Introduction
The term western blot was first coined by Dr. Burnette in 1981 after the eponymous Southern blot for DNA and the consequent coinage of the northern blot for RNA in 1977.[1][2] Western blotting separates, detects, and identifies one or more proteins in a complex mixture.[3] This process involves separating the individual proteins by polyacrylamide gel electrophoresis and then transferring or blotting onto an overlying strip of nitrocellulose or nylon membrane by electro-blotting.[4] Once the proteins are in the membrane, they can be detected using antibodies labeled with probes, such as radioactive isotopes or enzymes.[5] When such probes are used, the detection limits can be 10 to 100 times lower compared to those achieved through direct immunoprecipitation and protein staining methods.[6] Densitometric analysis of the bands on a Western blot enables researchers to quantitatively compare samples, such as evaluating the effects of treatments or time points.[7]
Centers for Disease Control and Prevention no longer recommends the western blot test as a diagnostic tool. This article remains for historical purposes and supports laboratories that still use the technique. However, it is no longer being actively updated.
Specimen Requirements and Procedure
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Specimen Requirements and Procedure
Principles of Western Blotting
The key principles of western blotting are equal loading of proteins, separation of proteins by molecular weight, electrophoretic transfer to a suitable membrane, and antibody probing.
Equal loading of proteins: Proper sample preparation for subsequent electrophoresis is crucial for downstream analysis. Western blot samples are first prepared by extracting proteins using specialized cell lysis buffers and protease and phosphatase inhibitors. There are various extraction methods, and the appropriate choice depends on the type of sample used. For instance, tissue preparation is typically performed through homogenization or sonication. In contrast, osmotic shock or detergent lysis is more suitable for efficiently lysed cells, such as erythrocytes or cultured cells. Furthermore, the cell lysis buffer used in extraction should align with the target protein cellular localization.[8] For example, radioimmunoprecipitation assay buffer is more adept for nuclear and mitochondrial proteins.
Although rare, some antibodies cannot detect denatured samples. In such cases, gentle lysis buffers without detergents should be used. PPIs are used to maintain the structure and phosphorylation state of the target protein from the activity of endogenous phosphatases upon cell lysis and exogenous phosphatases within the lysis microenvironment. These factors emphasize the importance of tailoring protein extraction methods based on the sample type and the target protein.[2]
Each western blot sample must have an equal protein concentration to ensure a valid experiment. Unequal protein loading across lanes can distort the results and compromise the accuracy of the analysis. Protein concentration is typically measured using a Bradford assay—a colorimetric protein assay that exploits the interaction between a dye and a protein.[9] In brief, the dye Coomassie brilliant blue G-250 binds to proteins to change color, and this absorbance shift is recorded using a spectrophotometer. By conducting this assay with known protein standards, a linear regression standard curve is generated to determine the concentrations of unknown protein extracts in the sample.[10]
All western blot samples have 3 elements—protein extract, cell lysis buffer, and Laemmli (sample) buffer. Protein extract is normalized with cell lysis buffer to the desired protein concentration, and an equal volume of Laemmli (sample) buffer is added. Therefore, a western blot sample always has a 1:1 volume ratio of normalized protein and Laemmli buffer. Laemmli buffer (60 mM Tris-HCl pH 6.8; 20% glycerol; 2% sodium dodecyl sulfate (SDS); 4% beta-mercaptoethanol; 0.01% bromophenol blue) is unique to western blot sample preparation, as each reagent is purposeful for sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE).[11] Glycerol increases the sample's density, allowing it to settle into the loading wells
Bromophenol blue is a nonreactive reagent that serves as a dye front for electrophoresis. SDS is a potent anionic detergent that coats denatured proteins with an equal anion-to-mass ratio, masking proteins' charge, shape, and size characteristics and rendering them solely a function of molecular weight.[12] Beta-mercaptoethanol is a reducing agent that acts on disulfide bonds; without beta-mercaptoethanol, proteins with disulfide bonds retain some shape and do not electrophorese consummately by molecular weight.[13] Tris-HCl pH 6.8 is an essential component of the discontinuous buffer system, which is explained in more detail below. Prepared samples are heated before loading to further denature proteins to their primary structure. Thus, proteins undergo electrophoresis by their monomeric weight.[14]
Separation of proteins by molecular weight: The separation of proteins by molecular weight is achieved through SDS-PAGE, which combines the use of a detergent and a discontinuous buffer system. Typically, PAGE is an analytical biochemistry method used to separate contents such as nucleic acids and proteins by electrophoretic mobility in a chemically inert gel; however, When SDS, a potent anionic detergent, is added, all denatured proteins are coated with an equal charge-to-mass ratio. Therefore, the rate of protein migration is proportional to weight. Indeed, larger proteins travel slower compared to smaller proteins due to retarding properties of the porous gel. A gel matrix is formed from the polymerization of acrylamide and the crosslinking of N,N'-methylenebisacrylamide. This matrix creates a molecular sieve that imbues retarding properties. The pore size of this sieve is alterable by adjusting the percentages of polyacrylamide and N,N'-methylenebisacrylamide as they are inversely proportional.[15]
Two sieves of different sizes used in PAGE are a stacking gel and a resolving gel. As the name suggests, the stacking gel stacks proteins into a narrow band to allow proteins to enter the resolving gel simultaneously, which is made possible due to its bigger pore size and acidity. With its much smaller pore size and basicity, the resolving gel is where proteins are separated.[16]
The Laemmli discontinuous buffer system is most commonly used in SDS-PAGE. This system uses a running buffer (25 mM Tris; 192 mM glycine; 0.1% SDS; pH~8.30) as the electrode buffer and tris-HCl to buffer an acidic stacking gel (pH~6.80) and a basic resolving gel (pH~8.80). The deliberate use of varied pHs exploits the charge properties of glycine.[17] In an acidic environment, glycine exists as a zwitterion, whereas in a basic environment, it appears as a glycinate anion. When electrophoresis begins, the current quickly draws glycinate into the stacking gel. The acidic gel protonates glycine into its zwitterionic form, significantly impeding its mobility.[18]
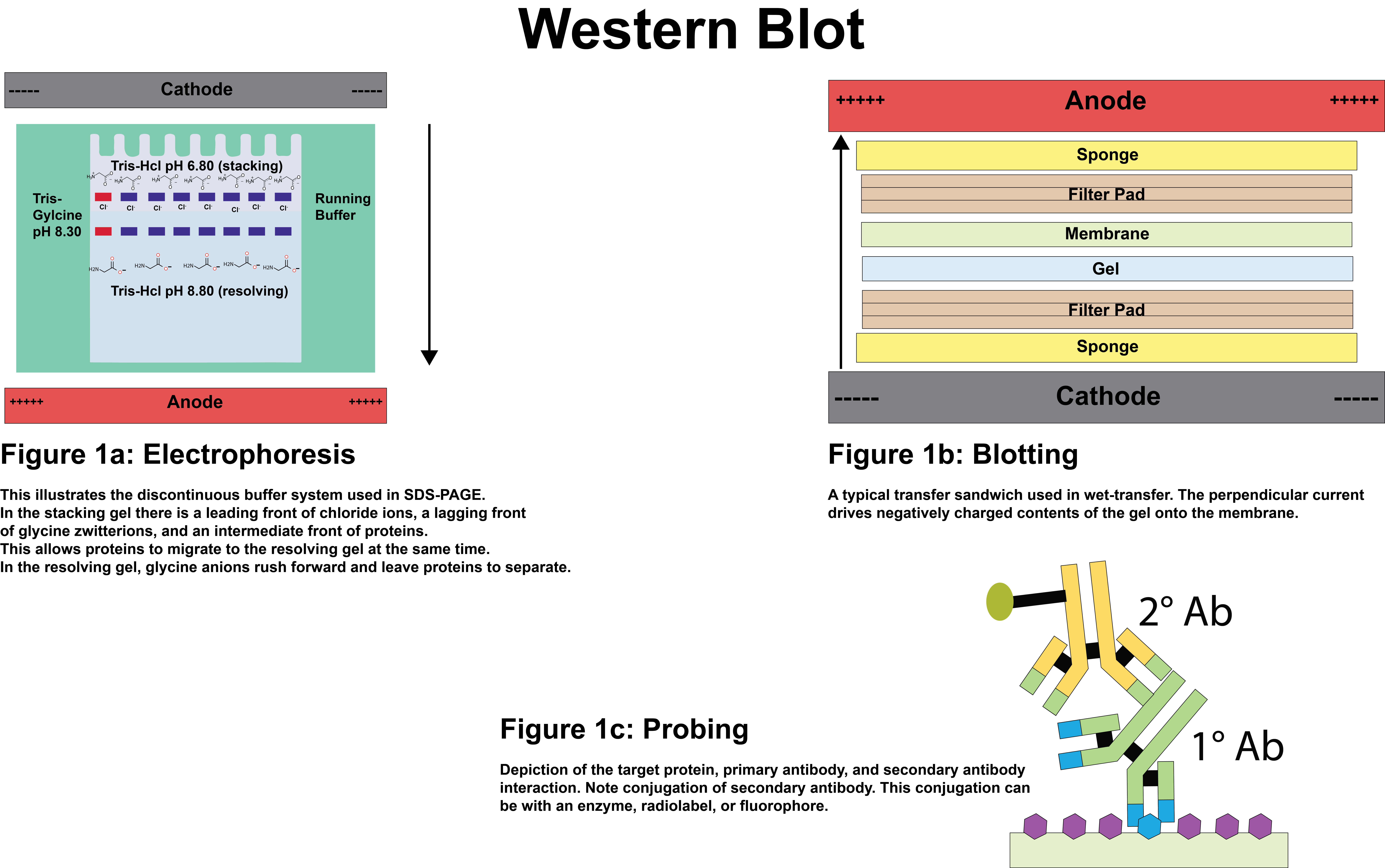
In contrast, chloride from the tris-HCl buffer disassociates from its counter ion and migrates quickly to the anode. Proteins are positioned between a trailing front of glycine and a leading front of chloride, resulting in all proteins simultaneously arriving at the resolving gel—a vital component for effective separation.[19] The basic pH of the resolving gel causes glycinate anions to reform at the interface between the stacking and resolving gels. From this interface, glycinate anions quickly migrate past the protein front. The proteins now hit the resolving gel in narrow bands without a zone of high voltage previously formed from the leading and trailing ions in the stacking gel.[16] Thus, this allows proteins to migrate down the resolving gel slower, which induces the separation of proteins due to the higher concentration of polyacrylamide (see Image. Electrophoresis).
The samples are run in their respective lanes alongside a molecular weight marker, often called a protein ladder. For example, a typical setup has the ladder in the first lane and the samples in the remaining lanes. The ladder establishes standard molecular weight bands that are then used to read the relative weight of proteins.[20]
Electrophoretic transfer (blotting): Blotting is the electrophoretic transfer of gel contents onto a suitable membrane. In western blotting, the contents are proteins. There are several methods of blotting in addition to numerous types of membranes. Despite the existence of various transfer systems, such as wet, semi-dry, and fast, the fundamental principle of electrophoretic transfer remains unchanged. Similar to electrophoresis, negatively charged samples migrate toward an anode; in blotting, a transfer sandwich with a slightly modified electrode buffer is used. Towbin buffer (25 mM Tris; 192 mM glycine; 20% methanol; pH 8.3) is the standard transfer buffer, although minor tweaks to this buffer are possible for the target protein.[21]
Methanol is important in blotting as it increases the hydrophobicity of proteins and facilitates the release of SDS, both of which increase the adsorption of proteins onto the membrane. From cathode to anode, the sandwich is organized as a filter paper, polyacrylamide gel, membrane, and filter paper.[22] In a wet transfer system, fiber pads or sponges are placed superficially on each side. The sandwich is subjected to a perpendicular current that drives gel contents onto the membrane (see Image. Blotting).
Equilibration of sandwich contents in the transfer buffer is crucial for increasing transfer efficiency. This process prevents the drying of the gel and membrane, washes electrophoretic contaminants off the gel, and reforms the original gel size.[23] As electrophoresis progresses, increasing voltage raises the temperature, causing the gel to expand. Thus, cold transfer buffer shrinks gels to the proper size. Interestingly, methanol in the transfer buffer also cools the gel during equilibration.[24]
Each transfer system has advantages, and the choice largely depends on the target protein and laboratory workflow. Among the various transfer apparatuses, the two most commonly used are wet and semi-dry systems.[22] The key differences between these systems lie in the volume of transfer buffer used and the transfer time. Wet transfer uses a tank transfer system that requires a large volume of transfer buffer, whereas semi-dry transfer systems typically only require the dampening of the sandwich.[25]
Semi-dry systems are also time-efficient, as blotting typically finishes within an hour, but a low voltage gets applied overnight in a wet transfer. Although semi-dry transfer seems to be the better option as there is a significant reduction in both the volume of transfer buffer and length of transfer time, it has its limitations.[26] Large proteins, such as membrane receptors, do not blot well, and overall transfer efficiency is lower. Wet transfer shines in its ability to yield high efficiency across a wide range of protein sizes, thus offering the most flexibility.[27]
When Drs Burnette and Towbin published their seminal studies, electrophoretic transfer was carried out on nitrocellulose membranes. These membranes remained the gold standard until the advent of polyvinylidene difluoride (PVDF) membranes. Concisely, PVDF membranes outcompete nitrocellulose membranes in their protein binding capacity, chemical resistance, and enhanced transfer efficiency in the presence of SDS.[28] PVDF promotes higher adsorption of proteins, and its chemical resistance allows for the stripping and reprobing of membranes. In addition, transfer efficiency improves by inserting a small percentage of SDS in the transfer buffer. However, noted protein sensitivity from PVDF could also increase the background signal for analysis.[29]
Methanol in transfer buffer can shrink nitrocellulose membranes and precipitate out large proteins. Both membranes come in different pore sizes, and membrane pore size is directly related to protein weight.[6] Smaller proteins require smaller pore sizes, although a pore size of 0.45 µm is suitable for most proteins.[30] Recent advancements have led to the development of unique membranes, such as those used for near-infrared detection systems. Therefore, the choice of membrane should be based on the target protein and the specific downstream detection method being used.[6]
Antibody probing: Upon completing the electrophoretic transfer, proteins are now positioned on the membrane, and 2 antibodies are used for probing and analysis. The primary antibody that binds a specific region on the target protein is used to detect its presence on the membrane. The secondary antibody conjugates with a component used for analysis.[31] This antibody indirectly binds the target protein by binding to the constant regions of the primary antibody (see Image. Probing).
As membranes have a high affinity for proteins, they are incubated in a buffer to coat the remaining surface area before probing. This blocking buffer includes a protein with a minimal binding affinity to the target protein and, consequently, the antibody. Typically, blocking buffer proteins include either casein from powdered milk or bovine serum albumin. Although casein is inexpensive and suitable for most proteins, bovine serum albumin is a better choice when the target protein is phosphorylated; there is a cross-reactivity between casein and phosphorylation-specific primary antibodies.[30] After blocking, the membrane is washed with TBS-T—a mixture of tris-buffered saline and tween-20. Tween-20 is a nonionic detergent that helps remove peripherally bound proteins on the membrane.[32]
Probing of primary and secondary antibodies is done by incubating the membrane in a probing buffer of either the primary or secondary antibody in TBS-T. The membrane is first incubated in the primary probing buffer, typically overnight in a cold room, and washed again with TBS-T.[33] The membrane is then incubated with the secondary probing buffer for about 1 hour and washed. These washing steps are crucial to reduce background noise in the analysis. After probing and washing, the membrane is ready for detection.[32]
The secondary antibody conjugates with a component specific to the type of analysis. Autoradiography was once a standard method for visualizing bands, but its popularity has declined due to the associated hazards. This technique uses a radiolabeled isotope that is conjugated to the secondary antibody.[34] More commonly, chemiluminescence is used. This method uses substrates that react with an enzyme-conjugated secondary antibody. These enzymes are horseradish peroxidase or alkaline phosphatase.[35]
The enzyme-mediated reaction produces light that is then recorded with an imaging system. More recently, secondary antibodies have been conjugated with fluorophores that can be detected without substrates.[7] This fluorescence-based detection is gaining popularity due to its capability of probing 2 target proteins using secondary antibodies with different wavelength fluorophores; this feature is a selective advantage for relative protein expression analysis, as housekeeping proteins are visible alongside a protein of interest.[6]
The visualization of bands can serve various analytical purposes. The presence of bands can verify the expression of a protein, whereas the density of bands can show comparative relative protein expression. A housekeeping protein is also probed to evaluate relative protein expression.[36] A housekeeping protein is a ubiquitous protein constitutively expressed in all cells. By normalizing the band densities of the target protein with those of the housekeeping protein, a statistically significant difference between sample types can be measured.[37]
Interfering Factors
Some limitations of the western blot technique are as follows:
- Western blot is a very delicate and time-consuming process. Even a minor imbalance at any stage can compromise the accuracy of the results.[38]
- The secondary antibody can occasionally react with a non-intended protein, leading to the labeling of incorrect proteins.[39]
- Insufficient transfer time can prevent larger proteins from transferring effectively onto the membrane, resulting in missing or erroneous bands.[38]
- Well-trained technicians are a must for this technique.[39]
- Western blot is semi-quantitative at best. Only an approximate estimation and not a precise measurement of the molecular weight of the protein is possible.[40]
- Primary antibody availability is crucial. If a primary antibody is not available for a specific protein, western blotting cannot be used to detect that protein.[7]
Clinical Significance
As highlighted earlier, a western blot involves numerous steps. This lengthy process drives up the time and cost required for accurate results. However, unlike an enzyme-linked immunosorbent assay (ELISA), the western blot is less likely to give false-positive results, especially in diagnosing HIV.[41]
Western blotting is commonly used to detect anti-HIV antibodies in human serum and urine samples.[42] Protein samples from a known HIV-infected individual are separated using electrophoresis and then blotted on the nitrocellulose membrane. A specific antibody is then affixed to detect the target protein.[43] Western blotting is typically performed after the ELISA test to confirm the diagnosis of HIV and is far more sensitive compared to the ELISA test.[44] More recently, in commercial HIV western blot kits, viral proteins come affixed to the membrane. Antibodies from human urine or serum samples bind to these proteins, and anti-HIV antibodies are used to detect bands alongside quality controls.[45]
In addition to HIV detection, western blotting is useful in diagnosing Lyme disease and atypical and typical bovine spongiform encephalopathy.[46][47]
Western blotting procedure determines the presence or absence, size, and abundance of target proteins in a sample, which is beneficial for various scientific reasons across many fields of study.[48] The technique is commonly used to evaluate protein-DNA interactions, protein-protein interactions, post-translational modifications, protein isoform detection, antibody characterization, epitope mapping, and subcellular protein localization.[38]
As western blotting is an antibody-based method, the technique often supports and produces reliable results for detecting non-infectious diseases using high throughput screening. For example, when determining cancers, incongruous isoforms of proteins can become potential markers of the pathology of the disease.[49] Autoantibodies may also indicate an autoimmune disease.[38]
Some proteins are engineered through molecular cloning to contain short sequences of amino acids that serve as a tag. Common tags include the HA-tag and the Myc-tag. These tags serve as a foreign protein epitope that does not naturally occur in the biological system being studied.[50] Thus, the tag makes the protein easier to detect compared to all other naturally occurring proteins. An antibody directed to the tag identifies the presence and amount of the tagged protein in the western blot.[7]
Quality Control and Lab Safety
Quality Controls
Like any experiment, quality controls should be used to validate findings. In western blotting, a positive control, negative control, loading control, and no-first-degree antibody control are all effective in achieving and maintaining robust experiments.[49] Controls are incorporated into dedicated lanes where the sample is specifically modified based on the control type. A positive control contains a sample known to include the target protein, whereas a negative control is a sample confirmed to lack the target protein. These distinctions can be as general as different organ types or as specific as different cellular localization. For example, if the goal is to analyze the expression of a nuclear protein, and subcellular fractionation is performed to isolate this region, a negative control evaluates the quality of fractionation, nonspecific binding of antibodies, and a false-positive.[38]
Positive controls are effective in verifying that the workflow is well-optimized, even in the absence of bands in sample lanes. In addition, a positive control can verify a negative result.[51] Loading control is a housekeeping protein, such as alpha-tubulin or beta-actin.[49] Probing with antibodies specific for a housekeeping protein checks for an equal amount of proteins per sample. A nonspecific secondary antibody can yield false positives. The specificity of a secondary antibody is evaluated by not incubating a membrane strip with the primary antibody.[38]
Troubleshooting
Due to its many steps and lengthy workflow, this method has many error arms. This review does not cover every possible error, its cause, or the solution. Instead, it describes the most common issues and how to troubleshoot them.
Smiling of Bands
When protein bands migrate unevenly down the gel, they may form a smiling pattern. This issue often arises due to air bubbles within the gel, excessive voltage during electrophoresis, or an excessive volume of the loading sample.[7] Air bubbles within the gel can distort the migration of bands. A constant voltage during electrophoresis is directly proportional to resistance, and as resistance and temperature are directly linked, a high voltage increases the temperature in the electrophoresis tank.[52] Heat pockets and an overall increase in the temperature of the running buffer can also alter migration. Before the buffer can warm up, a high voltage at the start of electrophoresis rushes bands and causes nonlinear migration. A large volume of loading samples can cause spillover into other lanes, and these large bands can skew into another lane.[49]
Absence of Bands
If no signal is detected across all lanes except for the molecular weight ladder, there are many possible causes. The first step is identifying where the error occurred within the workflow. Typically, the most common causes include poor transfer efficiency or poor probing.[7]
The membrane can be stained with Ponceau S, a membrane-safe red dye, to visualize bands. If bands are clearly visible on the membrane, especially where the target protein is expected, it suggests that transfer efficiency is not likely the cause.
If no bands are visible, transfer settings must be altered.[51] A washout of proteins can occur when proteins from the membrane migrate to the filter paper. The washout occurs when the transfer time is too high. To prevent this issue, reducing either the transfer voltage or transfer time can help by slowing down the process, ensuring that proteins stay bound to the membrane and do not wash out onto the filter paper.[7]
A poor transfer can also occur if little to no proteins are adsorbed on the membrane. To understand the directionality of transfer, the gel can be stained to reveal bands.[49] A significant visualization of bands can suggest that the actual transfer was poor rather than a high voltage or time. Rechecking the quality of the transfer buffer, increasing transfer settings, and ensuring proper contact between the gel and membrane can resolve this issue. If the target protein is small, a semi-dry system may be preferable.[38]
If a positive control lane is used and bands are absent, this can be due to a poor detection kit, poor antibodies, or even an incorrect antibody concentration. Antibody concentration is optimized by running titration experiments.[7]
Multiple Bands
Only a single row of bands should be visualized during detection. The presence of multiple bands suggests the nonspecific binding of antibodies.[52] This issue is often associated with polyclonal antibodies or high antibody concentrations. As mentioned earlier, titration experiments should be performed to optimize detection.[7]
High Background With or Without Splotches
Poor membrane blocking, excessive antibody concentration, and a dry membrane can result in high background signals. Increasing the period for blocking or changing the type of protein used in blocking buffer may solve this issue.[49] Titration experiments should be performed for antibody optimization. Insufficient washing can result in high background signals, a significant cause of splotches on the membrane. Membranes must be maintained wet throughout the experiment, and a dry membrane can give high background signals.[38]
Lab Safety
Standard laboratory safety protocols should always be followed. When casting gels, acrylamide is a potent neurotoxin; however, it is chemically inert once polymerized. Proper precautionary measures should be taken when handling this reagent to ensure safety.[53]
Enhancing Healthcare Team Outcomes
Interprofessional healthcare team members involved in treating and managing conditions where western blot testing is used must have a solid understanding of the test results and their implications for patient care. Laboratory technicians and nursing staff play critical roles in obtaining and preparing samples for testing, and their training is essential to ensure that samples meet the necessary quality standards for accurate results. This collaboration ensures that the test is performed properly, minimizing the risk of errors or invalid data that could lead to misdiagnosis or incorrect treatment.
Effective interprofessional training, coordination, and communication among team members are key to enhancing the clinical validity of western blot results. By fostering a well-coordinated approach, the healthcare team can ensure that test results are interpreted correctly and applied effectively in clinical decision-making. This integrated teamwork improves the accuracy of diagnostics and leads to better patient care and outcomes.
Media
(Click Image to Enlarge)
Major principles of a Western blot. Contributed by Kartheek Gavini, MS
References
Burnette WN. "Western blotting": electrophoretic transfer of proteins from sodium dodecyl sulfate--polyacrylamide gels to unmodified nitrocellulose and radiographic detection with antibody and radioiodinated protein A. Analytical biochemistry. 1981 Apr:112(2):195-203 [PubMed PMID: 6266278]
Level 3 (low-level) evidenceAlwine JC, Kemp DJ, Stark GR. Method for detection of specific RNAs in agarose gels by transfer to diazobenzyloxymethyl-paper and hybridization with DNA probes. Proceedings of the National Academy of Sciences of the United States of America. 1977 Dec:74(12):5350-4 [PubMed PMID: 414220]
Level 3 (low-level) evidenceHnasko TS, Hnasko RM. The Western Blot. Methods in molecular biology (Clifton, N.J.). 2015:1318():87-96. doi: 10.1007/978-1-4939-2742-5_9. Epub [PubMed PMID: 26160567]
Eslami A, Lujan J. Western blotting: sample preparation to detection. Journal of visualized experiments : JoVE. 2010 Oct 14:(44):. pii: 2359. doi: 10.3791/2359. Epub 2010 Oct 14 [PubMed PMID: 21189462]
Gallagher S, Winston Tank Transfer Systems SE, Fuller Tank Transfer Systems SA, Hurrell Tank Transfer Systems Reversible Staining Of Proteins JGR. Immunoblotting and immunodetection. Current protocols in immunology. 2008 Nov:Chapter 8():8.10.1-8.10.28. doi: 10.1002/0471142735.im0810s83. Epub [PubMed PMID: 19016449]
Hirano S. Western blot analysis. Methods in molecular biology (Clifton, N.J.). 2012:926():87-97. doi: 10.1007/978-1-62703-002-1_6. Epub [PubMed PMID: 22975958]
Pillai-Kastoori L, Schutz-Geschwender AR, Harford JA. A systematic approach to quantitative Western blot analysis. Analytical biochemistry. 2020 Mar 15:593():113608. doi: 10.1016/j.ab.2020.113608. Epub 2020 Jan 31 [PubMed PMID: 32007473]
Level 1 (high-level) evidencePeach M, Marsh N, Miskiewicz EI, MacPhee DJ. Solubilization of proteins: the importance of lysis buffer choice. Methods in molecular biology (Clifton, N.J.). 2015:1312():49-60. doi: 10.1007/978-1-4939-2694-7_8. Epub [PubMed PMID: 26043989]
Hammond JB, Kruger NJ. The bradford method for protein quantitation. Methods in molecular biology (Clifton, N.J.). 1988:3():25-32. doi: 10.1385/0-89603-126-8:25. Epub [PubMed PMID: 21400151]
Kruger NJ. The Bradford method for protein quantitation. Methods in molecular biology (Clifton, N.J.). 1994:32():9-15 [PubMed PMID: 7951753]
Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970 Aug 15:227(5259):680-5 [PubMed PMID: 5432063]
Harlow E, Lane D. Immunoblotting: preparing protein solutions. CSH protocols. 2006 Aug 1:2006(3):. pii: pdb.prot4298. doi: 10.1101/pdb.prot4298. Epub 2006 Aug 1 [PubMed PMID: 22485846]
Litovchick L. Preparing Protein Solutions for Immunoblotting. Cold Spring Harbor protocols. 2018 Jul 2:2018(7):. doi: 10.1101/pdb.prot098418. Epub 2018 Jul 2 [PubMed PMID: 29967277]
Heda GD, Omotola OB, Heda RP, Avery J. Effects of Reusing Gel Electrophoresis and Electrotransfer Buffers on Western Blotting. Journal of biomolecular techniques : JBT. 2016 Sep:27(3):113-8. doi: 10.7171/jbt.16-2703-004. Epub 2016 Aug 3 [PubMed PMID: 27582639]
HJERTEN S. "Molecular-sieve" electrophoresis in cross-linked polyacrylamide gels. Journal of chromatography. 1963 May:11():66-70 [PubMed PMID: 13954823]
Level 3 (low-level) evidenceGreen MR, Sambrook J. Polyacrylamide Gel Electrophoresis. Cold Spring Harbor protocols. 2020 Dec 1:2020(12):. doi: 10.1101/pdb.prot100412. Epub 2020 Dec 1 [PubMed PMID: 33262236]
Smith BJ. SDS polyacrylamide gel electrophoresis of proteins. Methods in molecular biology (Clifton, N.J.). 1994:32():23-34 [PubMed PMID: 7524943]
Maizel JV. SDS polyacrylamide gel electrophoresis. Trends in biochemical sciences. 2000 Dec:25(12):590-2 [PubMed PMID: 11116183]
Kurien BT, Aggarwal R, Scofield RH. Protein Extraction from Gels: A Brief Review. Methods in molecular biology (Clifton, N.J.). 2019:1855():479-482. doi: 10.1007/978-1-4939-8793-1_40. Epub [PubMed PMID: 30426441]
Brunelle JL, Green R. One-dimensional SDS-polyacrylamide gel electrophoresis (1D SDS-PAGE). Methods in enzymology. 2014:541():151-9. doi: 10.1016/B978-0-12-420119-4.00012-4. Epub [PubMed PMID: 24674069]
Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proceedings of the National Academy of Sciences of the United States of America. 1979 Sep:76(9):4350-4 [PubMed PMID: 388439]
Level 3 (low-level) evidenceKurien BT, Scofield RH. Western blotting: an introduction. Methods in molecular biology (Clifton, N.J.). 2015:1312():17-30. doi: 10.1007/978-1-4939-2694-7_5. Epub [PubMed PMID: 26043986]
Kurien BT, Scofield RH. Introduction to protein blotting. Methods in molecular biology (Clifton, N.J.). 2009:536():9-22. doi: 10.1007/978-1-59745-542-8_3. Epub [PubMed PMID: 19378040]
Kurien BT, Scofield RH. Western blotting. Methods (San Diego, Calif.). 2006 Apr:38(4):283-93 [PubMed PMID: 16483794]
Level 3 (low-level) evidenceJin Y, Cerletti N. Western blotting of transforming growth factor beta 2. Optimization of the electrophoretic transfer. Applied and theoretical electrophoresis : the official journal of the International Electrophoresis Society. 1992:3(2):85-90 [PubMed PMID: 1477118]
Garić D, Humbert L, Fils-Aimé N, Korah J, Zarfabian Y, Lebrun JJ, Ali S. Development of buffers for fast semidry transfer of proteins. Analytical biochemistry. 2013 Oct 15:441(2):182-4. doi: 10.1016/j.ab.2013.07.009. Epub 2013 Jul 16 [PubMed PMID: 23872007]
Silva JM, McMahon M. The fastest Western in town: a contemporary twist on the classic Western blot analysis. Journal of visualized experiments : JoVE. 2014 Feb 5:(84):e51149. doi: 10.3791/51149. Epub 2014 Feb 5 [PubMed PMID: 24561642]
Komatsu S. Western Blotting Using PVDF Membranes and Its Downstream Applications. Methods in molecular biology (Clifton, N.J.). 2015:1312():227-36. doi: 10.1007/978-1-4939-2694-7_24. Epub [PubMed PMID: 26044005]
Komatsu S. Western blotting/Edman sequencing using PVDF membrane. Methods in molecular biology (Clifton, N.J.). 2009:536():163-71. doi: 10.1007/978-1-59745-542-8_18. Epub [PubMed PMID: 19378055]
Kim B. Western Blot Techniques. Methods in molecular biology (Clifton, N.J.). 2017:1606():133-139. doi: 10.1007/978-1-4939-6990-6_9. Epub [PubMed PMID: 28501998]
Kurien BT, Danda D, Bachmann MP, Scofield RH. SDS-PAGE to Immunoblot in One Hour. Methods in molecular biology (Clifton, N.J.). 2015:1312():449-54. doi: 10.1007/978-1-4939-2694-7_45. Epub [PubMed PMID: 26044026]
Mahmood T, Yang PC. Western blot: technique, theory, and trouble shooting. North American journal of medical sciences. 2012 Sep:4(9):429-34. doi: 10.4103/1947-2714.100998. Epub [PubMed PMID: 23050259]
Liu ZQ, Mahmood T, Yang PC. Western blot: technique, theory and trouble shooting. North American journal of medical sciences. 2014 Mar:6(3):160. doi: 10.4103/1947-2714.128482. Epub [PubMed PMID: 24741558]
Madamanchi NR, Runge MS. Western blotting. Methods in molecular medicine. 2001:51():245-56. doi: 10.1385/1-59259-087-X:245. Epub [PubMed PMID: 21331721]
Taylor SC, Posch A. The design of a quantitative western blot experiment. BioMed research international. 2014:2014():361590. doi: 10.1155/2014/361590. Epub 2014 Mar 16 [PubMed PMID: 24738055]
ALmohaimeed HM, Mohammedsaleh ZM, Batawi AH, Balgoon MJ, Ramadan OI, Baz HA, Al Jaouni S, Ayuob NN. Synergistic Anti-inflammatory and Neuroprotective Effects of Cinnamomum cassia and Zingiber officinale Alleviate Diabetes-Induced Hippocampal Changes in Male Albino Rats: Structural and Molecular Evidence. Frontiers in cell and developmental biology. 2021:9():727049. doi: 10.3389/fcell.2021.727049. Epub 2021 Sep 8 [PubMed PMID: 34568337]
Bhakta A, Gavini K, Yang E, Lyman-Henley L, Parameshwaran K. Chronic traumatic stress impairs memory in mice: Potential roles of acetylcholine, neuroinflammation and corticotropin releasing factor expression in the hippocampus. Behavioural brain research. 2017 Sep 29:335():32-40. doi: 10.1016/j.bbr.2017.08.013. Epub 2017 Aug 8 [PubMed PMID: 28797603]
Meftahi GH, Bahari Z, Zarei Mahmoudabadi A, Iman M, Jangravi Z. Applications of western blot technique: From bench to bedside. Biochemistry and molecular biology education : a bimonthly publication of the International Union of Biochemistry and Molecular Biology. 2021 Jul:49(4):509-517. doi: 10.1002/bmb.21516. Epub 2021 Apr 13 [PubMed PMID: 33847452]
Lück C, Haitjema C, Heger C. Simple Western: Bringing the Western Blot into the Twenty-First Century. Methods in molecular biology (Clifton, N.J.). 2021:2261():481-488. doi: 10.1007/978-1-0716-1186-9_30. Epub [PubMed PMID: 33421009]
Ghosh R, Gilda JE, Gomes AV. The necessity of and strategies for improving confidence in the accuracy of western blots. Expert review of proteomics. 2014 Oct:11(5):549-60. doi: 10.1586/14789450.2014.939635. Epub 2014 Jul 25 [PubMed PMID: 25059473]
Torian LV, Forgione LA, Punsalang AE, Pirillo RE, Oleszko WR. Comparison of Multispot EIA with Western blot for confirmatory serodiagnosis of HIV. Journal of clinical virology : the official publication of the Pan American Society for Clinical Virology. 2011 Dec:52 Suppl 1():S41-4. doi: 10.1016/j.jcv.2011.09.017. Epub 2011 Oct 12 [PubMed PMID: 21995935]
Alexander TS. Human Immunodeficiency Virus Diagnostic Testing: 30 Years of Evolution. Clinical and vaccine immunology : CVI. 2016 Apr:23(4):249-53. doi: 10.1128/CVI.00053-16. Epub 2016 Apr 4 [PubMed PMID: 26936099]
Turner VF. HIV western blot test. The Medical journal of Australia. 1994 Jun 20:160(12):807-8 [PubMed PMID: 8208208]
Level 3 (low-level) evidenceCordes RJ, Ryan ME. Pitfalls in HIV testing. Application and limitations of current tests. Postgraduate medicine. 1995 Nov:98(5):177-80, 185-6, 189 [PubMed PMID: 7479453]
Houn HY, Pappas AA, Walker EM Jr. Status of current clinical tests for human immunodeficiency virus (HIV): applications and limitations. Annals of clinical and laboratory science. 1987 Sep-Oct:17(5):279-85 [PubMed PMID: 3314657]
Lloyd VK, Hawkins RG. Under-Detection of Lyme Disease in Canada. Healthcare (Basel, Switzerland). 2018 Oct 15:6(4):. doi: 10.3390/healthcare6040125. Epub 2018 Oct 15 [PubMed PMID: 30326576]
Porcario C, Hall SM, Martucci F, Corona C, Iulini B, Perazzini AZ, Acutis P, Hamir AN, Loiacono CM, Greenlee JJ, Richt JA, Caramelli M, Casalone C. Evaluation of two sets of immunohistochemical and Western blot confirmatory methods in the detection of typical and atypical BSE cases. BMC research notes. 2011 Sep 29:4():376. doi: 10.1186/1756-0500-4-376. Epub 2011 Sep 29 [PubMed PMID: 21958476]
Level 3 (low-level) evidenceMartins-Gomes C, Silva AM. Western Blot Methodologies for Analysis of In Vitro Protein Expression Induced by Teratogenic Agents. Methods in molecular biology (Clifton, N.J.). 2018:1797():191-203. doi: 10.1007/978-1-4939-7883-0_9. Epub [PubMed PMID: 29896693]
Bass JJ, Wilkinson DJ, Rankin D, Phillips BE, Szewczyk NJ, Smith K, Atherton PJ. An overview of technical considerations for Western blotting applications to physiological research. Scandinavian journal of medicine & science in sports. 2017 Jan:27(1):4-25. doi: 10.1111/sms.12702. Epub 2016 Jun 5 [PubMed PMID: 27263489]
Level 3 (low-level) evidenceMishra M, Tiwari S, Gomes AV. Protein purification and analysis: next generation Western blotting techniques. Expert review of proteomics. 2017 Nov:14(11):1037-1053. doi: 10.1080/14789450.2017.1388167. Epub 2017 Oct 13 [PubMed PMID: 28974114]
Geilfus CM, Mühling KH, Zörb C. A methodical approach for improving the reliability of quantifiable two-dimensional Western blots. Journal of immunological methods. 2010 Oct 31:362(1-2):89-94. doi: 10.1016/j.jim.2010.09.006. Epub 2010 Sep 15 [PubMed PMID: 20837019]
Begum H, Murugesan P, Tangutur AD. Western blotting: a powerful staple in scientific and biomedical research. BioTechniques. 2022 Jun:73(1):58-69. doi: 10.2144/btn-2022-0003. Epub 2022 Jul 1 [PubMed PMID: 35775367]
Ménard AD, Trant JF. A review and critique of academic lab safety research. Nature chemistry. 2020 Jan:12(1):17-25. doi: 10.1038/s41557-019-0375-x. Epub 2019 Nov 18 [PubMed PMID: 31740762]