and progressive cranial and peripheral neuropathy.[13] Symptoms arise from the accumulation of amyloid in nerve sheaths, basal membranes, vascular walls, and elsewhere. This alters the flow of nutrients and waste in the body and distorts the structural makeup of most tissues. Ocular symptoms include irritation, open-angle glaucoma, increased sensitivity to light, and eventual loss of visual acuity. Chronic open-angle glaucoma, peripheral neuropathy, meibomian gland dysfunction, and early cataract development have been described as well. Dermatological symptoms include laxity of skin, drooping of eyelids, skin fragility, easy bruising, and dry skin throughout the body. Neurological symptoms, derived from amyloid invasion of brain and nerves, include cognitive impairment, numbness, tingling, paresthesias, dysarthria, bilateral carpal tunnel syndrome, and loss of balance. Cardiac arrhythmias, hair loss, renal failure, and poor oral health have been associated with the disease as well.[3][5][6][7][13][23] In comparison with type I LCD, fewer and more delicate lattice lines are seen, oriented more peripherally, and sometimes entirely sparing the visual axis. Deposits are also less numerous with no epithelial erosions. Genetic analysis reveals the 9q34 locus of the GSN gene is affected.[3]
Avellino dystrophy, included here for the sake of facilitating differential diagnosis, shows superficial spots in the stroma with small “thorns,” linear inclusions, and superficial circular patches.[4] OCT shows large, dense, hyperreflective deposits in the epithelium and anterior stroma with irregular disruption of the Bowman layer. These deposits may also have a slightly lattice-like appearance; however, they rarely cross one another and therefore are less likely to form a true lattice.[4][6] GCD type II can be distinguished from LCD in two ways.[6]
- The linear deposits are whiter and more dash-like, while the lattice lines in LCD are more refractile.
- The linear deposits rarely cross or form the characteristic “lattice” for which LCD was named.
It is important to note that the size and appearance of lattice lines in any of these pathologies change with the patient’s age and progression of the disease.[6]
Treatment / Management
Penetrating keratoplasty is the treatment of choice for LCD once visual symptoms progress to the point that surgical intervention is warranted. Transplant is not usually necessary before the fourth decade, although it may be required as early as the second decade of life. The prognosis after PK is excellent; however, recurrence is quite common with amyloid deposits forming in the grafted cornea anywhere from 2to 14 years after transplant.[3][5] There are reports of delayed corneal epithelial wound healing in LCD after PK, and any patient undergoing PK is at risk of graft rejection and development of irregular astigmatism.[24][25][26](B2)
Deep anterior lamellar keratoplasty (DALK) has similar outcomes to PK. With recent advances in operative techniques and equipment, some studies now show that DALK may have better visual outcomes and lower risks of graft rejection. Because of these advantages, DALK is now considered first-line therapy for LCD in addition to PK.[24][27][28] Retaining the Descemet’s membrane and the endothelium reduces the risk of graft failure or rejection. It also maintains the structural stability of the eye and reduces some of the intraoperative and postoperative risks associated with PK.[26][28][29] One study found clinically significant recurrence at five years on average.[29] DALK includes the risk of intraoperative micro-perforation, irregular resection, hyperopic shift, and irregular astigmatism.[26][28][29](B2)
Phototherapeutic keratectomy (PTK) may also be used to improve vision.[30][31] It resolves lattice changes, erosions, and opacifications – these being the three defects common to LCD that tend to inhibit visual acuity. It is considered a second line intervention to keratoplasty, as it is unable to resolve the deeper lesions which are often present. It is also associated with hyperopic shift and stromal haze.[26] Improvement in vision is seen by three months.[30] PTK can be particularly useful in several settings:(B2)
- PTK can be used to delay the necessity of corneal grafting.[31][32]
- PTK can be used to treat the recurrence of lattice changes after corneal grafting. Recurrent opacities after grafting are often more superficial and within reach of PTK. Removing these opacities can delay or prevent the need for regrafting.[31]
- PTK is especially useful for epithelial erosions, which have a significant impact on visual acuity. Both the erosions and any associated opacifications are easily removed, being superficial and, therefore, easily within reach of PTK.[31]
- Childhood corneal dystrophy, particularly concerning due to the risk of amblyopia, is another indication for PTK. Corneal grafting is less successful in the very young, and PTK is well adapted to treating the early opacifications seen in this age. PTK also provides rapid visual improvement, particularly important in the avoidance of amblyopia.[31] (B2)
Recurrence is frequent, with the average recurrence occurring 9-10 years after the procedure.[30][33] These recurrences tend to be superficial, and the deposits are easily removed by additional rounds of PTK.[30] It is worth noting that these recurrence rates include patients undergoing PTK after previous PK, and thus the recurrence rate of PTK in isolation cannot be well ascertained.(B2)
Femtosecond laser-assisted lamellar keratectomy (FLK) removes a section of the anterior cornea, as does PTK. However, FLK can proceed to a greater depth than PTK (20% to 30% of corneal thickness). Because of this, FLK may be another second-line therapy in LCD when treating the anterior to middle depth of the cornea. OCT can accurately map the corneal deposits, and with the precision afforded by FLK, the deposits can be carefully resected with minimal damage to healthy structures. This results in fewer intraoperative and perioperative complications and is free of the risks of graft rejection, as seen in first-line therapies. While it is capable of removing opacifications deeper than PTK can reach, it remains limited to anterior disease and is often used as a temporizing measure. Eyes treated with FLK will most likely need eventual treatment with PK or DALK. Like PTK, FLK is particularly useful in young patients and recurrent LCD after a corneal graft. FLK is not as useful in patients with deep pathology or with notable corneal thinning in the area of the pathology. In one study, mild recurrence was seen at around two years of follow up.[26] Another study used PTK after FLK to smooth the cut surface and suppress keratocyte proliferation. They saw no recurrence in their LCD patients at two years of follow up and attribute this to the PTK.[34] Risks of FLK include irregular astigmatism, weakening of the cornea, and recurrence of pathology.[26](B2)
Femtosecond laser-assisted lamellar keratoplasty (FALK) is another modality that may be used to treat LCD. This method precisely removes diseased tissue via femtosecond laser, followed by the replacement of removed tissue with grafted corneal explant. The cornea is carefully mapped prior to the procedure via OCT, allowing for extreme precision. This process allows for increased depth (up to 500 um, as compared to less than 200 um in FLK) without dangerously thinning the cornea. It is similar to DALK, but with the femtosecond laser, there are the advantages of additional precision.[26][35] Because of these advantages, FALK may be another emerging first-line treatment for LCD. No data is currently available on recurrence rates for FALK. Myopic shift, irregular astigmatism, and graft rejection are risks in this procedure.[35](B2)
A note on the recurrence rate: Data is currently inadequate on the relative rates of recurrence in the various treatment modalities due to varying definitions of recurrence and often insufficient follow-up. In comparing the different treatment modalities, it can be assumed that recurrence is likely with any treatment due to the reaccumulation of amyloid after 5 to 10 years.
Other temporizing measures may be employed to delay the necessity of long-term treatments. These include corneal scraping, diamond burr polishing, topical antibiotic use, bandage contact lenses, and topical steroids. While none of these are curative, they may temporarily give some improvement in vision and allow for the delay of more invasive surgical treatments.
Treatment of type II LCD is broader due to the systemic nature of the disease. The corneal dystrophy itself is also treated with penetrating keratoplasty or PTK as above with excellent results. The dry eye symptoms are treated with topical lubricants, hydrophilic contact lenses, and lacrimal duct plugs. Symptoms related to increased skin laxity, including ectropion and deficient eyelid closure, can be treated with oculoplastic surgery to minimize symptoms.[7][14] Some locations, including Boston University, Cleveland Clinic, Stanford, University of Pennsylvania, University of Utah, Vanderbilt, and others, have amyloid centers or programs with multidisciplinary programs to treat the multi-system effects of systemic amyloidosis. Patients with type II LCD may benefit from treatment at one of these centers.(B3)
Gene therapies are also under development and may soon be the mainstay of treatment for corneal dystrophies. Significant progress has been made in identifying specific mutations associated with LCD. Because of this progress, therapies targeted at these specific mutations are becoming more feasible. Chaperone nanobodies, siRNA, CRISPER, and antisense oligonucleotides have all been suggested as possible treatments for LCD. So far, siRNA and nanobodies show promise for type I and type II LCD, respectively. Antisense oligonucleotides and CRISPR show promise in investigations for treatment of other corneal disorders, but have not yet been investigated for use in LCD.[36][37][38][39][40][41][42][43][44](B3)
Differential Diagnosis
Lattice corneal dystrophy is distinguished from other corneal dystrophies by comparing distinctive clinical features or through genetic analysis.
Type II LCD, being systemic amyloidosis, has a broader differential diagnosis. It is occasionally confused for Ehlers-Danlos syndrome due to the skin laxity found in both diseases. Dry eye symptoms are also common and can be confused for Sjögren syndrome.[7] It must also be differentiated from other systemic amyloidoses.
Prognosis
LCD is a progressive disease, resulting in the eventual loss of vision without treatment. For the subtypes limited to the cornea, patients tend to respond exceptionally well to surgical intervention, as described above. In these patients, symptoms may recur due to continued amyloid invasion of the grafted tissue, requiring further or repeated treatment.[3][31] With treatment, however, patients can live normal lives with minimal visual impairment.
In type II LCD, there is a broader range of symptoms that may have an impact on a patient’s life. While the ocular symptoms may be treated with a high degree of success, the neuropathy and facial paralysis are harder to treat and likely to cause long-term symptoms.[7]
Complications
Complications of LCD are primarily related to the lack of treatment, intraoperative and postoperative risks, and recurrence after treatment as described above.
Deterrence and Patient Education
An ophthalmologist should evaluate anyone with a strong family history of LCD. Early diagnosis is vital in children to avoid the development of amblyopia. Genetic counseling and testing may be helpful in diagnosis and family planning. Patients with diagnosed LCD should be followed closely by an ophthalmologist, and treatments should not be delayed. The patient’s pediatrician should be informed of the corneal findings so that appropriate workup for systemic amyloidosis may be completed if necessary.
Pearls and Other Issues
Lattice corneal dystrophy is an inherited disease of the eye characterized by amyloid deposits, corneal opacification, and recurrent corneal epithelial erosions.
Type II LCD is not corneal dystrophy, but systemic amyloidosis.
The symptoms of LCD can be treated with penetrating keratoplasty, DALK, PTK, FLK, and FALK, and has a good prognosis.
Enhancing Healthcare Team Outcomes
Patients diagnosed with LCD should be evaluated regularly by an ophthalmologist. Patients with type II LCD should be monitored for the development of the systemic signs of the disease. Any patient with a family history of LCD should be evaluated by an ophthalmologist and should consider genetic counseling.
Media
(Click Image to Enlarge)
LCD Types Table Contributed by William West
(Click Image to Enlarge)
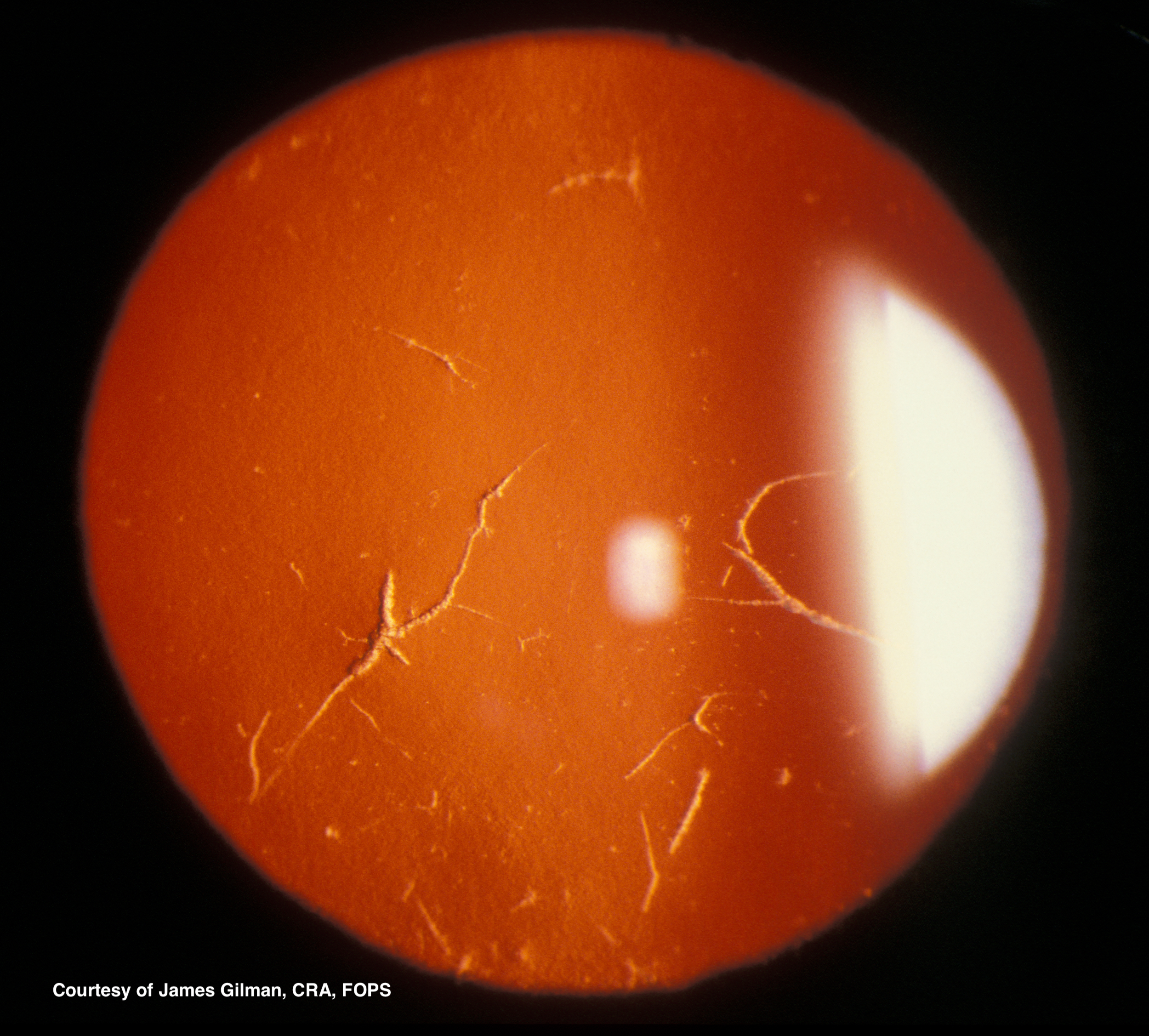
Figure 1 LCD Used with permission from James Gilman, CRA, FOPS
(Click Image to Enlarge)

Figure 2 LCD Used with permission from James Gilman, CRA, FOPS
(Click Image to Enlarge)
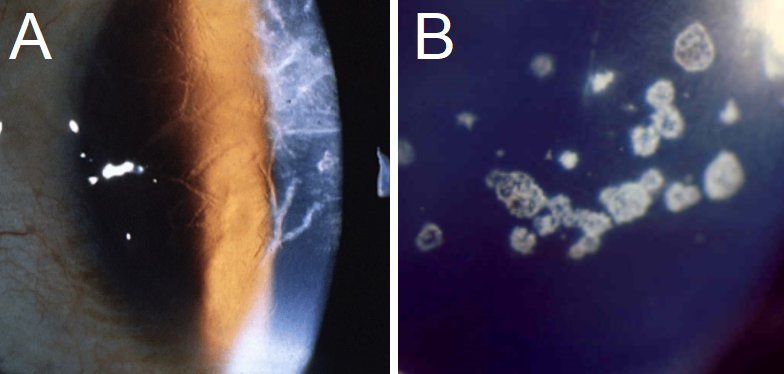
A: Typical branching opacities found in Lattice Corneal Dystrophy. B: A clear presentation of Granular Corneal Dystrophy, with crumb-like opacities separated by clear corneal surface space. Contributed by Majid Moshirfar, MD
References
Munier FL,Korvatska E,Djemaï A,Le Paslier D,Zografos L,Pescia G,Schorderet DF, Kerato-epithelin mutations in four 5q31-linked corneal dystrophies. Nature genetics. 1997 Mar; [PubMed PMID: 9054935]
Kannabiran C, Klintworth GK. TGFBI gene mutations in corneal dystrophies. Human mutation. 2006 Jul:27(7):615-25 [PubMed PMID: 16683255]
Klintworth GK. Corneal dystrophies. Orphanet journal of rare diseases. 2009 Feb 23:4():7. doi: 10.1186/1750-1172-4-7. Epub 2009 Feb 23 [PubMed PMID: 19236704]
Siebelmann S, Scholz P, Sonnenschein S, Bachmann B, Matthaei M, Cursiefen C, Heindl LM. Anterior segment optical coherence tomography for the diagnosis of corneal dystrophies according to the IC3D classification. Survey of ophthalmology. 2018 May-Jun:63(3):365-380. doi: 10.1016/j.survophthal.2017.08.001. Epub 2017 Aug 9 [PubMed PMID: 28801092]
Level 3 (low-level) evidenceKlintworth GK. The molecular genetics of the corneal dystrophies--current status. Frontiers in bioscience : a journal and virtual library. 2003 May 1:8():d687-713 [PubMed PMID: 12700042]
Weiss JS, Møller HU, Aldave AJ, Seitz B, Bredrup C, Kivelä T, Munier FL, Rapuano CJ, Nischal KK, Kim EK, Sutphin J, Busin M, Labbé A, Kenyon KR, Kinoshita S, Lisch W. IC3D classification of corneal dystrophies--edition 2. Cornea. 2015 Feb:34(2):117-59. doi: 10.1097/ICO.0000000000000307. Epub [PubMed PMID: 25564336]
Casal I, Monteiro S, Abreu C, Neves M, Oliveira L, Beirão M. Meretoja's Syndrome: Lattice Corneal Dystrophy, Gelsolin Type. Case reports in medicine. 2017:2017():2843417. doi: 10.1155/2017/2843417. Epub 2017 Jan 31 [PubMed PMID: 28250773]
Level 3 (low-level) evidenceAfshari NA, Mullally JE, Afshari MA, Steinert RF, Adamis AP, Azar DT, Talamo JH, Dohlman CH, Dryja TP. Survey of patients with granular, lattice, avellino, and Reis-Bücklers corneal dystrophies for mutations in the BIGH3 and gelsolin genes. Archives of ophthalmology (Chicago, Ill. : 1960). 2001 Jan:119(1):16-22 [PubMed PMID: 11146721]
Level 3 (low-level) evidencede la Chapelle A, Tolvanen R, Boysen G, Santavy J, Bleeker-Wagemakers L, Maury CP, Kere J. Gelsolin-derived familial amyloidosis caused by asparagine or tyrosine substitution for aspartic acid at residue 187. Nature genetics. 1992 Oct:2(2):157-60 [PubMed PMID: 1338910]
Stewart H, Black GC, Donnai D, Bonshek RE, McCarthy J, Morgan S, Dixon MJ, Ridgway AA. A mutation within exon 14 of the TGFBI (BIGH3) gene on chromosome 5q31 causes an asymmetric, late-onset form of lattice corneal dystrophy. Ophthalmology. 1999 May:106(5):964-70 [PubMed PMID: 10328397]
Kivelä T, Tarkkanen A, Frangione B, Ghiso J, Haltia M. Ocular amyloid deposition in familial amyloidosis, Finnish: an analysis of native and variant gelsolin in Meretoja's syndrome. Investigative ophthalmology & visual science. 1994 Sep:35(10):3759-69 [PubMed PMID: 8088963]
Kivelä T, Tarkkanen A, McLean I, Ghiso J, Frangione B, Haltia M. Immunohistochemical analysis of lattice corneal dystrophies types I and II. The British journal of ophthalmology. 1993 Dec:77(12):799-804 [PubMed PMID: 8110676]
Schmidt EK,Mustonen T,Kiuru-Enari S,Kivelä TT,Atula S, Finnish gelsolin amyloidosis causes significant disease burden but does not affect survival: FIN-GAR phase II study. Orphanet journal of rare diseases. 2020 Jan 17; [PubMed PMID: 31952544]
Nakamura T, Nishida K, Dota A, Adachi W, Yamamoto S, Maeda N, Okada M, Kinoshita S. Gelatino-lattice corneal dystrophy: clinical features and mutational analysis. American journal of ophthalmology. 2000 May:129(5):665-6 [PubMed PMID: 10844062]
Hida T,Tsubota K,Kigasawa K,Murata H,Ogata T,Akiya S, Clinical features of a newly recognized type of lattice corneal dystrophy. American journal of ophthalmology. 1987 Sep 15; [PubMed PMID: 3498366]
Level 3 (low-level) evidenceFujiki K,Hotta Y,Nakayasu K,Yokoyama T,Takano T,Yamaguchi T,Kanai A, A new L527R mutation of the betaIGH3 gene in patients with lattice corneal dystrophy with deep stromal opacities. Human genetics. 1998 Sep; [PubMed PMID: 9799082]
Level 3 (low-level) evidenceDighiero P, Niel F, Ellies P, D'Hermies F, Savoldelli M, Renard G, Delpech M, Valleix S. Histologic phenotype-genotype correlation of corneal dystrophies associated with eight distinct mutations in the TGFBI gene. Ophthalmology. 2001 Apr:108(4):818-23 [PubMed PMID: 11297504]
Level 2 (mid-level) evidenceSchmitt-Bernard CF, Guittard C, Arnaud B, Demaille J, Argiles A, Claustres M, Tuffery-Giraud S. BIGH3 exon 14 mutations lead to intermediate type I/IIIA of lattice corneal dystrophies. Investigative ophthalmology & visual science. 2000 May:41(6):1302-8 [PubMed PMID: 10798644]
Eifrig DE Jr, Afshari NA, Buchanan HW 4th, Bowling BL, Klintworth GK. Polymorphic corneal amyloidosis: a disorder due to a novel mutation in the transforming growth factor beta-induced (BIGH3) gene. Ophthalmology. 2004 Jun:111(6):1108-14 [PubMed PMID: 15177960]
Zenteno JC, Correa-Gomez V, Santacruz-Valdez C, Suarez-Sanchez R, Villanueva-Mendoza C. Clinical and genetic features of TGFBI-linked corneal dystrophies in Mexican population: description of novel mutations and novel genotype-phenotype correlations. Experimental eye research. 2009 Aug:89(2):172-7. doi: 10.1016/j.exer.2009.03.004. Epub 2009 Mar 18 [PubMed PMID: 19303004]
Takács L, Losonczy G, Matesz K, Balogh I, Sohajda Z, Tóth K, Fazakas F, Vereb G, Berta A. TGFBI (BIGH3) gene mutations in Hungary--report of the novel F547S mutation associated with polymorphic corneal amyloidosis. Molecular vision. 2007 Oct 18:13():1976-83 [PubMed PMID: 17982422]
Pihlamaa T, Salmi T, Suominen S, Kiuru-Enari S. Progressive cranial nerve involvement and grading of facial paralysis in gelsolin amyloidosis. Muscle & nerve. 2016 May:53(5):762-9. doi: 10.1002/mus.24922. Epub 2016 Feb 26 [PubMed PMID: 26422119]
Arora R. Deep anterior lamellar keratoplasty or penetrating keratoplasty in lattice corneal dystrophy. Indian journal of ophthalmology. 2018 May:66(5):673-674. doi: 10.4103/ijo.IJO_393_18. Epub [PubMed PMID: 29676313]
Kawamoto K, Morishige N, Yamada N, Chikama T, Nishida T. Delayed corneal epithelial wound healing after penetrating keratoplasty in individuals with lattice corneal dystrophy. American journal of ophthalmology. 2006 Jul:142(1):173-4 [PubMed PMID: 16815275]
Level 2 (mid-level) evidenceSteger B, Romano V, Biddolph S, Willoughby CE, Batterbury M, Kaye SB. Femtosecond Laser-Assisted Lamellar Keratectomy for Corneal Opacities Secondary to Anterior Corneal Dystrophies: An Interventional Case Series. Cornea. 2016 Jan:35(1):6-13. doi: 10.1097/ICO.0000000000000665. Epub [PubMed PMID: 26509759]
Level 2 (mid-level) evidenceKawashima M, Kawakita T, Den S, Shimmura S, Tsubota K, Shimazaki J. Comparison of deep lamellar keratoplasty and penetrating keratoplasty for lattice and macular corneal dystrophies. American journal of ophthalmology. 2006 Aug:142(2):304-9 [PubMed PMID: 16876513]
Level 2 (mid-level) evidenceShousha MA, Yoo SH, Kymionis GD, Ide T, Feuer W, Karp CL, O'Brien TP, Culbertson WW, Alfonso E. Long-term results of femtosecond laser-assisted sutureless anterior lamellar keratoplasty. Ophthalmology. 2011 Feb:118(2):315-23. doi: 10.1016/j.ophtha.2010.06.037. Epub [PubMed PMID: 20869117]
Level 2 (mid-level) evidenceUnal M, Arslan OS, Atalay E, Mangan MS, Bilgin AB. Deep anterior lamellar keratoplasty for the treatment of stromal corneal dystrophies. Cornea. 2013 Mar:32(3):301-5. doi: 10.1097/ICO.0b013e31825718ca. Epub [PubMed PMID: 22790186]
Level 2 (mid-level) evidenceDinh R, Rapuano CJ, Cohen EJ, Laibson PR. Recurrence of corneal dystrophy after excimer laser phototherapeutic keratectomy. Ophthalmology. 1999 Aug:106(8):1490-7 [PubMed PMID: 10442892]
Level 2 (mid-level) evidenceFagerholm P. Phototherapeutic keratectomy: 12 years of experience. Acta ophthalmologica Scandinavica. 2003 Feb:81(1):19-32 [PubMed PMID: 12631015]
Orndahl M, Fagerholm P, Fitzsimmons T, Tengroth B. Treatment of corneal dystrophies with excimer laser. Acta ophthalmologica. 1994 Apr:72(2):235-40 [PubMed PMID: 8079631]
Hieda O, Kawasaki S, Yamamura K, Nakatsukasa M, Kinoshita S, Sotozono C. Clinical outcomes and time to recurrence of phototherapeutic keratectomy in Japan. Medicine. 2019 Jul:98(27):e16216. doi: 10.1097/MD.0000000000016216. Epub [PubMed PMID: 31277131]
Level 2 (mid-level) evidenceLee J, Kim JH, Lee D, Chang JW, Shin JY, Seo JW, Seo MH, Moon NJ. Long-term clinical outcome of femtosecond laser-assisted lamellar keratectomy with phototherapeutic keratectomy in anterior corneal stromal dystrophy. The British journal of ophthalmology. 2018 Jan:102(1):31-36. doi: 10.1136/bjophthalmol-2017-310189. Epub 2017 Jun 13 [PubMed PMID: 28611133]
Level 2 (mid-level) evidenceLu Y, Yang L, Ge Y, Chen X, Huang Z. Femtosecond laser-assisted anterior lamellar keratoplasty for the treatment of stromal corneal pathology. BMC ophthalmology. 2015 Mar 1:15():15. doi: 10.1186/s12886-015-0009-z. Epub 2015 Mar 1 [PubMed PMID: 25884506]
Friedhofer H,Vassiliadis AH,Scarpa MB,Luitgards BF,Gemperli R, Meretoja Syndrome: General Considerations and Contributions of Plastic Surgery in Surgical Treatment. Aesthetic surgery journal. 2017 Dec 13; [PubMed PMID: 29149274]
Courtney DG, Atkinson SD, Moore JE, Maurizi E, Serafini C, Pellegrini G, Black GC, Manson FD, Yam GH, Macewen CJ, Allen EH, McLean WH, Moore CB. Development of allele-specific gene-silencing siRNAs for TGFBI Arg124Cys in lattice corneal dystrophy type I. Investigative ophthalmology & visual science. 2014 Feb 18:55(2):977-85. doi: 10.1167/iovs.13-13279. Epub 2014 Feb 18 [PubMed PMID: 24425855]
Williams KA, Irani YD. Gene Therapy and Gene Editing for the Corneal Dystrophies. Asia-Pacific journal of ophthalmology (Philadelphia, Pa.). 2016 Jul-Aug:5(4):312-6. doi: 10.1097/APO.0000000000000215. Epub [PubMed PMID: 27488074]
Van Overbeke W, Verhelle A, Everaert I, Zwaenepoel O, Vandekerckhove J, Cuvelier C, Derave W, Gettemans J. Chaperone nanobodies protect gelsolin against MT1-MMP degradation and alleviate amyloid burden in the gelsolin amyloidosis mouse model. Molecular therapy : the journal of the American Society of Gene Therapy. 2014 Oct:22(10):1768-78. doi: 10.1038/mt.2014.132. Epub 2014 Jul 15 [PubMed PMID: 25023329]
Level 3 (low-level) evidenceMohan RR, Tovey JC, Sharma A, Tandon A. Gene therapy in the cornea: 2005--present. Progress in retinal and eye research. 2012 Jan:31(1):43-64. doi: 10.1016/j.preteyeres.2011.09.001. Epub 2011 Sep 28 [PubMed PMID: 21967960]
Mehta JS, Kocaba V, Soh YQ. The future of keratoplasty: cell-based therapy, regenerative medicine, bioengineering keratoplasty, gene therapy. Current opinion in ophthalmology. 2019 Jul:30(4):286-291. doi: 10.1097/ICU.0000000000000573. Epub [PubMed PMID: 31045881]
Level 3 (low-level) evidenceGibson DJ, Tuli SS, Schultz GS. Dual-Phase Iontophoresis for the Delivery of Antisense Oligonucleotides. Nucleic acid therapeutics. 2017 Aug:27(4):238-250. doi: 10.1089/nat.2016.0654. Epub 2017 Apr 4 [PubMed PMID: 28375679]
Berdugo M, Valamanesh F, Andrieu C, Klein C, Benezra D, Courtois Y, Behar-Cohen F. Delivery of antisense oligonucleotide to the cornea by iontophoresis. Antisense & nucleic acid drug development. 2003 Apr:13(2):107-14 [PubMed PMID: 12804037]
Level 3 (low-level) evidenceSchoch KM, Miller TM. Antisense Oligonucleotides: Translation from Mouse Models to Human Neurodegenerative Diseases. Neuron. 2017 Jun 21:94(6):1056-1070. doi: 10.1016/j.neuron.2017.04.010. Epub [PubMed PMID: 28641106]
Taketani Y, Kitamoto K, Sakisaka T, Kimakura M, Toyono T, Yamagami S, Amano S, Kuroda M, Moore T, Usui T, Ouchi Y. Repair of the TGFBI gene in human corneal keratocytes derived from a granular corneal dystrophy patient via CRISPR/Cas9-induced homology-directed repair. Scientific reports. 2017 Dec 1:7(1):16713. doi: 10.1038/s41598-017-16308-2. Epub 2017 Dec 1 [PubMed PMID: 29196743]