Introduction
Alkalosis is a pathophysiological condition characterized by the buildup of excess base or alkali in the body, leading to an abnormally high serum pH (arterial pH >7.45), termed alkalemia. This condition represents one end of the acid-base disorder spectrum. There is generally a loss of hydrogen ions (H+) or an excess of bicarbonate ions (HCO3−), with various factors potentially causing either of these mechanisms. Alkalosis can be as life-threatening as acidosis, and severe electrolyte derangements can accompany alkalosis due to transcellular shifts, potentially resulting in rare but severe clinical disorders.
Alkalosis can originate from either respiratory or metabolic causes, with metabolic alkalosis being more common. The renal and pulmonary systems are the primary regulators of serum pH levels. Respiratory alkalosis results in compensatory metabolic acidosis, whereas metabolic alkalosis results in compensatory respiratory acidosis. However, the pH does not typically correct to the normal value of 7.40. Pulmonary compensation is typically immediate, whereas renal compensation can take hours to days.[1]
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
The etiology of alkalosis can be divided into metabolic and respiratory causes.
Metabolic Causes of Alkalosis
Excess loss of hydrogen ions: This primarily occurs due to gastric losses—such as gastric lavage; excessive emesis of gastric contents, including emesis secondary to pyloric stenosis; chronic diarrhea; or congenital chloridorrhea.[2] As part of the digestive process, the gastric parietal cells secrete hydrogen ions, which are combined with bicarbonate secreted by the small intestine. When the secreted hydrogen ions are lost, the secreted bicarbonate becomes part of the extracellular fluid compartment. The bicarbonate is excreted by the kidneys as NaHCO3, leading to volume depletion. The volume depletion activates the renin-angiotensin-aldosterone system, promoting potassium secretion. Low extracellular potassium leads to the exchange of intracellular potassium for extracellular hydrogen, further enhancing alkalosis. In this situation, urine chloride is low, and urine sodium is high due to the secretion of NaHCO3.[3][4]
Increased bicarbonate in the extracellular compartment: This occurs due to excess enteral intake of bicarbonate or alkali, such as in milk-alkali syndrome, or increased parenteral intake of citrate, acetate, or lactate, which can be converted to bicarbonate. Commonly ingested bicarbonate-containing medications include baking soda, sodium bicarbonate tablets, potassium bicarbonate tablets, and potassium citrate tablets. Sodium citrate is also used as an anticoagulant in fresh frozen plasma; however, transfusing large amounts can lead to metabolic alkalosis.[5][6]
Increased renal reabsorption of bicarbonate: Increased renal reabsorption of bicarbonate can also contribute to metabolic alkalosis. Conditions such as primary hyperaldosteronism, renin-secreting tumors, renal artery stenosis, exogenous mineralocorticoids, congenital adrenal hyperplasia, Liddle syndrome, Cushing syndrome, and Cushing disease can all act in this manner.
Classification of Metabolic Alkalosis Causes
Metabolic alkalosis can be examined by classifying its causes into 2 categories—intravascular volume depletion with hypochloremia and intravascular volume expansion with hypokalemia.
Hypovolemic states
- Gastric losses: Gastric losses lead to the loss of HCl and volume contraction, activating the renin-angiotensin-aldosterone system.
- Vomiting
- Nasogastric tube drainage
- Intestinal losses: Intestinal losses result in the loss of potassium and chloride through stool, along with fluid loss.
- Ileostomy output
- Villous adenoma
- Diarrhea, particularly secretory diarrhea
- Congenital chloridorrhea is a rare congenital disorder causing loss of the chloride/bicarbonate transporter and bicarbonate retention.[3]
- Diuretic use: Loop diuretics block the Na+/K+/2Cl− cotransporter (NKCC2) in the thick ascending limb of the loop of Henle in the kidneys, reducing sodium and chloride reabsorption and increasing the excretion of water, sodium, potassium, and chloride. Thiazide diuretics block the Na+/Cl− cotransporter (NCC) in the distal convoluted tubule.[5][7][8]
- Bartter syndrome: This autosomal recessive condition is primarily caused by mutations in the NKCC2 and ROMK transporters in the thick ascending loop of Henle.[9] Excessive distal delivery of sodium results in enhanced distal convoluted tubule sodium reabsorption and exchange with the positively charged potassium or hydrogen ion, which leads to increased loss of potassium in urine and increased hydrogen secretion. Bicarbonate is increased secondary to decreased hydrogen ion secretion due to hyperaldosteronism. In addition, loss of the electropositivity of the thick ascending loop of Henle results in the inability to reabsorb magnesium and calcium through claudin channels, leading to nephrocalcinosis. About 50% of cases present in the first year of life and can present as failure to thrive, polyuria, polydipsia, and vomiting.[10] There are 5 known variants. Please see StatPearls' companion reference, "Bartter Syndrome," for more details.
- Gitelman syndrome: This autosomal recessive syndrome affects the NCC transporter in the distal convoluted tubule. Gitelman syndrome is characterized by metabolic alkalosis, renal potassium wasting, hypokalemia, hypomagnesemia, hypochloremia, and hypocalciuria. Gitelman syndrome is also referred to as familial hypokalemia-hypomagnesemia and generally presents in late childhood or adulthood.[9] The renin-aldosterone axis is highly activated in both Bartter and Gitelman syndromes, leading to hypokalemic metabolic alkalosis. Please see StatPearl's companion reference, "Gitelman Syndrome," for more details.
- Cystic fibrosis: Patients with cystic fibrosis have downregulated pendrin receptors in the B intercalated cells of the collecting tubules, leading to bicarbonate retention and chloride secretion. The renal chloride loss and bicarbonate retention significantly exacerbate metabolic alkalosis in the context of volume depletion.
Hypervolemic states
- Aldosterone excess, renin-secreting tumors, Cushing syndrome, and congenital adrenal hyperplasia: Increased sodium absorption in the collecting ducts results in the excretion of hydrogen and potassium. Potassium loss can worsen metabolic alkalosis by causing potassium to shift from the intracellular to the extracellular space, resulting in an intracellular hydrogen shift. Cortisol binds to aldosterone receptors with high affinity, mainly if not activated by 11-β-dehydrogenase. Defects in 11-β-dehydrogenase or large amounts of natural licorice, which contains glycyrrhizic acid that inhibits this enzyme, can lead to the abovementioned symptoms.
- Liddle syndrome: This rare autosomal dominant condition is characterized by a gain of function in the epithelial sodium channel in the collecting tubule. Affected patients typically present with hypertension, hypokalemia, and metabolic alkalosis—findings that are similar to those observed in other disorders caused by mineralocorticoid excess. Liddle syndrome is characterized by highly resistant hypertension. Please see StatPearls' companion reference, "Liddle Syndrome," for more details.
Please see StatPearls' companion reference, "Hypokalemic Metabolic Alkalosis," for more details (see image. Metabolic Alkalosis Diagram). Notably, the abuse of the above medications, such as diuretics, steroids, or unregulated supplements that may contain unknown ingredients, can mimic these symptoms.
Respiratory Causes of Alkalosis
Carbon dioxide (CO2) is a by-product of aerobic metabolism, and respiratory alkalosis is almost always induced by a process causing hyperventilation. Hypothermia and myxedema coma are two rare, unusual cases of decreased CO2 production. Even in these rare cases, ventilation, such as mechanical ventilation, often has to be fixed for hypocapnia to occur.[1] The vast majority of respiratory alkalosis cases are due to hyperventilation.
Hyperventilation may be due to central, hypoxemic, cardiopulmonary, iatrogenic, or physiologic causes.[1][11]
Central causes:
- Head injury
- Stroke Hyperthyroidism
- Anxiety
- Pain
- Fear
- Drugs and toxins, such as salicylates, beta-agonists, and endotoxin
Hypoxemic causes:
- High altitude
- Pulmonary disease [12]
Cardiopulmonary causes:
- Pulmonary embolism
- Pneumothorax
- Pneumonia
- Acute asthma
- Chronic obstructive pulmonary disease exacerbations
- Cardiogenic edema
Iatrogenic causes:
- Hyperventilation in intubated patients on mechanical ventilation
- Extracorporeal membranous oxygenation
Physiologic causes:
- Pregnancy
- Hyperthermia, including fever
Hypoxic stimulation leads to hyperventilation in an attempt to correct hypoxia at the expense of CO2 loss.[11] Physiological causes of hyperventilation include pregnancy and fever, which increase metabolic rates and lead to hyperventilation. High altitude and accompanying hypoxia can also stimulate respiratory alkalosis.[12] Another physiological cause is hyperthermia, which can cause increased minute ventilation and hypocapnia at rest and during exercise, although the exact mechanisms by which this occurs are not fully elucidated.
Leigh syndrome is a rare, genetic neurodegenerative disorder that causes respiratory alkalosis through centrally driven hyperventilation, and the condition can be exacerbated by oxygen supplementation. This disorder typically presents in infancy or childhood, but late-onset cases have been described. Respiratory alkalosis causes compensatory lactate accumulation. Several studies have shown the adverse effects of alkalosis on the central nervous system, and alkalosis also causes cerebral vasoconstriction. Although this is a rare disease, it provides insight into the physiological effects of respiratory alkalosis, particularly on the brain.[12][13]
Respiratory alkalosis may be an acute process or a chronic process, which can be determined based on the level of metabolic compensation for the respiratory disease. Excess HCO3 levels are buffered to reduce levels and maintain a physiological pH through decreased renal hydrogen ion secretion and increasing HCO3 secretion; however, this metabolic process occurs for days, whereas respiratory disease can adjust CO2 levels in minutes to hours. Therefore, acute respiratory alkalosis is associated with high bicarbonate levels, as there has not been sufficient time to lower the HCO3 levels. Chronic respiratory alkalosis is associated with low to normal HCO3 levels.[14][15]
Epidemiology
Metabolic alkalosis is the most common acid-base disturbance in hospitalized patients and tends to worsen over the hospital course. A large Norwegian study involving intensive care patients found that although most patients were not alkalemic at admission, up to 85% developed alkalosis during their hospital course. Other large studies have also shown metabolic alkalosis to be the most prevalent acid-base disorder, including those with mixed acid-base disorders, and this disorder is highly associated with mortality.[3] Gender distribution appears to be equal, except in cases of infantile pyloric stenosis, where there is an overwhelming male predominance noted.[16]
Pathophysiology
The intracellular pH is between 7.0 and 7.35, making it more acidic than the extracellular blood. The extracellular and intracellular pH levels are assiduously maintained within a narrow range by active mechanisms. The body has a robust buffering system that minimizes pH changes in the initial stages of acid-base derangements. When these buffering systems are overwhelmed, alkalosis may result.[3]
The fundamental pH buffer system in the human body is the bicarbonate/carbon dioxide (HCO3−/CO2) chemical equilibrium system, represented by the following equation: [1]
- H+ + HCO3− <----> H2CO3 <----> CO2 + H2O
This reaction is catalyzed by carbonic anhydrase. HCO3− acts as an alkalotic substance by accepting a hydrogen ion, whereas H2CO3 (carbonic acid) serves as an acidic substance by releasing a hydrogen ion. Therefore, increased bicarbonate or decreased carbon dioxide—resulting in lower levels of H2CO3—can lead to alkalemia. The opposite is also true, where a decrease in bicarbonate or an increase in carbon dioxide induces acidemia. Carbon dioxide levels are physiologically regulated by the pulmonary system through respiration, whereas bicarbonate levels are regulated by the renal system through reabsorption. Therefore, respiratory alkalosis results from a decrease in serum CO2. Although it is theoretically possible to have decreased CO2 production, this illness results from hyperventilation, where CO2 is expelled from the body.[3][17][18]
The kidneys maintain normal acid-base balance through 2 primary mechanisms—bicarbonate reabsorption in the proximal tubule and bicarbonate production in the distal nephron. About 85% to 90% of filtered bicarbonate occurs in the proximal tubule, with the remaining 10% to 15% absorbed in the thick ascending loop of Henle or distal convoluted tubule and 3% to 5% in the distal collecting duct. Although considerably less bicarbonate is absorbed in the distal nephron, this is where a significant portion of regulation and fine-tuning occurs.[7]
Reabsorption of bicarbonate in the proximal tubule is mediated by a Na+/H+ antiporter and the hydrogen-ATPase. The secreted hydrogen ion (H+) combines with luminal bicarbonate (HCO3−), dissociating into carbon dioxide and water catalyzed by luminal carbonic anhydrase. Carbon dioxide enters the proximal tubular cell, where it is catalyzed by cytosolic carbonic anhydrase to produce HCO3. This bicarbonate is then transported into the serum with sodium through the Na+/HCO3− cotransporter (NBCe1).[7]
The thick ascending loop of Henle is the location of a key regulatory channel known as the Na+/K+/2Cl− cotransporter (NKCC2). This transporter absorbs sodium, potassium, and chloride from the lumen into the blood and serves as the site of action for loop diuretics. This segment also contains the Na+/H+ exchanger 3 (NHE3), which absorbs sodium into the cell and the blood and secretes hydrogen into the urine. These processes are powered by the basolateral Na+/K+ ATPase.
Na+ and Cl− typically coexist in a ratio of 1.4:1. In metabolic alkalosis, this balance is disrupted, and chloride is secreted in exchange for bicarbonate. This regulation primarily occurs in the cortical collecting ducts.[4] The cortical collecting ducts contain 3 cell types—type A intercalated cells, type B intercalated cells, and principal cells. The functions are as follows:
- Type A intercalated cells: These cells secrete H+ through hydrogen-ATPases, generating new HCO3− by cytosolic carbonic anhydrase. This new HCO3− is transported into the blood by the basolateral HCO3−/Cl− anion exchange protein 1 (AE1).
- Type B intercalated cells: These cells secrete HCO3− into the tubular lumen in exchange for lumenal Cl− through the apical HCO3−/Cl− exchanger called pendrin. The resultant acid is reabsorbed into the blood through the basolateral hydrogen-ATPase.
- Principal cells: These cells reabsorb sodium through the epithelial sodium channel, and water is absorbed along with sodium through aquaporin 2 channels. Principal cells also secrete potassium. The primary site of action for aldosterone is in these principal cells.
Ammoniagenesis is also part of bicarbonate metabolism. Ammonia (NH3) is a weak base, and, at a physiologic pH, it can acquire H+ from H2O to become ammonium (NH4+), which is eventually secreted in the distal collecting duct type A intercalated cell through the apical hydrogen-ATPase; this antiporter is upregulated by aldosterone. Hypokalemia increases ammoniagenesis, increases the activity of the hydrogen-ATPase, and decreases the activity of type B intercalated cells in the collecting tubule.
Effective arterial blood volume, glomerular filtration rate, serum chloride, and serum potassium concentrations influence bicarbonate reabsorption. In conditions resulting in respiratory alkalosis, the kidney decreases both bicarbonate reabsorption and bicarbonate production as a compensatory mechanism. This process helps maintain the pH of the extracellular compartment to neutralize the effect of low pCO2, which is the primary derangement of respiratory alkalosis. However, the kidneys' complex buffering mechanisms may take several days to achieve full effect, with an expected decrease in bicarbonate levels of 4 to 5 mmol/L for every 10 mm Hg reduction in pCO2.
Conversely, respiratory depression resulting in increased PaCO2 occurs promptly and predictably to buffer the alkalemia resulting from metabolic conditions. Although exact values may vary, expectations are that there is a 0.5 mm Hg increase in PaCO2 per 1 mmol/L increase in HCO3. Alkalemia also causes a shift in the oxyhemoglobin dissociation curve towards the left, thus increasing hemoglobin's affinity for oxygen and decreasing oxygen release to the tissues.[19][20]
Suboptimal potassium intake can correlate with metabolic alkalosis due to rising intracellular sodium and hydrogen levels (in exchange for transporting potassium extracellularly) and a consequent depression in aldosterone levels. When protons shift into the cellular compartment, metabolic alkalosis develops, followed by respiratory center depression of respiratory drive and, ultimately, the purging of bicarbonate by the kidney.
Histopathology
There are no specific histopathological features that are pathognomonic for alkalosis. However, the primary cause of alkalosis may be established by histopathological studies, especially when related to kidney or adrenal disorders.
History and Physical
Alkalosis can present with a wide range of signs and symptoms based on the etiology of alkalosis (respiratory versus metabolic) and the primary condition leading to alkalosis. The exact history and physical examination findings are highly variable, as many pathologies induce alkalosis. In metabolic alkalosis, history may include vomiting, diarrhea, known endocrine disorders, licorice ingestion, or hypertension of unknown cause. In respiratory alkalosis, the history may include mechanical ventilation, trauma, central line catheter insertion, recent surgery, thromboembolic disease, asthma, chronic obstructive pulmonary disease, aspirin ingestion, or a central neurological process.
Symptoms may include dyspnea, fever, chills, peripheral edema, orthopnea, weakness, confusion, light-headedness, dizziness, anxiety, chest pain, numbness, paresthesia, abdominal pain, nausea, vomiting, or tinnitus. Patients with respiratory alkalosis can have associated syncope, tremors, and signs of hyperventilation, along with chest pain and dyspnea.[21]
The primary sign of respiratory alkalosis is an increased respiratory rate. Although a normal respiratory rate is often defined as 20 or fewer breaths/minute, the normal respiratory rate is 12 to 15 breaths/minute.[22] As hyperventilation is the primary cause of all respiratory alkalosis etiologies, many patients complain of shortness of breath.
Alkalosis can lead to significant central nervous system manifestations, ranging from confusion to coma or peripheral neuropathic symptoms such as tremors, tingling, numbness, muscle weakness, twitching, and positive Trousseau and Chvostek signs. Arrhythmias may also present, particularly when associated with hypokalemia and hypocalcemia.[23] Normochloremic metabolic alkalosis is associated with hypertension and is typically the result of syndromes of excess mineralocorticoid production. These generally correlate with signs of volume expansion, hypertension, and hypokalemia.[24]
Failure to thrive in infants or children when associated with polyuria, polydipsia, and vomiting should raise the suspicion of Bartter or Giteleman syndrome. Due to the rarity of these syndromes, they can often be missed. A detailed family history can be helpful, but many cases are sporadic. These patients are sometimes diagnosed with the more common cystic fibrosis. One differentiating factor is that urinary chloride is low in cystic fibrosis.[10] Another specific example of which to be alert is persistent and projectile, non-bilious emesis in a 2 to 6-week-old, otherwise well-appearing infant, which is a hallmark presentation of pyloric stenosis. In addition, evidence shows that infants with metabolic alkalosis who undergo surgery to fix the pyloric stenosis are more likely to have perioperative complications than those without metabolic alkalosis.[2]
Evaluation
A blood gas analysis, preferably arterial, is required to determine alkalosis and whether it appears to be metabolic or respiratory in origin. Ancillary blood tests are necessary, including serum chemistries with electrolytes, blood urea nitrogen, and creatinine. Although elevated bicarbonate levels can suggest metabolic alkalosis, they do not indicate whether this is a primary disturbance or secondary to respiratory acidosis. Hence, a blood gas estimate of pH and pCO2 is also needed.[24] Although arterial blood gas testing is the preferable method, it can be inconvenient or painful for patients if done frequently. As a result, venous blood gases are sometimes used to estimate pCO2. A normal PCO2 value on venous blood gas is 40 to 50 mm Hg.[3]
As noted above, compensatory respiratory acidosis is expected with metabolic alkalosis. Many formulas exist to estimate the expected respiratory compensation. Even with respiratory or metabolic compensation, the initial inciting alkalosis should not normalize to a pH of 7.4. If the pH is close to 7.4, it suggests a mixed-acid base disorder. For example, a patient with severe chronic obstructive pulmonary disease who has vomiting has a loss of acid, creating a metabolic alkalosis; however, the patient may also have an underlying respiratory acidosis. Whether the pH is lower or higher than 7.4 determines the primary disorder. In metabolic alkalosis, the PaCO2 rarely is higher than 55 mm Hg without underlying lung pathology.[3]
In metabolic alkalosis, exact values may vary, but an estimate is that there is about a 0.5 mm Hg increase in PaCO2 per 1 mmol/L increase in HCO3. A more precise formula is that for every 1 mmol/L rise in HCO3 above 24 mmol/L, there should be a 0.6 mm Hg rise in PaCO2 (arterial blood gas) as per the following equation: [3]
PaCO2 in mm Hg = 40 + 0.6 × (HCO3 − 24 mmol/L)
For example, if the measured serum bicarbonate is 30 mmol/L, the expected PaCO2 is 43.6 mm Hg. If the PaCO2 is significantly higher than that, then there is a primary metabolic alkalosis with accompanying respiratory acidosis due to inadequate respiratory compensation. If the PaCO2 is significantly lower, there is a primary metabolic alkalosis with a secondary respiratory alkalosis. Several acid-base disorders can be present at the same time, making calculations very complicated.
Associated electrolyte abnormalities also need to be identified, including hypochloremia, hypokalemia, and hypocalcemia. An electrocardiogram may be necessary to evaluate for arrhythmias. Urine chemistry is required to assess the kidney's response to alkalosis. Hypertension requires assessment and other tests for hyperaldosteronism or hypercortisolism when indicated. Volume depletion also requires evaluation as a coexisting condition.
When associated with hypoxia or an increased alveolar-arterial gradient, respiratory alkalosis requires a search for the cause of hypoxia. However, pulmonary embolism may cause respiratory alkalosis without associated hypoxia and must be ruled out before attributing respiratory alkalosis to hyperventilation to pain or anxiety.[25][26] Notably, patients with needle phobia or who hyperventilate in response to phlebotomy may show temporary respiratory alkalosis related to the pain of a blood draw.
Treatment / Management
The appropriate management of alkalosis depends on the prompt identification of its underlying cause, followed by management of the primary etiology of the alkalosis and the type of the alkalosis, such as metabolic, respiratory, or mixed. Specific etiologies such as pyloric stenosis need surgical correction, whereas excessive ingestion of alkali responds to restriction of excess intake. Alkalosis associated with conditions of excess aldosterone may need hormonal correction or replacement along with the treatment of associated hypertension. Correction of chloride-responsive alkalosis caused by volume depletion is possible by replenishing extracellular volume, typically with isotonic saline. Electrolyte disturbances associated with alkalosis, such as hypokalemia and hypocalcemia, are the chief causes of clinical deterioration in the patient and must undergo correction before the onset of life-threatening complications. Slow acid administration or dialysis with low bicarbonate baths may be necessary for emergencies.[27]
Treatment for respiratory alkalosis primarily targets correcting hyperventilation, such as primary or iatrogenic. In addition to addressing anxiety and pain, this may sometimes involve adjusting mechanical ventilation to induce intentional hypercapnia.[21](B3)
Differential Diagnosis
Given the wide range of manifestations, the differential diagnosis of alkalosis can be complex, especially when electrolyte imbalances such as hypochloremia, hypokalemia, and hypocalcemia are also present.
The differential diagnosis for respiratory alkalosis includes the following:
- Asthma exacerbation
- Atrial fibrillation
- Atrial flutter
- Atrial tachycardia
- Bacterial sepsis
- Community-acquired pneumonia
- Chronic obstructive pulmonary disease exacerbation
- Head trauma
- Heatstroke
- Hyperthyroidism and thyrotoxicosis
- Idiopathic pulmonary fibrosis
- Meningitis
- Metabolic acidosis
- Myocardial infarction
- Panic disorder
- Pneumothorax
- Pneumonia
- Pulmonary edema
- Pulmonary embolism
- Salicylate toxicity
- Theophylline toxicity
Prognosis
Whether respiratory or metabolic, alkalosis is typically compensated by the body's innate buffering mechanisms in the acute and subacute phases. When the alkalosis is uncorrected or chronic, the buffering mechanisms may become overwhelmed, potentially leading to a poor prognosis. Prognosis varies based on the primary cause of alkalosis and depends on associated factors such as volume depletion, electrolyte disturbances, and hormonal imbalances.
Studies indicate that patients with metabolic alkalosis have been found to have longer intensive care unit stays, more days on mechanical ventilation, and higher hospital mortality. An increase of 5 mEq/L in the serum bicarbonate level over 30 mEq/L correlated with an odds ratio of 1.21 for hospital mortality. The association between metabolic alkalosis and mortality occurs independently of the etiology of alkalosis.[28]
Complications
Alkalosis can lead to life-threatening arrhythmias, such as atrial and ventricular tachyarrhythmias, especially when associated with hypokalemia and hypocalcemia. These associated electrolyte abnormalities can cause carpopedal spasms, muscle weakness, and altered mental status.[29][30]
Consultations
Depending on the primary etiology of the alkalosis, consultation with various subspecialties may be necessary. A nephrology consult can assist with managing blood pressure and correcting hormonal imbalances in cases of hyperaldosteronism. For severe electrolyte imbalances, the patient may require admission under the care of an intensivist. Infants with pyloric stenosis need surgical evaluation and intervention.
Deterrence and Patient Education
Patients should be educated on the significance of severe alkalosis and its underlying cause. Patients and families need to understand the link between causes of alkalosis, such as anxiety disorders, severe emesis or excessive alkali ingestion, and resultant alkalosis.
Enhancing Healthcare Team Outcomes
Effective management of alkalosis requires an interprofessional team of healthcare professionals, including nurses, laboratory technologists, pharmacists, and several physicians in different specialties.
Upon identifying alkalosis, the primary clinician is responsible for coordinating the care, including the following:
- Order serial blood gas analysis, blood, and urine chemistries.
- Monitor the patient for signs and symptoms of neuromuscular depression, cardiac arrhythmias, hypertension, and volume depletion.
- Perform various maneuvers to help limit the severity of alkalosis and boost the body's buffering mechanisms.
- Consult the pharmacist about the correct dose and method for correcting electrolyte abnormalities.
- Consult with a nephrologist on further management, which may include dialysis.
- Consult with the radiologist about imaging tests to evaluate the causes of persistent emesis.
- Consult with the intensivist regarding care and monitoring in the intensive care unit during the hospital stay.
- Nurses monitoring the patient with metabolic alkalosis should be aware of the potential complications, including arrhythmias, and promptly inform the team members.[31]
The management of alkalosis does not stop with its correction. After the patient reaches a stable state, it is essential to determine its causes and identify risk factors for its recurrence. The morbidity and mortality of alkalosis in hospitalized patients are significant.
Reducing the morbidity of alkalosis requires an interprofessional team approach. Nursing staff play a crucial role in ongoing monitoring, promptly notifying the supervising clinician of patient status changes. Pharmacists are vital in medication reconciliation and assisting with selecting, ordering, and administering fluids, bicarbonate, and other pharmaceutical interventions to correct alkalosis. Their expertise supports clinicians in a collaborative, interprofessional effort to achieve optimal patient outcomes.
Media
(Click Image to Enlarge)
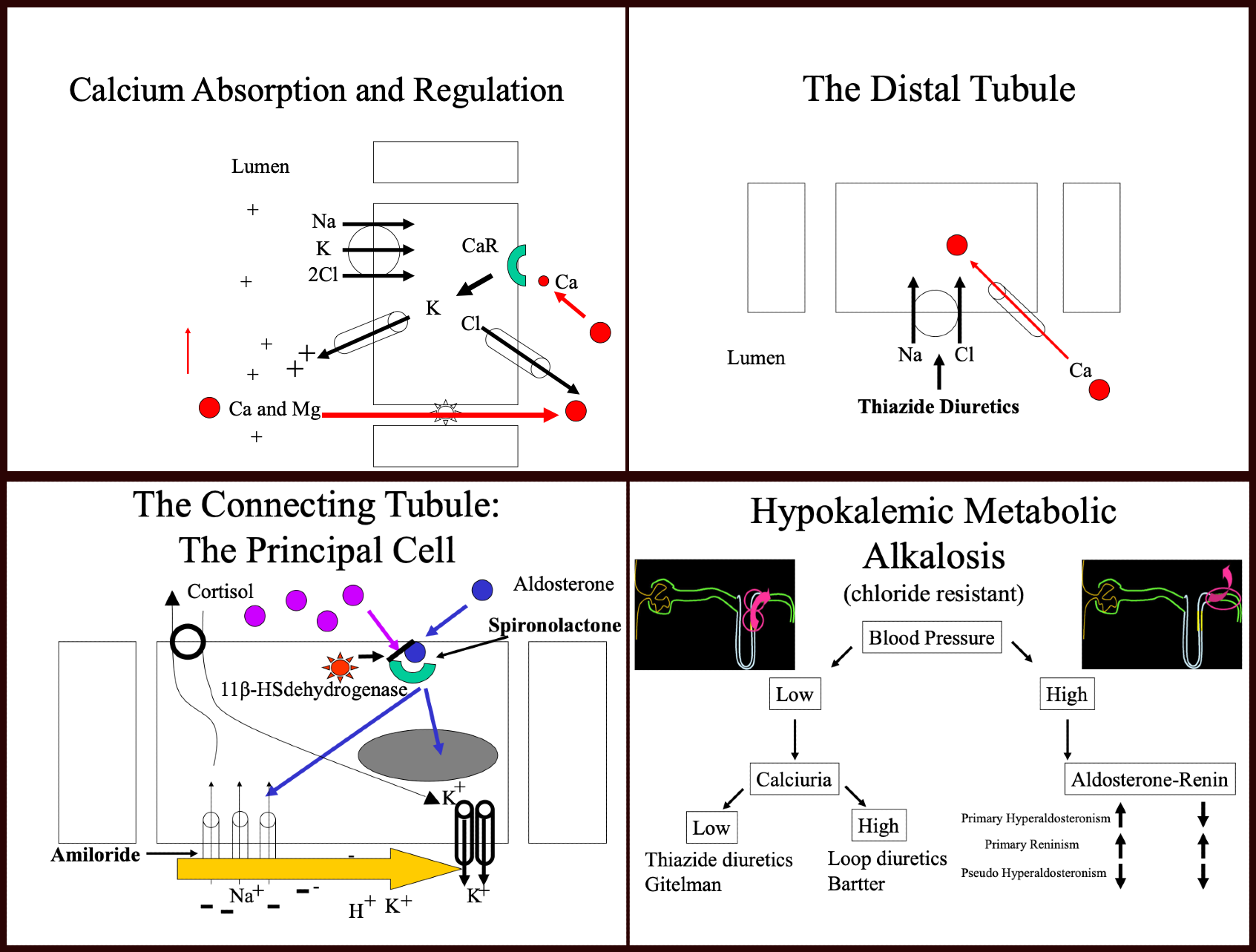
Metabolic Alkalosis Diagram
Contributed by Farah Leclercq, MD
References
Palmer BF, Clegg DJ. Respiratory Acidosis and Respiratory Alkalosis: Core Curriculum 2023. American journal of kidney diseases : the official journal of the National Kidney Foundation. 2023 Sep:82(3):347-359. doi: 10.1053/j.ajkd.2023.02.004. Epub 2023 Jun 21 [PubMed PMID: 37341662]
van den Bunder FAIM, van Woensel JBM, Stevens MF, van de Brug T, van Heurn LWE, Derikx JPM. Respiratory problems owing to severe metabolic alkalosis in infants presenting with hypertrophic pyloric stenosis. Journal of pediatric surgery. 2020 Dec:55(12):2772-2776. doi: 10.1016/j.jpedsurg.2020.05.041. Epub 2020 Jun 6 [PubMed PMID: 32641249]
Tinawi M. Pathophysiology, Evaluation, and Management of Metabolic Alkalosis. Cureus. 2021 Jan 21:13(1):e12841. doi: 10.7759/cureus.12841. Epub 2021 Jan 21 [PubMed PMID: 33628696]
Emmett M. Metabolic Alkalosis: A Brief Pathophysiologic Review. Clinical journal of the American Society of Nephrology : CJASN. 2020 Dec 7:15(12):1848-1856. doi: 10.2215/CJN.16041219. Epub 2020 Jun 25 [PubMed PMID: 32586924]
Rout P, Hashmi MF, Patel C. Milk-Alkali Syndrome. StatPearls. 2024 Jan:(): [PubMed PMID: 32491432]
Patel V, Mehra D, Ramirez B, Lindo A, Suarez M. Milk-Alkali Syndrome as a Cause of Hypercalcemia in a Gentleman With Acute Kidney Injury and Excessive Antacid Intake. Cureus. 2021 Feb 1:13(2):e13056. doi: 10.7759/cureus.13056. Epub 2021 Feb 1 [PubMed PMID: 33680598]
Do C, Vasquez PC, Soleimani M. Metabolic Alkalosis Pathogenesis, Diagnosis, and Treatment: Core Curriculum 2022. American journal of kidney diseases : the official journal of the National Kidney Foundation. 2022 Oct:80(4):536-551. doi: 10.1053/j.ajkd.2021.12.016. Epub 2022 May 5 [PubMed PMID: 35525634]
Bokhari SRA, Zulfiqar H, Mansur A. Bartter Syndrome. StatPearls. 2025 Jan:(): [PubMed PMID: 28723048]
Besouw MTP, Kleta R, Bockenhauer D. Bartter and Gitelman syndromes: Questions of class. Pediatric nephrology (Berlin, Germany). 2020 Oct:35(10):1815-1824. doi: 10.1007/s00467-019-04371-y. Epub 2019 Oct 29 [PubMed PMID: 31664557]
R A, Upendra Bhatia S, Narayan K, Mohammed S, Yelkur P. An Unusual Presentation of Failure to Thrive in a Toddler: Bartter Syndrome. Cureus. 2024 Aug:16(8):e67289. doi: 10.7759/cureus.67289. Epub 2024 Aug 20 [PubMed PMID: 39301344]
Gardner WN. The pathophysiology of hyperventilation disorders. Chest. 1996 Feb:109(2):516-34 [PubMed PMID: 8620731]
Pronicka E. Hypocapnic hypothesis of Leigh disease. Medical hypotheses. 2017 Apr:101():23-27. doi: 10.1016/j.mehy.2017.01.016. Epub 2017 Feb 1 [PubMed PMID: 28351484]
Piekutowska-Abramczuk D, Rutyna R, Czyżyk E, Jurkiewicz E, Iwanicka-Pronicka K, Rokicki D, Stachowicz S, Strzemecka J, Guz W, Gawroński M, Kosierb A, Ligas J, Puchala M, Drelich-Zbroja A, Bednarska-Makaruk M, Dąbrowski W, Ciara E, Książyk JB, Pronicka E. Leigh syndrome in individuals bearing m.9185T}C MTATP6 variant. Is hyperventilation a factor which starts its development? Metabolic brain disease. 2018 Feb:33(1):191-199. doi: 10.1007/s11011-017-0122-1. Epub 2017 Nov 7 [PubMed PMID: 29116603]
Castro D, Patil SM, Zubair M, Keenaghan M. Arterial Blood Gas. StatPearls. 2025 Jan:(): [PubMed PMID: 30725604]
Hopkins E, Sanvictores T, Sharma S. Physiology, Acid Base Balance. StatPearls. 2024 Jan:(): [PubMed PMID: 29939584]
To T, Wajja A, Wales PW, Langer JC. Population demographic indicators associated with incidence of pyloric stenosis. Archives of pediatrics & adolescent medicine. 2005 Jun:159(6):520-5 [PubMed PMID: 15939849]
Bae K, Jee D. Hyperventilation Syndrome and Sustained Hyperchloremia After Kidney Transplant: Time-Sequence Swing of Acid-Base Interpretation. Experimental and clinical transplantation : official journal of the Middle East Society for Organ Transplantation. 2018 Dec:16(6):754-756. doi: 10.6002/ect.2018.0099. Epub 2018 Aug 17 [PubMed PMID: 30119620]
Raphael KL, Murphy RA, Shlipak MG, Satterfield S, Huston HK, Sebastian A, Sellmeyer DE, Patel KV, Newman AB, Sarnak MJ, Ix JH, Fried LF, Health ABC Study. Bicarbonate Concentration, Acid-Base Status, and Mortality in the Health, Aging, and Body Composition Study. Clinical journal of the American Society of Nephrology : CJASN. 2016 Feb 5:11(2):308-16. doi: 10.2215/CJN.06200615. Epub 2016 Jan 14 [PubMed PMID: 26769766]
Brimioulle S, Kahn RJ. Effects of metabolic alkalosis on pulmonary gas exchange. The American review of respiratory disease. 1990 May:141(5 Pt 1):1185-9 [PubMed PMID: 2339841]
Hess W. [Affinity of oxygen for hemoglobin--its significance under physiological and pathological conditions]. Der Anaesthesist. 1987 Sep:36(9):455-67 [PubMed PMID: 3318547]
Hopper K. Respiratory Acid-Base Disorders in the Critical Care Unit. The Veterinary clinics of North America. Small animal practice. 2017 Mar:47(2):351-357. doi: 10.1016/j.cvsm.2016.09.006. Epub 2016 Nov 24 [PubMed PMID: 27890436]
Level 3 (low-level) evidenceNatarajan A, Su HW, Heneghan C, Blunt L, O'Connor C, Niehaus L. Measurement of respiratory rate using wearable devices and applications to COVID-19 detection. NPJ digital medicine. 2021 Sep 15:4(1):136. doi: 10.1038/s41746-021-00493-6. Epub 2021 Sep 15 [PubMed PMID: 34526602]
Dhondup T, Qian Q. Acid-Base and Electrolyte Disorders in Patients with and without Chronic Kidney Disease: An Update. Kidney diseases (Basel, Switzerland). 2017 Dec:3(4):136-148. doi: 10.1159/000479968. Epub 2017 Oct 5 [PubMed PMID: 29344508]
Level 2 (mid-level) evidenceSeifter JL, Chang HY. Disorders of Acid-Base Balance: New Perspectives. Kidney diseases (Basel, Switzerland). 2017 Jan:2(4):170-186. doi: 10.1159/000453028. Epub 2016 Dec 10 [PubMed PMID: 28232934]
Level 3 (low-level) evidenceLum LC. Hyperventilation and anxiety state. Journal of the Royal Society of Medicine. 1981 Jan:74(1):1-4 [PubMed PMID: 6780688]
Marini C, Di Ricco G, Formichi B, Michelassi C, Bauleo C, Monti S, Giuntini C. Arterial base deficit in pulmonary embolism is an index of severity and diagnostic delay. Internal and emergency medicine. 2010 Jun:5(3):235-43. doi: 10.1007/s11739-010-0354-0. Epub 2010 Mar 16 [PubMed PMID: 20232176]
Level 2 (mid-level) evidencePahari DK, Kazmi W, Raman G, Biswas S. Diagnosis and management of metabolic alkalosis. Journal of the Indian Medical Association. 2006 Nov:104(11):630-4, 636 [PubMed PMID: 17444063]
Libório AB, Noritomi DT, Leite TT, de Melo Bezerra CT, de Faria ER, Kellum JA. Increased serum bicarbonate in critically ill patients: a retrospective analysis. Intensive care medicine. 2015 Mar:41(3):479-86. doi: 10.1007/s00134-015-3649-9. Epub 2015 Jan 20 [PubMed PMID: 25600192]
Level 2 (mid-level) evidenceSalanova-Villanueva L, Bernis-Carro C, Alberto-Blazquez L, Sanchez-Tomero JA. Severe arrhythmia due to hypokalemia. Influence from diuretic substances. Nefrologia : publicacion oficial de la Sociedad Espanola Nefrologia. 2015:35(3):334-6. doi: 10.1016/j.nefro.2015.05.008. Epub 2015 Jun 18 [PubMed PMID: 26299178]
Espay AJ. Neurologic complications of electrolyte disturbances and acid-base balance. Handbook of clinical neurology. 2014:119():365-82. doi: 10.1016/B978-0-7020-4086-3.00023-0. Epub [PubMed PMID: 24365306]
Batlle D, Chin-Theodorou J, Tucker BM. Metabolic Acidosis or Respiratory Alkalosis? Evaluation of a Low Plasma Bicarbonate Using the Urine Anion Gap. American journal of kidney diseases : the official journal of the National Kidney Foundation. 2017 Sep:70(3):440-444. doi: 10.1053/j.ajkd.2017.04.017. Epub 2017 Jun 7 [PubMed PMID: 28599903]