Introduction
Juvenile myoclonic epilepsy (JME), otherwise known as Janz syndrome and impulsive petit mal, is an idiopathic, hereditary, and generalized form of epilepsy. It was first described by Herpin in 1867, later on by Janz and Christian in 1957 as 'impulsive petit mal,' and by Lund in 1975 as JME. Its characteristics are the presence of absence, myoclonic, and generalized tonic-clonic seizures.
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
JME falls into the classification of an idiopathic as well as hereditary (positive family history in approximately 50% of cases) disorder.
Epidemiology
JME is one of the most common childhood/juvenile epilepsy syndromes accounting for approximately 5%-10% of all cases of epilepsy.[1][2] JME is seen in both sexes equally, although some studies have reported a higher incidence in females.[3] JME usually manifests between 12 and 18 years of age.[4]
Pathophysiology
The pathophysiology of JME has both idiopathic and hereditary components. The genetics of inheritance is not fully understood, but a multifactorial mechanism is suspected. CACNB4, EFHC1, GABRA1 are some of the genes that carry known associations with JME.[5][6] Although magnetic resonance imaging (MRI) of the brain is unremarkable in classic JME cases, there are reports of structural defects as a possible cause of JME.[7][8][9]
History and Physical
The majority of patients with JME start having seizures between the age of 12 to 18 with a mean age of onset around 15 years. But, the onset can be early or later with an age range between 5-34 years.[4][10] As mentioned in the introduction, JME is characterized by the presence of absence, myoclonic, and generalized tonic-clonic (GTC) seizures. All patients with JME have myoclonic seizures, 85%-90% of patients have GTC seizures, and about 20-40% of patients have absence seizures.[10][11][12]
Myoclonic seizures are the hallmark of this condition and are necessary for the diagnosis of JME. Only occasionally they will manifest as the only seizure type. They are described as brief, usually bilateral, jerking movements of the arms and sometimes legs with preservation of consciousness. The misdiagnosis of myoclonic seizures is possible as twitches or anxiety/nervousness. Many a time, increasing intensity and frequency of myoclonic jerks in clusters evolve into GTC seizures. GTC seizures usually occur a few months after the onset of myoclonic seizures. Absence seizures are generally the first seizures to manifest in JME. They tend to occur 3 to 5 years before the onset of myoclonic or GTC seizures, sometimes as early as 5 to 6 years of age. The early and isolated occurrence of absence seizures frequently leads to a diagnosis of childhood or juvenile absence epilepsy. Seizures with JME occur 30 minutes to an hour after morning awakening, especially the myoclonic seizures. Usual triggers for JME are sleep deprivation, alcohol intake, emotional stress, anxiety, fatigue. A large proportion (30%-40%) of JME patients exhibit photosensitivity and flashing lights, sunlight, TV, and computer can trigger seizures. Photosensitive patients tend to have an earlier onset of seizures.
Physical examination remains normal, and cognition well preserved. Studies suggest an increased prevalence of psychiatric disorders, including anxiety, mood disorder, and personality disorders in patients with JME.[13][14][15]
Evaluation
History provides most of the clues necessary for a diagnosis of JME. Physical examination is generally unremarkable. An electroencephalogram (EEG) provides supporting evidence for diagnosing JME. An interictal EEG is abnormal in the majority of patients with JME.[16] If the routine EEG is normal, an overnight and sleep-deprived EEG is abnormal in almost every patient. The abnormalities are seen mostly during the transition from sleep to awakening.[17] The typical EEG in JME shows diffuse, symmetric, bilateral 4 to 6 hertz (Hz) polyspike and wave discharges with a fronto-central predominance. The background has normal alpha rhythm, and approximately half of the patient population will have focal or asymmetric abnormalities.[18] Ictal or continuous EEG recording shows 10-16 Hz polyspike discharges with myoclonic jerks. The spikes correlate with myoclonic jerks.[19] The EEG findings in GTC (low voltage fast activity with spike and wave discharges) and absence seizures (generalized 3 Hz spike and wave discharges) associated with JME are similar to those seen with other epilepsies. Since it is known to be photosensitive epilepsy, photic stimulation provides enhanced yield on EEG in JME.
If history, examination, and EEG are indicative of JME, imaging (with magnetic resonance imaging-MRI) is usually unnecessary as it tends to be normal.[20] Although, there are reports/studies about structural abnormalities in the cortex and subcortical gray matter in JME patients. There have been reports on JME patients showing abnormalities on electrophysiological tests and advanced imaging techniques such as magnetic resonance spectroscopy (MRS) and positron emission tomography (PET) as well.
Treatment / Management
JME is relatively easy to control with anti-epilepsy drugs (AEDs), and most patients respond to monotherapy. Valproic acid is the most effective medication and drug of choice for treatment of JME.[21] It is a broad-spectrum medication and treats all seizure types of JME. Its use requires a great deal of caution in women of childbearing age, given its well known teratogenic potential.[22] Other treatment options include levetiracetam, lamotrigine, topiramate, and zonisamide.[23][24][25][26][27] Lamotrigine can lead to a worsening in myoclonic seizures but is still widely used for the treatment of JME as it controls the other seizure types.[23][28] If only absence seizures are present, ethosuximide can be a therapy choice. Clonazepam is effective against myoclonic jerks. Contraindicated agents include carbamazepine, oxcarbazepine, phenytoin (sodium channel blocking agents), given their potential to exacerbate myoclonic and absence seizures. But, they can be useful for the treatment of GTC seizures in JME.[29] Other medications to avoid include vigabatrin, tiagabine, gabapentin, pregabalin, and primidone.[30] JME is intractable in only a few patients, and combination therapy with or without vagus nerve stimulation (VNS) is an option in such cases.[31](A1)
Differential Diagnosis
Many other epilepsy syndromes have similar features and resemble JME at various stages of their progression. Differential diagnosis includes childhood or juvenile absence epilepsy, eyelid myoclonia with absences, progressive myoclonic epilepsy, photosensitive occipital epilepsy, epilepsy with grand mal seizures upon awakening, hypnogogic myoclonus (hypnic jerk), and non-epileptic seizures.
Prognosis
Seizures are usually well controlled with medications in a great majority of patients with JME and generally improve after the fourth decade of life.[32] But, JME requires lifelong treatment (even after a long seizure-free interval), as the chances of relapse are high if medications are discontinued.[33]
Enhancing Healthcare Team Outcomes
While JME management is chiefly under a neurologist, the follow up is usually under the primary caregiver or nurse practitioner. The key to the prevention of seizures is patient education by the interprofessional team. Pharmacists should educate patients and their families about the importance of compliance and side effects. Neuroscience nurses monitor patients, educate, and keep the team informed as to changes in status. [Level 5]
Avoidance of precipitating factors/triggers should be a strong emphasis. These include avoiding alcohol use, fatigue, stress, sleep deprivation, flashing lights in any form, medication non-compliance, hunger, extremes of temperature, drug use. Patients should also be advised/educated with precautions regarding driving, operating heavy machinery, swimming, avoiding heights/being near fires alone. The treatment is usually life long (even after a long seizure-free interval), as the chances of relapse are high if medications are discontinued.[33] As with any epilepsy syndrome, risks versus benefits of medications during pregnancy should be a point of discussion in detail with the patient.
Media
(Click Image to Enlarge)
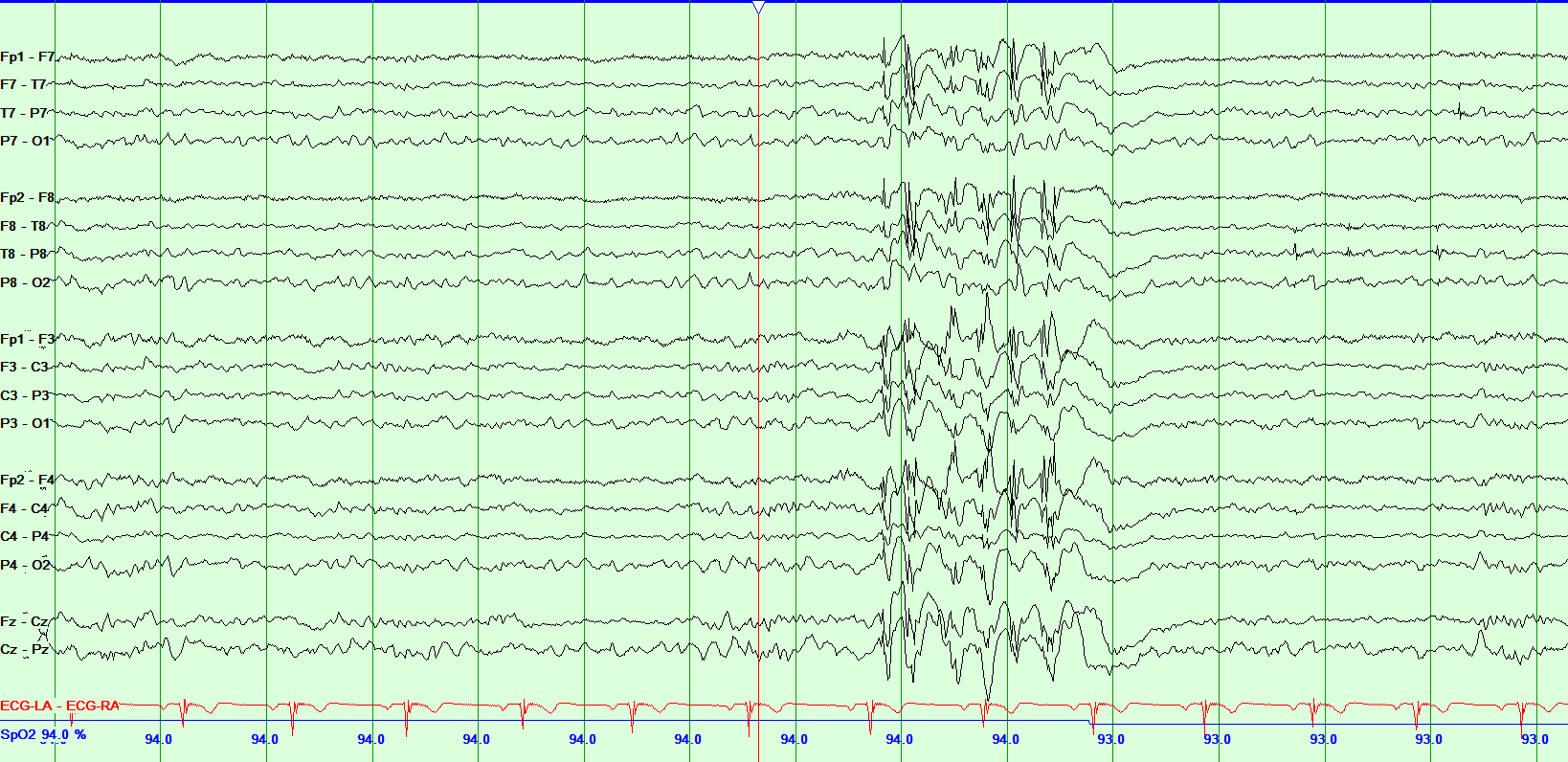
Juvenile Myoclonic Epilepsy Contributed by Chaitanya Amrutkar, MD
References
Panayiotopoulos CP, Obeid T, Tahan AR. Juvenile myoclonic epilepsy: a 5-year prospective study. Epilepsia. 1994 Mar-Apr:35(2):285-96 [PubMed PMID: 8156946]
Hauser WA. The prevalence and incidence of convulsive disorders in children. Epilepsia. 1994:35 Suppl 2():S1-6 [PubMed PMID: 8275976]
Janz D. Epilepsy with impulsive petit mal (juvenile myoclonic epilepsy). Acta neurologica Scandinavica. 1985 Nov:72(5):449-59 [PubMed PMID: 3936330]
Canevini MP,Mai R,Di Marco C,Bertin C,Minotti L,Pontrelli V,Saltarelli A,Canger R, Juvenile myoclonic epilepsy of Janz: clinical observations in 60 patients. Seizure. 1992 Dec [PubMed PMID: 1344779]
Buraei Z, Yang J. The ß subunit of voltage-gated Ca2+ channels. Physiological reviews. 2010 Oct:90(4):1461-506. doi: 10.1152/physrev.00057.2009. Epub [PubMed PMID: 20959621]
Level 3 (low-level) evidenceDelgado-Escueta AV, Koeleman BP, Bailey JN, Medina MT, Durón RM. The quest for juvenile myoclonic epilepsy genes. Epilepsy & behavior : E&B. 2013 Jul:28 Suppl 1():S52-7. doi: 10.1016/j.yebeh.2012.06.033. Epub [PubMed PMID: 23756480]
Level 3 (low-level) evidenceLiu M, Concha L, Beaulieu C, Gross DW. Distinct white matter abnormalities in different idiopathic generalized epilepsy syndromes. Epilepsia. 2011 Dec:52(12):2267-75. doi: 10.1111/j.1528-1167.2011.03313.x. Epub 2011 Nov 16 [PubMed PMID: 22092238]
Tae WS, Kim SH, Joo EY, Han SJ, Kim IY, Kim SI, Lee JM, Hong SB. Cortical thickness abnormality in juvenile myoclonic epilepsy. Journal of neurology. 2008 Apr:255(4):561-6. doi: 10.1007/s00415-008-0745-6. Epub 2008 Jan 31 [PubMed PMID: 18227991]
Level 2 (mid-level) evidenceMory SB, Betting LE, Fernandes PT, Lopes-Cendes I, Guerreiro MM, Guerreiro CA, Cendes F, Li LM. Structural abnormalities of the thalamus in juvenile myoclonic epilepsy. Epilepsy & behavior : E&B. 2011 Aug:21(4):407-11. doi: 10.1016/j.yebeh.2011.05.018. Epub 2011 Jun 23 [PubMed PMID: 21700499]
Vijai J, Cherian PJ, Stlaja PN, Anand A, Radhakrishnan K. Clinical characteristics of a South Indian cohort of juvenile myoclonic epilepsy probands. Seizure. 2003 Oct:12(7):490-6 [PubMed PMID: 12967578]
Level 2 (mid-level) evidenceJayalakshmi SS, Srinivasa Rao B, Sailaja S. Focal clinical and electroencephalographic features in patients with juvenile myoclonic epilepsy. Acta neurologica Scandinavica. 2010 Aug:122(2):115-23. doi: 10.1111/j.1600-0404.2009.01270.x. Epub 2009 Oct 19 [PubMed PMID: 19845556]
Level 2 (mid-level) evidenceMurthy JM, Rao CM, Meena AK. Clinical observations of juvenile myoclonic epilepsy in 131 patients: a study in South India. Seizure. 1998 Feb:7(1):43-7 [PubMed PMID: 9548225]
Level 2 (mid-level) evidenceTrinka E, Kienpointner G, Unterberger I, Luef G, Bauer G, Doering LB, Doering S. Psychiatric comorbidity in juvenile myoclonic epilepsy. Epilepsia. 2006 Dec:47(12):2086-91 [PubMed PMID: 17201708]
Level 2 (mid-level) evidenceMoschetta S, Fiore LA, Fuentes D, Gois J, Valente KD. Personality traits in patients with juvenile myoclonic epilepsy. Epilepsy & behavior : E&B. 2011 Aug:21(4):473-7. doi: 10.1016/j.yebeh.2011.03.036. Epub 2011 Jun 17 [PubMed PMID: 21683658]
Somayajula S, Vooturi S, Jayalakshmi S. Psychiatric disorders among 165 patients with juvenile myoclonic epilepsy in India and association with clinical and sociodemographic variables. Epilepsy & behavior : E&B. 2015 Dec:53():37-42. doi: 10.1016/j.yebeh.2015.09.024. Epub 2015 Oct 28 [PubMed PMID: 26519664]
Baise-Zung C, Guilhoto LM, Grossmann RM. Juvenile myoclonic epilepsy: non-classic electroencephalographical presentation in adult patients. European journal of neurology. 2006 Feb:13(2):171-5 [PubMed PMID: 16490048]
Level 2 (mid-level) evidenceDhanuka AK, Jain BK, Daljit S, Maheshwari D. Juvenile myoclonic epilepsy: a clinical and sleep EEG study. Seizure. 2001 Jul:10(5):374-8 [PubMed PMID: 11488650]
Létourneau K, Cieuta-Walti C, Deacon C. Epileptiform asymetries and treatment response in juvenile myoclonic epilepsy. The Canadian journal of neurological sciences. Le journal canadien des sciences neurologiques. 2010 Nov:37(6):826-30 [PubMed PMID: 21059546]
Level 2 (mid-level) evidenceHrachovy RA, Frost JD Jr. The EEG in selected generalized seizures. Journal of clinical neurophysiology : official publication of the American Electroencephalographic Society. 2006 Aug:23(4):312-32 [PubMed PMID: 16885706]
Anderson J, Hamandi K. Understanding juvenile myoclonic epilepsy: contributions from neuroimaging. Epilepsy research. 2011 May:94(3):127-37. doi: 10.1016/j.eplepsyres.2011.03.008. Epub 2011 Apr 8 [PubMed PMID: 21482074]
Level 3 (low-level) evidenceChowdhury A, Brodie MJ. Pharmacological outcomes in juvenile myoclonic epilepsy: Support for sodium valproate. Epilepsy research. 2016 Jan:119():62-6. doi: 10.1016/j.eplepsyres.2015.11.012. Epub 2015 Dec 1 [PubMed PMID: 26675554]
Tomson T, Battino D, Perucca E. Valproic acid after five decades of use in epilepsy: time to reconsider the indications of a time-honoured drug. The Lancet. Neurology. 2016 Feb:15(2):210-218. doi: 10.1016/S1474-4422(15)00314-2. Epub 2015 Dec 5 [PubMed PMID: 26655849]
Bodenstein-Sachar H, Gandelman-Marton R, Ben-Zeev B, Chapman J, Blatt I. Outcome of lamotrigine treatment in juvenile myoclonic epilepsy. Acta neurologica Scandinavica. 2011 Jul:124(1):22-7. doi: 10.1111/j.1600-0404.2010.01472.x. Epub 2011 Jan 6 [PubMed PMID: 21208196]
Level 2 (mid-level) evidenceVerrotti A, Cerminara C, Coppola G, Franzoni E, Parisi P, Iannetti P, Aloisi P, Tozzi E, Cusmai R, Vigevano F, Chiarelli F, Curatolo P. Levetiracetam in juvenile myoclonic epilepsy: long-term efficacy in newly diagnosed adolescents. Developmental medicine and child neurology. 2008 Jan:50(1):29-32. doi: 10.1111/j.1469-8749.2007.02009.x. Epub [PubMed PMID: 18173626]
Levisohn PM, Holland KD. Topiramate or valproate in patients with juvenile myoclonic epilepsy: a randomized open-label comparison. Epilepsy & behavior : E&B. 2007 Jun:10(4):547-52 [PubMed PMID: 17482520]
Level 3 (low-level) evidenceLiu J, Wang LN, Wang YP. Topiramate monotherapy for juvenile myoclonic epilepsy. The Cochrane database of systematic reviews. 2015 Dec 23:(12):CD010008. doi: 10.1002/14651858.CD010008.pub2. Epub 2015 Dec 23 [PubMed PMID: 26695884]
Level 1 (high-level) evidenceKothare SV, Valencia I, Khurana DS, Hardison H, Melvin JJ, Legido A. Efficacy and tolerability of zonisamide in juvenile myoclonic epilepsy. Epileptic disorders : international epilepsy journal with videotape. 2004 Dec:6(4):267-70 [PubMed PMID: 15634623]
Level 2 (mid-level) evidenceBiraben A, Allain H, Scarabin JM, Schück S, Edan G. Exacerbation of juvenile myoclonic epilepsy with lamotrigine. Neurology. 2000 Dec 12:55(11):1758 [PubMed PMID: 11113246]
Knott C, Panayiotopoulos CP. Carbamazepine in the treatment of generalised tonic clonic seizures in juvenile myoclonic epilepsy. Journal of neurology, neurosurgery, and psychiatry. 1994 Apr:57(4):503 [PubMed PMID: 8164005]
Level 3 (low-level) evidenceMantoan L, Walker M. Treatment options in juvenile myoclonic epilepsy. Current treatment options in neurology. 2011 Aug:13(4):355-70. doi: 10.1007/s11940-011-0131-z. Epub [PubMed PMID: 21494841]
Kostov H, Larsson PG, Røste GK. Is vagus nerve stimulation a treatment option for patients with drug-resistant idiopathic generalized epilepsy? Acta neurologica Scandinavica. Supplementum. 2007:187():55-8 [PubMed PMID: 17419830]
Baykan B, Altindag EA, Bebek N, Ozturk AY, Aslantas B, Gurses C, Baral-Kulaksizoglu I, Gokyigit A. Myoclonic seizures subside in the fourth decade in juvenile myoclonic epilepsy. Neurology. 2008 May 27:70(22 Pt 2):2123-9. doi: 10.1212/01.wnl.0000313148.34629.1d. Epub [PubMed PMID: 18505992]
Martínez-Juárez IE, Alonso ME, Medina MT, Durón RM, Bailey JN, López-Ruiz M, Ramos-Ramírez R, León L, Pineda G, Castroviejo IP, Silva R, Mija L, Perez-Gosiengfiao K, Machado-Salas J, Delgado-Escueta AV. Juvenile myoclonic epilepsy subsyndromes: family studies and long-term follow-up. Brain : a journal of neurology. 2006 May:129(Pt 5):1269-80 [PubMed PMID: 16520331]