Introduction
Urea cycle disorders (UCDs) are inborn errors of metabolism (IEMs) resulting from defects in any 1 of the six enzymes or 2 transporters involved in the hepatic removal of ammonia from the bloodstream by conversion to urea, which is excreted by the kidneys [1] IEMs fall into two very broad categories: deficiencies in specific enzymes needed to convert fat or carbohydrate into energy, or deficiencies in specific enzymes needed to break down amino acids or other metabolites thereby allowing them to accumulate and become toxic if not treated. UCDs fall into the second category. The turnover of proteins releases amino acids, and their deamination generates ammonia.
-2H
+H3N-CHR-COO- --> O=CR-COO- + NH4+
+H2O
Ammonia is extremely toxic, particularly to the central nervous system (CNS). Newborns with severe mutations in any one of the first four enzymes of the urea cycle (see figure 1 and below) can become catastrophically ill within 36 to 48 hours of birth despite appearing normal at birth. It is possible, therefore, that a newborn can be discharged from the hospital before signs of UCDs develop and subsequently develop signs that go unrecognized at home. Hyperammonemia is key in diagnosing UCDs, and treatment should not be delayed while trying to reach a final diagnosis.
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
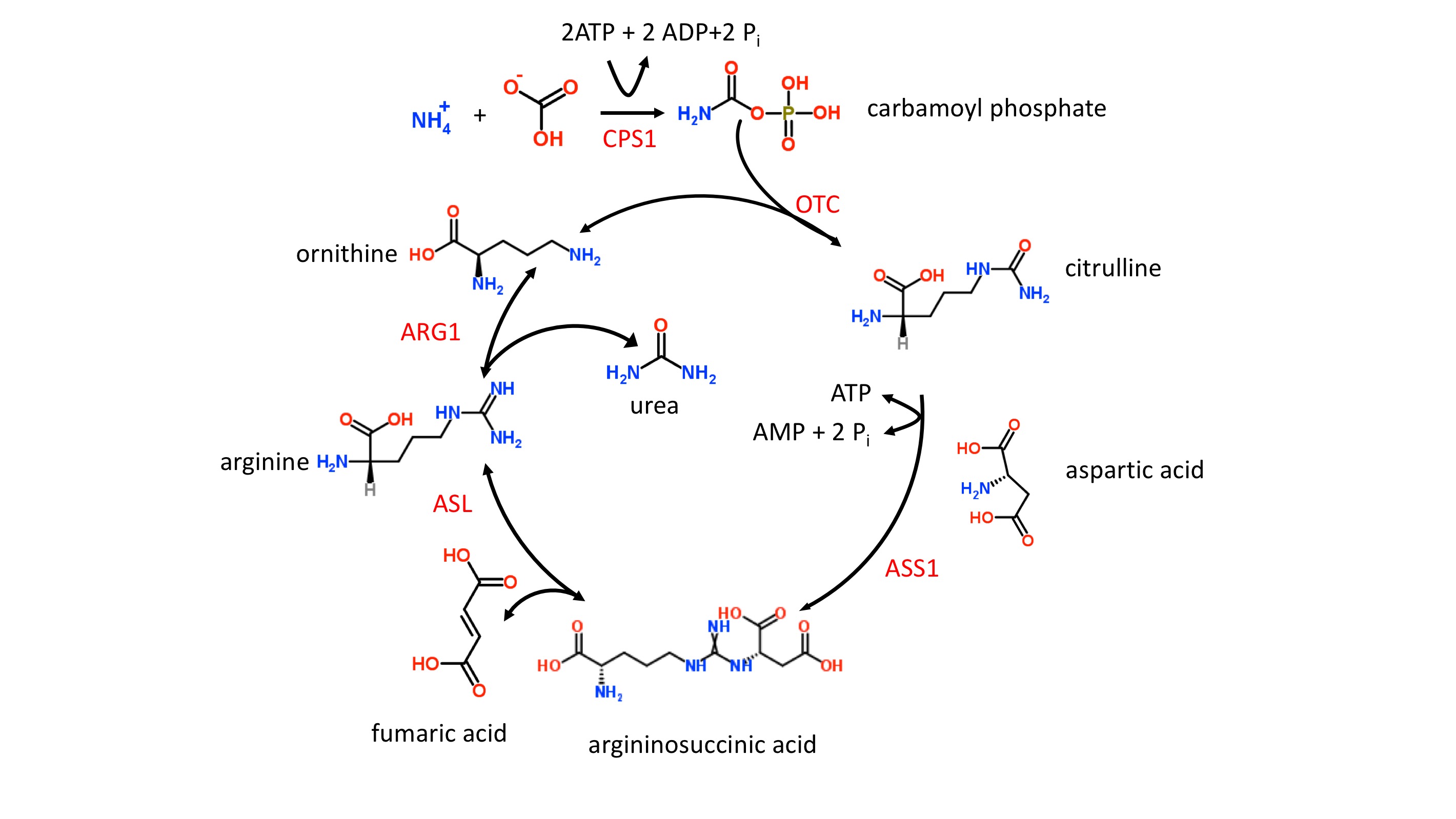
The etiology of UCDs is complex since multiple proteins and 2 subcellular compartments are involved, i.e., the mitochondrial matrix and cytoplasm [2]. The first 2 enzymatic steps in the urea cycle take place in the mitochondrial matrix. In step 1, carbamoylphosphate synthetase I (CPS1) ligates ammonia with bicarbonate, forming carbamoyl phosphate, which enters the urea cycle (figure 1). Stressors such as sepsis can easily initiate defects in CPS1, resulting in the most severe form of UCDs and hyperammonemia. It should be noted that N-acetylglutamate is an allosteric activator of CPS1. N-acetylglutamate is formed from acetyl-CoA and glutamate in a reaction catalyzed by N-acetylglutamate synthase (NAG), a mitochondrial enzyme. Defects in NAG, although very rare, mimic CPS1 UCDs and result in hyperammonemia.
In step 2, the liver enzyme ornithine transcarbamylase (OTC) catalyzes the reaction of carbamoyl phosphate with ornithine to form citrulline (figure 1). The OTC gene (SLC25A15) is located on the X chromosome, and OTC defects are inherited in an X-linked fashion. Defects in OTC are the most common cause of UCDs. OTC deficiency in males results in a severe form of UCD. In females, the severity is variable ranging from asymptomatic to severe depending upon the random nature of X chromosome inactivation. The ornithine needed for OTC must be transported into the mitochondrial matrix, and this task is accomplished by the mitochondrial ornithine translocase (ORNT1) protein. Defective ORNT1, although very rare, can result in hyperammonemia, which often manifests itself later in life.
Step 3 and all remaining enzymatic steps take place in the cytoplasm. In step 3, argininosuccinate synthase 1 (ASS1) catalyzes the formation of argininosuccinate from citrulline and aspartic acid. Defects in ASS1 result in both citrullinemia and hyperammonemia. The second mitochondrial transporter, citrin, contributes to the cytoplasmic pool of aspartic acid by transporting aspartic out of the mitochondria and into the cytoplasm, where it is needed for the urea cycle. Citrin also transports glutamate from the cytoplasm into the mitochondria. Gene SLC25A13 codes for citrin, and defects in this gene can result in both citrullinemia and hyperammonemia.
Step 4 of the urea cycle (figure 1) involves the cleavage of argininosuccinate to form fumaric acid and arginine, and this reaction is catalyzed by argininosuccinate lyase (ASL). Defects in ASL can cause a severe UCD in neonates as well as an adult-onset form. In contrast to the adult-onset form in which hyperammonemia is episodic, the neonatal form results in persistent hyperammonemia if left untreated. Elevated plasma levels of both citrulline and argininosuccinate are characteristic of ASL deficiency.
In the last step of the urea cycle (figure 1), the arginine from step 4 is cleaved into urea and ornithine by the arginase enzyme, which is coded for by the ARG1 gene. Defects in arginase can result in both argininemia and hyperammonemia. The signs and symptoms of arginase deficiency usually appear by 3 years of age and without a rapid onset of hyperammonemia.
Epidemiology
Overall, the incidence of UCDs has recently been determined to be 1 in 35,000 births. Most state screening laboratories currently detect only 2 of all the conditions that cause UCDs. About two-thirds of all USDs are due to mutations in OTC, one-fifth due to ASS1, and one-tenth due to ASL[3].
Pathophysiology
There has been considerable progress in understanding the pathophysiology of UCDs, which may ultimately promote better medical interventions.[4] Hyperammonemia is a key etiological factor in UCDs and plays a key role in CNS toxicity. Although complex, some important concepts are emerging. The toxic effects of ammonia on the CNS are more severe in the developing brain than in the adult brain. Whether the toxic effects of hyperammonemia are reversible or not depends upon the exposure time, the dose of hyperammonemia, and the stage of neurological development. In the neonatal brain, hyperammonemia results in edema caused by swelling of astrocytes. The subsequent frequency and degree of swelling are key factors in determining the severity of CNS neurological disorders such as seizures, coma, and cognitive/motor deficits.
While OTC deficiency (OTCD) is inherited in an X-linked manner, all other UCDs follow an autosomal recessive pattern. Moreover, there is evidence that even asymptomatic female carriers of OTCD can develop cognitive defects as a result of episodic hyperammonemia.
History and Physical
Newborns with UCDs typically appear normal at birth and shortly after that can present with nonspecific signs and symptoms similar to many IEMs or an infection, ie, lethargy, poor appetite, vomiting, and irritability. IEMs, in general, should be considered in the differential diagnosis of any infant with such signs. Except for arginase deficiency, most infants with severe UCDs rapidly develop cerebral edema and subsequent neurological problems resulting from acute hyperammonemia. Unless treated, hyperammonemia from severe UCDs can progress to coma and death. In cases of mild UCDs, hyperammonemia can result from a variety of stressors, such as illness and surgery [4].
Evaluation
A key biomarker for UCDs is hyperammonemia (> 150 micromolar for a neonate and > 100 micromolar for an infant) in the absence of high anion gap and with a normal plasma glucose level. In addition to plasma ammonia, it is useful to obtain plasma amino acids and plasma lactic acid if metabolic acidosis is present. The laboratory finding characteristic of UCDs includes (a) elevated levels of plasma glutamine and alanine, (b) decreased plasma arginine (except for arginase deficiency), and (c) high or low plasma citrulline levels.
Treatment / Management
Treatment for UCDs, or any IEM, should be done in consultation with an appropriate genetics/metabolic expert. For neonates with hyperammonemia, the immediate treatment goal is a rapid lowering of ammonia and reducing dietary protein intake. Hemodialysis is very effective at reducing plasma ammonia and should immediately be initiated if elevated hyperammonemia is observed. Ammonia scavenger medications such as Ammonul IV are also useful. Ammonul IV acts by removing glycine and glutamate from plasma thereby their reducing contribution to ammonia formation.
N-carbamylglutamate (Carbaglu) is an analog of N-acetylglutamate, and small clinical trials have found it to improve ureagenesis in NAGS deficiency and late-onset CPS1 deficiency [5].[6][7] More extensive clinical trials are underway.
Differential Diagnosis
- Neonatal sepsis
- Liver failure
- Congenital infection
- Transient hyperammonemia
- Exogenous toxins
- Medication reaction
- Reye syndrome
- fatty acid oxidation defects
- Lactic acidemia
- Citrin deficiency
- Transient hyperammonemia of the newborn
- Organic aciduria
- Fatty acid oxidation disorder
Enhancing Healthcare Team Outcomes
The diagnosis and management of urea cycle disorders are complex and best done with an interprofessional team that includes a geneticist, pathologist, pediatrician, nephrologist, internist, biochemist, and dialysis nurse/nurse practitioner. Treatment for UCDs, or any IEM, should be done in consultation with an appropriate genetics/metabolic expert. For neonates with hyperammonemia, the immediate treatment goal is a rapid lowering of ammonia and reducing dietary protein intake. Hemodialysis is very effective at reducing plasma ammonia and should immediately be initiated if elevated hyperammonemia is observed. Ammonia scavenger medications such as an IV nitrogen binding agent are also useful. These agents act by removing glycine and glutamate from plasma, thereby reducing their contribution to ammonia formation.
Media
(Click Image to Enlarge)
Urea Cycle Disorder Contributed by William Stone, MD
References
Braissant O. Current concepts in the pathogenesis of urea cycle disorders. Molecular genetics and metabolism. 2010:100 Suppl 1():S3-S12. doi: 10.1016/j.ymgme.2010.02.010. Epub 2010 Feb 14 [PubMed PMID: 20227314]
Level 3 (low-level) evidenceAdam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, Ah Mew N, Simpson KL, Gropman AL, Lanpher BC, Chapman KA, Summar ML. Urea Cycle Disorders Overview. GeneReviews(®). 1993:(): [PubMed PMID: 20301396]
Level 3 (low-level) evidenceSummar ML, Koelker S, Freedenberg D, Le Mons C, Haberle J, Lee HS, Kirmse B, European Registry and Network for Intoxication Type Metabolic Diseases (E-IMD). Electronic address: http://www.e-imd.org/en/index.phtml, Members of the Urea Cycle Disorders Consortium (UCDC). Electronic address: http://rarediseasesnetwork.epi.usf.edu/ucdc/. The incidence of urea cycle disorders. Molecular genetics and metabolism. 2013 Sep-Oct:110(1-2):179-80. doi: 10.1016/j.ymgme.2013.07.008. Epub 2013 Jul 18 [PubMed PMID: 23972786]
Waisbren SE, Gropman AL, Members of the Urea Cycle Disorders Consortium (UCDC), Batshaw ML. Improving long term outcomes in urea cycle disorders-report from the Urea Cycle Disorders Consortium. Journal of inherited metabolic disease. 2016 Jul:39(4):573-84. doi: 10.1007/s10545-016-9942-0. Epub 2016 May 23 [PubMed PMID: 27215558]
Daniotti M, la Marca G, Fiorini P, Filippi L. New developments in the treatment of hyperammonemia: emerging use of carglumic acid. International journal of general medicine. 2011 Jan 7:4():21-8. doi: 10.2147/IJGM.S10490. Epub 2011 Jan 7 [PubMed PMID: 21403788]
Allegri G, Deplazes S, Rimann N, Causton B, Scherer T, Leff JW, Diez-Fernandez C, Klimovskaia A, Fingerhut R, Krijt J, Kožich V, Nuoffer JM, Grisch-Chan HM, Thöny B, Häberle J. Comprehensive characterization of ureagenesis in the spf(ash) mouse, a model of human ornithine transcarbamylase deficiency, reveals age-dependency of ammonia detoxification. Journal of inherited metabolic disease. 2019 Nov:42(6):1064-1076. doi: 10.1002/jimd.12068. Epub 2019 Mar 13 [PubMed PMID: 30714172]
Buerger C, Garbade SF, Dietrich Alber F, Waisbren SE, McCarter R, Kölker S, Burgard P, Urea Cycle Disorders Consortium. Impairment of cognitive function in ornithine transcarbamylase deficiency is global rather than domain-specific and is associated with disease onset, sex, maximum ammonium, and number of hyperammonemic events. Journal of inherited metabolic disease. 2019 Mar:42(2):243-253. doi: 10.1002/jimd.12013. Epub 2019 Jan 22 [PubMed PMID: 30671983]
Cho H. Renal replacement therapy in neonates with an inborn error of metabolism. Korean journal of pediatrics. 2019 Feb:62(2):43-47. doi: 10.3345/kjp.2018.07143. Epub 2018 Nov 7 [PubMed PMID: 30404428]
Vara R, Dhawan A, Deheragoda M, Grünewald S, Pierre G, Heaton ND, Vilca-Melendez H, Hadžić N. Liver transplantation for neonatal-onset citrullinemia. Pediatric transplantation. 2018 Jun:22(4):e13191. doi: 10.1111/petr.13191. Epub 2018 May 3 [PubMed PMID: 29726081]