Introduction
Congenital poikiloderma, also known as Rothmund-Thomson syndrome (RTS), is a rare genodermatosis of autosomal recessive (AR) inheritance characterized by a typical erythema facial (poikiloderma) of early-onset, associated with different clinical features including short stature, sparse scalp hair, absent or sparse eyelashes or eyebrows, juvenile cataracts, skeletal abnormalities, juvenile cataract, premature aging, and susceptibility to osteosarcoma. See Image. Poikiloderma Congenitale. There are 2 types of RTS. Type 1 is characterized by rapidly progressive, bilateral, juvenile cataracts. In contrast, congenital bone abnormalities and an increased risk of osteosarcoma in childhood and squamous cell carcinoma at an older age characterize type 2.[1][2]
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
The gene responsible for type 1 RTS has not yet been found. Type 2 RTS is due to homozygous or heterozygous composite mutations of the RECQL4 gene of the RecQ-helicase family (8q24.3, detected in 60% to 65% of patients). The RECQL4 gene encodes a protein DNA helicase RecQ involved in the replication and repair of DNA and telomeres.[2]
Epidemiology
The prevalence of congenital poikiloderma is unknown, but about 300 cases have been described in the literature, and type 2 RTS accounts for almost two-thirds of cases. Given the AR inheritance, most reported cases were isolated, but cases involving more than 1 person in the same family were also reported. The gender ratio equals 1, but a male or female predominance has been noted in some series. Finally, congenital poikiloderma has been reported in all ethnic groups.[2]
Pathophysiology
RECQL4, whose mutation is responsible for most congenital poikiloderma cases, belongs to the RECQ DNA helicase family. In this family, gene mutations RECQL2 (BLM), WRN, and RECQL4 are responsible for Bloom, Werner, and type 2 RTS, respectively. The origin of type 1 RTS is still unknown. The protein product of RECQL4 plays an essential role in maintaining the integrity of the human genome. Indeed, it is essential in DNA replication, damage repair, homologous recombination, and maintenance of mitochondrial and telomere DNA integrity. The alteration of these functions is at the origin of the poikiloderma, premature age, and different clinical features that characterize congenital poikiloderma. Apart from type 2 RTS, the mutation of the same gene RECQL4 is at the origin of 2 other rare syndromes, namely Baller-Gerold syndrome and RAPADILINO syndrome, which have common signs.[1][2]
Histopathology
In cases of histological examination of a skin specimen, one finds the usual signs of poikiloderma, namely hyperkeratosis, epidermal atrophy, vacuolization of the basal layer, rare apoptotic bodies, many telangiectatic vessels, pigment incontinence with dermal melanophages, and an inflammatory infiltrate of the superficial dermis. Biopsy of verrucous lesions shows significant hyperkeratosis, normal or thickened epidermis, and dyskeratotic cells.[2]
History and Physical
The dermatologist generally suspects the diagnosis of congenital poikiloderma since the first clinical signs of this syndrome affect organs of ectodermal and mesodermal origin, namely skin, hair, nails, and teeth. The involvement of other organs is rarer and later.
Skin Features
The skin is usually normal at birth, but erythema of the cheeks appears between the third and sixth month of life and then extends to the extremities and possibly to the buttocks. The trunk and abdomen are usually spared. In the case of intense erythema, swelling and blistering can appear. The evolution is towards punctate skin atrophy with reticular hypo- and hyperpigmentation and the development of permanent telangiectasias, realizing the typical appearance of poikiloderma that persists throughout life. Recognition of the age of onset, evolution, and stabilization of this poikiloderma may be an important argument for differentiating RTS from other syndromes that include poikilothermic features. Later, there may be latte milk cafe au lait spots. Premature aging of the skin has been reported in some subjects. One-third of individuals had hyperkeratotic palmoplantar lesions. Sparse, thin, or absent hair on the scalp, eyebrows, or eyelashes are common signs. Nail anomalies such as dystrophy or pachyonychia are also frequent. Dental abnormalities are usual and include microdontia, rudimentary teeth, crown formations, and an increased incidence of caries.
Ocular Involvement
Bilateral cataracts are the most common ocular sign and develop in the first years of life. They are subcapsular, can lead to blindness, and characterize type 1 RTS. Other ocular abnormalities have been reported, including corneal atrophy, exophthalmia, congenital bilateral glaucoma, retinal atrophy, strabismus, photophobia, and blue sclerae.
Skeletal Abnormalities
They are common (68% in the literature), including saddle noses, frontal bossing, and abnormalities of the long bones. All patients with bone abnormalities had a RECQL4 mutation that explains the predisposition of this mutation to osteosarcoma development.
Systemic Manifestations
They include growth anomalies with low birth weight and short stature, mental retardation, and sensorineural deafness, anomalies of the gastrointestinal system such as pyloric stenosis and anal atresia, bronchiectasis with an increased incidence of lower respiratory tract infections, hematological signs such as progressive leukopenia and chronic hypochromic anemia.[1][3][4][5][6]
Malignancies
The major risk in congenital poikiloderma is the development of neoplasia, which is life-threatening. Bone (30%) and skin (5%) cancers are the most common. Osteosarcoma is the most common mesenchymal cancer. It has the same characteristics as the sporadic bone but of earlier onset (14 versus 17 years). Cases of multiple osteosarcomas have been reported with a more frequent incidence than sporadic osteosarcoma (17.9 to 25.6% versus 0.4% to 10%). The most common skin cancer is squamous cell carcinoma. Other skin cancers (basal cell carcinoma, Bowen disease, melanoma), hematologic (myelodysplasia, Hodgkin and non-Hodgkin lymphomas, acute myeloid leukemia), and visceral (fibrosarcoma, gastric carcinoma) have been reported rarely.[7][8][9][10]
Evaluation
Histological examination of the cutaneous lesions is not necessary. This diagnostic contribution is modest. Radiological investigation of skeletal abnormalities should be systematic because clinical examination may not detect them. Data from the anamnesis and clinical examination guide the rest of the biological and radiological explorations.
Treatment / Management
The management of congenital poikiloderma requires long-term follow-up, which is performed by an interprofessional team, including a dermatologist, an ophthalmologist, an oncologist, and an orthopedic surgeon. The surveillance is done at least twice a year, looking for skin changes, a cataract, or a bone tumor. Treating skin manifestations must be preventive by effective external photoprotection, including wearing long sleeves, applying sunscreens with a high protection index, and adopting healthy behavior to avoid maximum sun exposure. The telangiectasias already shown can be treated by the pulsed dye laser. Cataract treatment is surgical. The orthopedic surgeon and the oncologist should discuss and manage bone tumors. Other abnormalities that can be seen in the RTS, namely stomatological and respiratory abnormalities, require specialized and early management.[7][8][9](B2)
Differential Diagnosis
The diagnosis of congenital poikiloderma is not always obvious. Some genodermatoses may have a fairly similar clinical presentation with a poikiloderma. The differential diagnosis includes:
- Bloom syndrome (AR)
- Werner syndrome (AR)
- Cockayne syndrome
- Xeroderma pigmentosum (AR)
- Fanconi anemia (AR)
Prognosis
The prognosis is variable. Life expectancy is normal if there is no cancer; whereas, the evolution of patients with malignant diseases depends on the quality and frequency of cancer screening and treatment. Patients with type 2 RTS should be monitored regularly because of the increased risk of developing cancer. Management includes laser treatment of telangiectatic lesions, annual ophthalmologic examination, and radiological examination for bone pain, lameness, or fracture suggestive of osteosarcoma. Increased sensitivity to the effects of chemotherapy is suspected, with a risk of secondary malignancy (risk of 5% of developing skin cancer).[7][9]
Complications
The primary complication accompanying RTS is cancer susceptibility.[11]
Deterrence and Patient Education
Patients with congenital poikiloderma need to understand that sun exposure increases their risk of skin cancer. Therefore, sun exposure protection is crucial, including remaining in full shade, wearing clothing that covers the exposed skin, and using broad-spectrum sunscreens. Blindness secondary to cataract development is another potential complication. Genetic counseling is also a consideration for family members.
Pearls and Other Issues
There is a problem of genotype/phenotype correlation in the congenital poikiloderma or RTS group. The mutation of the RECQL4 gene was not detected in type 1 RTS, and it was only detected in two-thirds of type 2 RTS cases. The risk of osteosarcoma is associated with type 2 RTS. According to many studies, it is directly associated with at least 1 mutation of the RECQL4 gene since this mutation is not detected in a third of cases. It is now known that there are other syndromes associated with a mutation of the same gene, namely the RAPADILINO syndrome, in which the risk of cancer is much lower. Cataract characterizes type 1 RTS, but the mutation related to this association is not yet identified.
Enhancing Healthcare Team Outcomes
Congenital poikiloderma is a rare genetic disease that requires early diagnosis and adequate multispecialty management. The dermatologist's role is important because the poikiloderma is a fundamental sign that allows them to suspect the diagnosis. The advice on effective and long-term photoprotection is of major importance. The early detection of cataracts and their adequate management by the ophthalmologist in type 1 RTS is necessary to avoid the evolution towards blindness with its heavy consequences both medically and socially. Also, screening for bone abnormalities in type 2 RTS is fundamental. This has consequences on the musculoskeletal system, but above all, a predictive value on the occurrence of osteosarcoma is the major risk in type 2 RTS. Diagnosing this neoplasia and its treatment by orthopedists must be early and adequate. Psychological care is also critical since the disease can alter the patient's quality of life. Nurses and primary care clinicians must be aware of the disease and treatment to support the patient and their families appropriately.
Media
(Click Image to Enlarge)
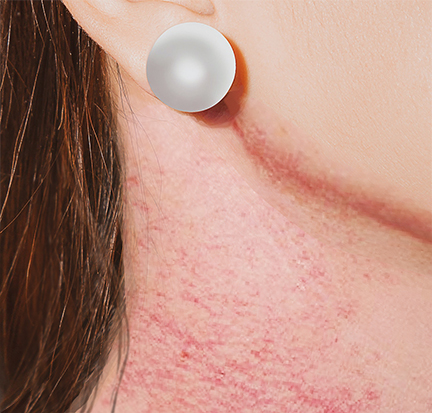
Poikiloderma Congenitale
Contributed by K Humphreys
References
Colombo EA, Locatelli A, Cubells Sánchez L, Romeo S, Elcioglu NH, Maystadt I, Esteve Martínez A, Sironi A, Fontana L, Finelli P, Gervasini C, Pecile V, Larizza L. Rothmund-Thomson Syndrome: Insights from New Patients on the Genetic Variability Underpinning Clinical Presentation and Cancer Outcome. International journal of molecular sciences. 2018 Apr 6:19(4):. doi: 10.3390/ijms19041103. Epub 2018 Apr 6 [PubMed PMID: 29642415]
Manavi S, Mahajan VK. Rothmund-Thomson syndrome. Indian dermatology online journal. 2014 Oct:5(4):518-9. doi: 10.4103/2229-5178.142533. Epub [PubMed PMID: 25396146]
Salih A, Inoue S, Onwuzurike N. Rothmund-Thomson syndrome (RTS) with osteosarcoma due to RECQL4 mutation. BMJ case reports. 2018 Jan 23:2018():. pii: bcr-2017-222384. doi: 10.1136/bcr-2017-222384. Epub 2018 Jan 23 [PubMed PMID: 29367366]
Level 3 (low-level) evidenceChinmayee JT, Meghana GR, Prathiba RK, Ramesh TK. Ophthalmic manifestations in Rothmund-Thomson syndrome: Case report and review of literature. Indian journal of ophthalmology. 2017 Oct:65(10):1025-1027. doi: 10.4103/ijo.IJO_89_17. Epub [PubMed PMID: 29044077]
Level 3 (low-level) evidenceYang JY, Sohn YB, Lee JS, Jang JH, Lee ES. Rare presentation of Rothmund-Thomson syndrome with predominantly cutaneous findings. JAAD case reports. 2017 May:3(3):172-174. doi: 10.1016/j.jdcr.2017.01.023. Epub 2017 Apr 14 [PubMed PMID: 28443301]
Level 3 (low-level) evidenceAdam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, Wang LL, Plon SE. Rothmund-Thomson Syndrome. GeneReviews(®). 1993:(): [PubMed PMID: 20301415]
Miranda AF, Rivera-Monge MD, Farias CC. Rothmund-Thomson syndrome and ocular surface findings: case reports and review of the literature. Arquivos brasileiros de oftalmologia. 2016 May-Jun:79(3):186-8. doi: 10.5935/0004-2749.20160053. Epub [PubMed PMID: 27463631]
Level 3 (low-level) evidenceZils K, Klingebiel T, Behnisch W, Mueller HL, Schlegel PG, Fruehwald M, Suttorp M, Simon T, Werner M, Bielack S. Osteosarcoma in patients with Rothmund-Thomson syndrome. Pediatric hematology and oncology. 2015 Feb:32(1):32-40. doi: 10.3109/08880018.2014.987939. Epub 2014 Dec 31 [PubMed PMID: 25551679]
Level 2 (mid-level) evidenceGuerrero-González GA, Martínez-Cabriales SA, Hernández-Juárez AA, de Jesús Lugo-Trampe J, Espinoza-González NA, Gómez-Flores M, Ocampo-Candiani J. Rothmund-thomson syndrome: a 13-year follow-up. Case reports in dermatology. 2014 May:6(2):176-9. doi: 10.1159/000365625. Epub 2014 Jul 11 [PubMed PMID: 25120469]
Level 3 (low-level) evidenceLarizza L, Roversi G, Volpi L. Rothmund-Thomson syndrome. Orphanet journal of rare diseases. 2010 Jan 29:5():2. doi: 10.1186/1750-1172-5-2. Epub 2010 Jan 29 [PubMed PMID: 20113479]
Narayan S, Fleming C, Trainer AH, Craig JA. Rothmund-Thomson syndrome with myelodysplasia. Pediatric dermatology. 2001 May-Jun:18(3):210-2 [PubMed PMID: 11438000]
Level 3 (low-level) evidence