Introduction
Tuberous sclerosis complex (TSC) is a genetic disorder inherited in an autosomal dominant fashion and is characterized by an increased predisposition to hamartoma formation.[1] TSC results from mutations in the genes TSC1 and TSC2 and is known for causing neurological disorders, including epilepsy and intellectual disability.[2] Other organ systems involved include pulmonary, renal, dermatologic, and cardiac. TSC is usually diagnosed in childhood or infancy, and the affected individuals may present with developmental delay, skin manifestations, or seizures. However, it may also be diagnosed earlier or later based on various clinical manifestations.[3]
Some manifestations, such as cardiac rhabdomyomas or cortical tubers, may be present prenatally. Other signs, including renal and pulmonary lesions, are commonly diagnosed in adulthood.[2][3] The presentation of the disease will vary depending on the individual's developmental stage. While skin lesions are detected in 90% of patients of all ages, hypopigmented macules are usually found in early childhood. Ungual fibromas appear near puberty, and facial angiofibromas are more common in adolescence.[2] This disease has a highly variable clinical course. The prognosis may be uncertain, and follow-up requires a comprehensive evaluation, often in specialized institutions. This disorder may be overwhelming for some patients and family members; thus, education and counseling play a vital role.[1][4]
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
Tuberous sclerosis complex arises from mutations in the genes TSC1 (9q34) or TSC2 (16p13.3), encoding hamartin and tuberin, respectively.[1][5] A broad spectrum of mutations has been described in both genes and while no particular regions seem more liable to mutations, the frequency is consistently higher for TSC2 than TSC1.[2] Among patients who meet the clinical criteria for tuberous sclerosis, approximately 15% have no identifiable genetic mutations.[6]
TSC1 and TSC2 Gene Mutations
There are 2 specific gene mutations:
- TSC1 mutation occurs on chromosome 9 and is related to hamartin protein production.
- TSC2 is on chromosome 16 and affects tuberin protein production.
These genes are thought to cause the characteristic tumors of the condition. The gene mutation may be inherited or may occur spontaneously. Most cases present sporadically, with no known family history, but approximately 1 in 3 patients inherit a defective TSC1 or TSC2 gene. If a parent has tuberous sclerosis, their children will carry a 50% risk of inheriting the disease. Because it follows an autosomal dominant inheritance pattern, and men and women are equally affected.
Hamartin and Tuberin Protein
Hamartin and tuberin play a role in a complex that controls cell growth and division in the body. These proteins modulate gene transcription and suppress tumor growth.[7]
Role in Central Nervous System Development
TSC1/TSC2 mutations inactivate the inhibitory TSC protein complex, allowing aberrant activation of the mTOR pathways. The role of mTOR is to regulate cell growth and metabolism through the multimeric complexes mTORC1 and mTORC2. The complex mTORC1 integrates inputs from growth factors, amino acids, energy status, and hypoxia stressors to phosphorylate p70S6K and 4E-BP1 and promote cell proliferation. Without the TSC protein complex, the RAS homolog Rheb hyperactivates mTORC1. This reprogramming inhibits autophagy and favors anaerobic glycolysis. Therefore, mTORC1 aids solid tumor formation.[8][14]
The mTOR pathway is especially significant in the central nervous and dermatologic systems. This pathway involves neurocortical lamination, neuronal migration, dendrite arborization, and neuronal polarity. Abnormalities in these processes contribute to the epilepsy and neurocognitive issues found in TSC. The hyperactivity of mTOR also inhibits melanogenesis, leading to hypopigmented areas.[12][14] Tuberous sclerosis is also associated with vascular epithelial growth factor D, VEGF-D. Higher levels have especially been associated with lymphangioleiomyomatosis and renal angiomyolipomas, and levels of VEGF-D have been shown to decrease with mTOR inhibition. However, this is not commonly measured outside of research settings.[14]
Epidemiology
Tuberous sclerosis complex affects approximately 1 in 6000 to 1 in 10,000 live births, with an overall prevalence of 1 in 20,000. Clinical presentation is extremely variable, usually affecting multiple organs and involving all racial groups.[1] In a longitudinal study involving 125 patients, the median age of presentation was 7 months.[9] Pulmonary lymphangioleiomyomatosis is usually diagnosed in women with TSC around age 35.[7]
Pathophysiology
Tuberous sclerosis involves mutations in genes TSC1 or TSC2, which are responsible for the production of proteins that regulate cell division and growth in the body. When gene mutations disrupt this balance, hamartomas may grow in the brain, skin, heart, kidney, liver, and lungs, affecting the function of these organs.[10][11][12] (See Figure. Tuberous Sclerosis Complex.)
Affected Areas and Organs
Tuberous sclerosis causes tumors to grow in multiple locations, resulting in a variation in the severity of the condition between individuals.
- Dermatologic manifestations include skin and nail lesions, which usually do not cause serious problems but can result in unwanted aesthetic effects.[13]
- Skin lesions include facial angiofibromas (75%), shagreen patches (>50%), ungual fibromas (20%–80%), focal hypopigmentation (90%), and fibrous cephalic plaques (25%).[14]
- In addition, retinal astrocytic hamartomas were observed in 40% to 50% of patients with TSC, especially those with gene mutations.[15]
- Oral manifestations include gingival fibromas and dental enamel pits. The incidences are 20% to 50% and 90%, respectively.[14]
- Brain tumors often cause seizures or developmental delays. About 80% to 90% of TSC patients suffer from seizures.[14]
- Cortical, subcortical, and subependymal tubers are common; several studies have documented an association between the number of tubers with cognitive impairment and seizures.[16][17][18]
- Subependymal nodules are seen in 80% to 90% of patients, while about 10% to 20% develop subependymal giant cell astrocytomas (SEGAs).[14]
- Neurodevelopmental delays and behavioral issues are seen in up to 90% of patients with TSC and are further discussed in the history and physical section.[14]
- Renal angiomyolipoma and cysts may affect renal function; complications include pain, bleeding, and possible renal failure.
- Angiomyolipomas are very common, affecting up to 75% of patients. Please see StatPearls’ accompanying reference, “Renal Angiomyolipoma,” for further information.
- More severe but less common renal manifestations include polycystic kidney disease (PKD) and renal cell carcinoma, with incidences of about 5% and 3%, respectively.[14] The TSC2 and PKD1 genes are adjacently located; therefore, a TSC2 mutation can be partially or completely accompanied by PKD1 mutation. This is called the contiguous gene syndrome. The renal phenotype in these patients is often more severe, with early progression to renal failure. In addition, some TSC patients suffer from simple cystic disease without a PKD1 mutation, and phenotypically, these patients are often indistinguishable.[14][19]
- Cardiac rhabdomyomas are present in the majority of patients with TSC prenatally. Although they usually spontaneously regress by early childhood, cardiac rhabdomyomas can cause neonatal death. They can also re-present or occur de novo in adolescence, especially in girls. About 20% of adults with TSC are thought to have cardiac rhabdomyomas, but these are usually asymptomatic.[14]
- Pulmonary cysts and lymphangioleiomyomatosis (LAM) can affect the lungs and are the most common cause of fatality for patients who exhibit evidence of this condition. The incidence of LAM in patients with tuberous sclerosis is about 30%, and the median age of diagnosis is about 35.[7]
- This complication presents almost exclusively in women of childbearing age and worsens with high estrogen exposure, such as during pregnancy. Considered a low-grade neoplasm, it can also recur in transplanted lungs.[20] One study using screening by lung computed tomography (CT) scan of males with TSC found cystic lung changes in 10% to 38% of patients (men and women); however, only a small minority were symptomatic.[21] Please see StatPearls companion reference, “Lymphangioleimyomatosis,” for further review.
- Another possible pulmonary complication is multifocal micronodular pneumocyte hyperplasia, which affects males and females equally and has an incidence of about 40% to 60%, although the vast majority are asymptomatic. This disease is characterized by small type II pneumocyte nodular lung deposits.[21]
- Hepatic angiomyolipomas, cysts, and other benign lesions are seen in about 30% of patients with TSC, and some lesions grow significantly over time. However, these lesions are largely asymptomatic.[22]
Histopathology
Pulmonary pathology
Lymphangioleiomyomatosis demonstrates diffuse, bilateral proliferation of smooth muscle-like cells in the lungs expressing melanocyte lineage markers, including gp100. There are 2 types of LAM cells: small spindle-shaped cells with eosinophilic cytoplasm and epithelioid-like cells with abundant cytoplasm.[19] The gold standard for the pathologic diagnosis of LAM is immunoreactivity with the HMB-45 antibody, which recognizes gp100. These LAM cells exist within microscopic nodules containing other cell types, including lymphatic vessels and lymphocytes.[20] Both small spindle-shaped cells and epithelioid-like cells react with antibodies against smooth-muscle cell-specific antigens (eg, α-actin, desmin, and vimentin). The cystic destruction of elastic fibers and collagen in the basal membrane of LAM cells is mediated by the secretion of matrix metalloproteinases. Sex hormone receptors are always present in LAM cells. Interestingly, the muscle component of renal angiomyolipomas has an identical structure and immunohistochemical profile as LAM cells.[19]
Brain pathology
Cortical tubers include giant dysplastic neurons and astrocytes. Microtubers may also be present in normal-appearing white matter. Subependymal nodules are structures primarily arising along the lateral and third ventricle walls and are present in over 80% of patients with TSC. Approximately 5% to 15% of these tubers transform into subependymal giant cell astrocytomas (SEGAs). SEGAs are comprised of ganglion-like giant cells expressing neuronal and astrocytic markers.[21]
Skin lesions
Of the many skin manifestations seen in TSC, some common patterns emerge: concentric fibrosis/thickened collagen, vascular hyperplasia, fibroblast proliferation, and decreased elastic fibers. The most indicative of TSC is perifollicular fibrosis with atrophy and compression of the skin adnexa. Cascarino et al have methodically detailed the cutaneous findings as follows.[15]
Hypomelannotic (ash-leaf) lesions demonstrate a normal density of active melanocytes but significantly decreased epidermal melanin pigment in the epidermis. Electron microscopy exhibits a reduced number of immature, smaller, and less melanized melanosomes in melanocytes and keratinocytes due to a lack of mTOR pathway inhibition (which normally aids melanogenesis). Hypopigmented macules have been microscopically studied after topical rapamycin application. Topical rapamycin substantially improved hypopigmented macules and normalized some of the melanosome abnormalities in the treated skin.
Angiofibromas present as dome-shaped lesions with normal or sometimes hyperplastic epidermis. In the dermis, the collagen tissue and vascular structures expand hypertrophically. In older lesions, the collagen becomes more dense and sclerotic. Around the skin adnexa, a perifollicular arrangement of collagen compresses these structures, sometimes replacing them with concentric collagen bundles. Other characteristics include dilated venules throughout, decreased or absent elastic tissue, and increased stellate cells clustering around the dilated blood vessels. Sometimes, multinucleated giant cells are visualized. See Image. Tuberous Sclerosis Angiofibroma.
Periungual Fibromas have pathology similar to angiofibromas but with increased vascularity and dense collagen that can extend to the hypodermis. Stellate cells are also present. See Figure. Periungual Fibroma of Tuberous Sclerosis.
Fibrous cephalic plaques (forehead plaques) have pathological features similar to angiofibromas. Distinguishing features are more prominent vascular dilatation and more sclerosis and hyalinization of collagen with concentric perifollicular fibrosis causing atrophy and compression of the follicle.
- Shagreen patches are similar to collagenomas (or collagenic hamartomas). The dermis is replaced by hyaline collagen, which is dense and mostly acellular extending down to the subcutaneous fat.
- As with fibromas and forehead plaques, follicles show concentric perifollicular collagen with follicular atrophy and compression. Elastic fibers are thin or absent.[15] See Figure. Shagreen Patch of Tuberous Sclerosis.
History and Physical
The clinical presentation of TSC is highly variable, and manifestations will continue developing over a patient’s lifetime.[5] Central nervous system manifestations are common. As noted above, seizures are present in up to 90% of patients with TSC, usually presenting before age 3.[14] Subependymal giant cell astrocytomas are present in up to 20% of patients with TSC, usually presenting in the first 2 decades of life; they can cause mass effect, hydrocephalus, and even death.[16][22] Neuropsychiatric manifestations are present in up to 90% of patients with TSC, including intellectual disability, behavioral difficulties, attention deficit hyperactivity disorder, obsessive-compulsive disorder, and autism spectrum disorders.[1][14] About 50% of patients with TSC show signs of having autism spectrum disorder. Some evidence shows improvement of all these entities with the use of mTOR inhibitors.[22]
Skin manifestations are very common, and various skin manifestations are included in the major criteria for diagnosing tuberous sclerosis. Facial angiofibromas present in 75% of patients.[2][3] Hypomelanotic macules are the most common dermatological manifestation, present in approximately 90% of patients as lighter patches of skin (ie, ash leaf or confetti lesions). Fibrous cephalic plaques present most commonly on the forehead, but they can be anywhere on the face or scalp. These plaques appear as smooth or bumpy lesions, skin-colored, pink, red, or brown.[23] Facial angiofibromas commonly follow a butterfly or malar distribution [24], while Shagreen patches are present in areas of thicker skin as a leathery lesion with a pebbly texture.[3] Periungual fibromas are also common and can appear after trauma; about 90% are on the toes. Please see StatPearls’ companion reference, “Cutaneous Angiofibromas,” for further information. See Image. Tuberous Sclerosis Ash Leaf.
Lymphangioleiomyomatosis (LAM) occurs primarily in women of childbearing age; studies have shown a wide range of patients with TSC who have radiographic imaging evidence of LAM, ranging from 30% to 80%, with rates increasing with age. Fortunately, the vast of patients with LAM are asymptomatic and have only radiographic evidence of LAM.[14][28] Presenting symptoms may be chest pain or dyspnea, which, when sudden, could represent a spontaneous pneumothorax.[20] Chylothorax is also a possible presentation and can present with progressive dyspnea or chyloptysis.[29]
Cardiac lesions may be present in 50% to 70% of patients at some time. As noted above the lesions can cause neonatal death, but they often regress by early childhood.[2] A case series involving 125 patients reported that cardiac rhabdomyomas were the second most common presentation of TSC in early childhood or infancy (second to seizures).[9] Renal angiomyolipomas may manifest in 55% to 75% of patients.[6][30] Incidence increases with age, and a longitudinal study showed a 75% prevalence of renal angiomyolipomas by the age of 10.5 years.[30] These angiomyolipomas are often asymptomatic, but symptoms can include flank pain, hemorrhage, hematuria, and a tender abdominal mass.[20][31] The diagnosis of tuberous sclerosis, discussed below, consists of major and minor clinical criteria.[2] The diagnosis is often first made during childhood, when several dermatological features may become apparent.[32]
Evaluation
Tuberous sclerosis is mainly diagnosed based on clinical criteria, but it can also be diagnosed with genetic testing. Genetic testing is not required if a patient fulfills the clinical criteria, although it may provide useful information for other family members.[5] Genetic testing of TSC1 and TSC2 is positive for mutations in 75% to 95% of individuals affected with TSC. Currently, reliable screening tests for TSC1 and TSC2 detect pathogenic mutations causing inactivation of the TSC1 or TSC2 proteins, leading to the loss of inhibition of mTOR.[25][26]
As per the recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference, the diagnostic criteria for tuberous sclerosis include the following major and minor features:
Major Features
- Hypomelanotic macules (>2 at least 5 mm in diameter)
- Angiofibromas (>2) or a fibrous cephalic plaque
- Ungual fibromas (>1)
- Shagreen patch
- Multiple retinal hamartomas
- Cortical dysplasias
- Subependymal nodules
- Subependymal giant cell astrocytoma
- Cardiac rhabdomyoma
- Lymphangioleiomyomatosis
- Angiomyolipomas (>1)
Minor Features
- Confetti skin lesions
- Dental enamel pits (>3)
- Intraoral fibromas (>1)
- Retinal achromic patch
- Multiple renal cysts
- Nonrenal hamartomas [5][27]
Definitive diagnosis is established in patients with 2 major features or 1 major feature with at least 2 minor features, while “possible diagnosis” is recognized in patients with 1 major feature or at least 2 minor features.[5][27] The appropriate diagnosis should be made using either clinical criteria or genetic testing. As most patients present with dermatological features, the physical examination should include a careful skin examination using a Wood’s lamp to locate hypomelanotic lesions.[13] In addition, a thorough funduscopic examination is warranted to evaluate for retinal hamartomas. Imaging tests prove useful in identifying lesions such as cardiac rhabdomyomas through echocardiography. Intracranial tubers and subependymal tumors can be visualized through magnetic resonance imaging (MRI) or CT. Renal angiomyolipomas can be identified by either ultrasonography, CT, or MRI, but they generally require CT or MRI to rule out malignancy and fully characterize the lesion.[2]
Treatment / Management
The 2012 TSC consensus recommended that patients undergo lifetime surveillance to monitor for common manifestations.[5] For children with tuberous sclerosis, an every 1- or 3-year lifetime surveillance, including imaging studies, has been suggested.[4][27] As discussed, the follow-up for TSC should be comprehensive, and it commonly presents a wide variety of multi-systemic complications. A comprehensive dermatologic evaluation may aid in the early recognition of angiofibromas that may eventually cause cosmetic disfigurations requiring laser therapy or surgical removal.[2] Skin and dermatologic manifestations are also often responsive to mTOR inhibitors. Topical rapamycin has also been used with good effect for neural angiofibromas and seems to be more efficacious in children than adults with fewer adverse effects than systemic mTOR administration.[14][15](B3)
Anticonvulsants are often required for TSC-associated epilepsy, especially vigabatrin for infantile spasms. However, about one-third of patients have treatment-refractory seizures, and surgery may be needed. Emergent neurosurgery may also be required for complications like hydrocephalus.[22] As recommended by the 2012 consensus for tuberous sclerosis, treatment of acutely symptomatic subependymal giant cell astrocytomas (SEGA) (primarily complications of obstructive hydrocephalus) is surgical resection.[27] Surgical resection for symptomatic SEGA, however, is associated with a substantial increase in mortality and comorbidities, including hemiparesis, bleeding, infection, and cognitive decline. Asymptomatic SEGA is usually treated with mTOR inhibitors, including sirolimus, everolimus, and rapamycin.[28][29](B3)
For renal angiomyolipomas larger than 3.5 cm, arterial embolization is recommended to avoid total nephrectomy and decrease the incidence of renal complications. Despite this effort, however, the incidence of complications, including chronic kidney disease, is similar in TSC patients who undergo partial versus complete nephrectomy.[30][31] For lesions measuring larger than 3 cm, treatment with mTOR inhibitors is considered first-line therapy. Bleeding from renal angiomyolipomas is a cause of mortality in TSC patients, and for those presenting with acutely bleeding renal angiomyolipoma, arterial embolization followed by corticosteroids is considered the treatment of choice.[1] Post-embolization recurrence is common, and both arterial embolization and surgical removal (including nephron-sparing surgery) are second-line options if mTOR inhibition is not sufficient.(B2)
Renal angiomyolipomas are present in 50% to 75% of patients with TSC. Indications for treatment are a size larger than 6 cm diameter, associated clinical symptoms, women desiring pregnancy, and other high-risk situations. Please see StatPearls accompanying reference, "Renal Angiomyolipoma," for further information. Both sirolimus and everolimus are used for renal angiomyolipoma treatment and and considered first-line treatments, preferable to surgical removal; these mTOR inhibitors are also used intermittently for maintenance therapy to prevent regrowth of the angiomyolipoma.[1][32] The use of mTOR inhibition has also been studied for associated polycystic kidney disease and renal cystic disease, and it is thought that mTOR inhibitors are effective in reducing cyst size and number.[33]
Lymphangiomyomatosis (LAM) can also be treated with mTOR inhibitors.[34] European Respiratory System guidelines recommend screening all females for LAM starting at age 18 and then every 5 to 7 years until menopause thereafter, or if they develop respiratory symptoms, or in males with respiratory symptoms.[13][35] For patients with LAM, some sources recommend follow-up with an annual pulmonary function test to monitor for lung function deterioration.[2] Chylothorax occurs in about 7% to 10% of patients with LAM and has traditionally been treated with a fat-free or low-fat diet, which can be restrictive and place patients at risk for malnutrition. Thoracentesis, pleurodesis, and thoracic duct ligation are surgical techniques that have also been used. However, multiple recent studies have shown sirolimus effective in controlling this rare complication.[36](A1)
Differential Diagnosis
The differential diagnosis will vary and depend on each case's clinical manifestations. Some possible conditions in the differential diagnosis include the following: TSC and neurofibromatosis type 1 (NF1) are both neurocutaneous disorders with autosomal dominant inheritance. NF1 is not usually associated with neurocognitive problems and seizures, both of which are extremely common with TSC. The morbidity of NF1 is primarily related to central nervous system and nerve sheath tumors. NF1 is diagnosed with multiple cafe-au-lait spots and may be associated with bone deformities. Other neurocutaneous syndromes with similar symptoms include neurofibromatosis type 2, ataxia telangiectasia, von Hippel Lindau syndrome, and Sturge-Weber syndrome. Please see the associated references for further review.[43][44] Birt-Hogg-Dubé syndrome and multiple endocrine neoplasia type 1 (MEN1) can present with facial angiofibromas, but these usually develop at a later age than in TSC.[5]
Prognosis
The prognosis for TSC includes the following:
- Each patient will present different manifestations of the disease and will, therefore, follow different clinical courses.[4] The morbidity of TSC is quite significant for patients and caregivers. In addition, mortality is significantly higher than in patients without TSC, and various long-term studies report mortality at 4.8% to 8.3% at a follow-up range of 8 to 17.4 years.[37]
- Mortality is most often caused by complications of seizures or renal angiomyolipomas.[37]
- Other causes of mortality include bronchopneumonia, complications of LAM, and neonatal heart failure related to rhabdomyomas.[14]
- The 5-year survival rate of patients receiving lung transplants due to LAM is about 65%.[36]
Complications
Tuberous sclerosis affects multiple systems and may manifest in various tissues within a lifetime. As detailed above, renal manifestations are common in patients with tuberous sclerosis complex, especially renal angiomyolipomas. Other common renal lesions include polycystic kidney disease, renal cysts, and renal cell carcinomas (RCC). The latter has a similar lifetime risk to the general population; however, they usually present at a younger age.[2]
Up to 56% of patients with lymphangioleiomyomatosis (LAM) may develop pneumothorax, and of those who do, recurrence is likely, with an average recurrence of 4.4 times. For this reason, pleurodesis is recommended in patients who develop pneumothorax, although even with this procedure, the recurrence rate is between 18% and 32%.[36] A newer surgical technique called total pleural covering uses oxidized regenerated cellulose mesh to wrap the visceral pleura and has shown promising outcomes.[36][38]
In addition, patients with LAM are more likely to develop pneumothorax with air travel (which involves changes in barometric pressure). One study found the rate at 1.1% per flight in LAM patients.[36][20] Subependymal giant cell astrocytomas may cause obstructive hydrocephalus due to their location near Monro's foramen and size. As previously discussed, surgery for these lesions also carries a substantial risk of mortality and other complications.[39][40] Seizures are common in tuberous sclerosis and often refractory to medical treatment; for this reason, they may need to be treated surgically.[3]
Although mTOR inhibitors have greatly improved the prognosis and decreased morbidity and mortality of TSC, they still have associated adverse effects, including stomatitis, increased infections, and menstrual irregularities. The most serious adverse event associated with everolimus in one long-term study was pneumonia; results also found adverse effects were more severe in children than adults.[41] Less common adverse effects include skin rash, hyperlipidemia, hyperglycemia, myelosuppression, and proteinuria. Less severe adverse effects can usually be treated with temporary cessation or dose reduction.[38]
Consultations
The following healthcare professions can provide consults for this condition:
- Pediatrics
- Nephrology
- Neurology
- Neurosurgery
- Cardiology
- Ophthalmology
- Dermatology
Deterrence and Patient Education
Tuberous sclerosis is a disease with a highly variable clinical course and has a wide range of possible manifestations. As such, the uncertainty and complexity of this disease may be daunting, particularly for family members.[1][4] Genetic counseling is useful for patients and parents of children with tuberous sclerosis complex. Those parents affected should be advised that the risk of having an affected child is approximately 50%.[2] For parents who are not affected by the disease, the risk of having another child with TSC is around 1% to 2%. Parents may also be tested as they present the disease after their children (parents may present mosaicism).[42]
As noted above, estrogen exposure (through birth control pills or pregnancy) can worsen LAM symptoms, and women should be counseled in advance of pregnancy regarding the use of birth control pills with estrogen. Patients with LAM are also at risk of spontaneous pneumothorax, especially with a history of pneumothorax or with air travel, and should be educated about these risks. They should also receive appropriate pneumovax and other vaccines for upper respiratory infections.[20] Patients should be counseled on using sunscreen, especially on hypomelanotic lesions.[13][35]
Pearls and Other Issues
About 75% to 90% of patients with SC carry a mutation in TSC1 or TSC2, which encodes the proteins hamartin and tuberin, which are inhibitor complexes considered to be tumor suppressor genes. Mutations in these proteins cause benign brain, skin, lung, heart, liver, and kidney hamartomas. Neurologic and dermatologic involvement are the most prominent early symptoms. About 90% of patients will have neurologic symptoms, including epilepsy and neurocognitive issues; infantile spasms requiring treatment with vigabatrin are especially common. About 90% will have dermatologic manifestations, with focal hypomelanotic areas (ash leaf lesions) being the most common. Other common dermatologic manifestations are facial angiofibromas, shagreen patches, ungual fibromas, and fibrous cephalic plaques. In addition, retinal astrocytic hamartomas are seen in 40% to 50% of patients with TSC, especially those with gene mutations.
Renal angiomyolipomas are present in about 75% of patients by age 10. The likelihood of bleeding or other complications is related to size, vascularity, and other factors. Mortality is highly linked to complications of seizures and renal angiomyolipomas. Lymphangioleiomyomatosis is the presence of smooth muscle tissue in the lungs associated with cystic loss of lung parenchyma. It is most common in women of childbearing age, and while many TSC patients have radiographic evidence of LAM, symptomatic LAM is much less common. Many experts recommend against estrogen-containing birth control pills as this could exacerbate LAM progression in women. Progesterone-only pills do not seem to have the same exacerbating effect. Pregnancy must be undertaken cautiously for the same reason, and pregnant women require frequent monitoring. The recognition that mTOR inhibitors (rapamycin, sirolimus, everolimus) greatly slow the progression of hamartomas in many tissues, including the brain, kidney, lung, and skin, has greatly changed the management of TSC. Surgical management is still warranted in some cases, and genetic counseling is advised, given the autosomal dominant nature of the disease.
Enhancing Healthcare Team Outcomes
Patients with tuberous sclerosis are best managed with an interprofessional team approach. Specialties include primary care providers, pediatrics, dermatology, nephrology, neurology, neurosurgery, cardiology, ophthalmology, radiology, surgery, and interventional radiology. Primary care providers and pediatricians should provide annual examinations for patients with tuberous sclerosis, focusing on the common manifestations and complications of tuberous sclerosis, and conduct a thorough multisystemic history and physical. Several clinics in the United States specialize in the follow-up and care of patients with tuberous sclerosis. Referral of patients to these centers should ensure that they will receive a comprehensive follow-up. When this is impossible, individual specialty referral should be conducted for the different manifestations, such as epilepsy, lymphangioleiomyomatosis, and renal angiomyolipomas.
A strategic approach is equally crucial, involving evidence-based strategies to optimize treatment plans and minimize adverse effects. Ethical considerations must guide decision-making, ensuring informed consent and respecting patient autonomy in treatment choices. Each healthcare professional must know their responsibilities and contribute their unique expertise to the patient’s care plan, fostering a multidisciplinary approach. Effective interprofessional communication is paramount, allowing seamless information exchange and collaborative decision-making among the team members. Care coordination plays a pivotal role in ensuring that the patient’s journey from diagnosis to treatment and follow-up is well-managed, minimizing errors and enhancing patient safety. By embracing these principles of skill, strategy, ethics, responsibilities, interprofessional communication, and care coordination, healthcare professionals can deliver patient-centered care, ultimately improving patient outcomes and enhancing team performance in the management of tuberous sclerosis.
Media
(Click Image to Enlarge)
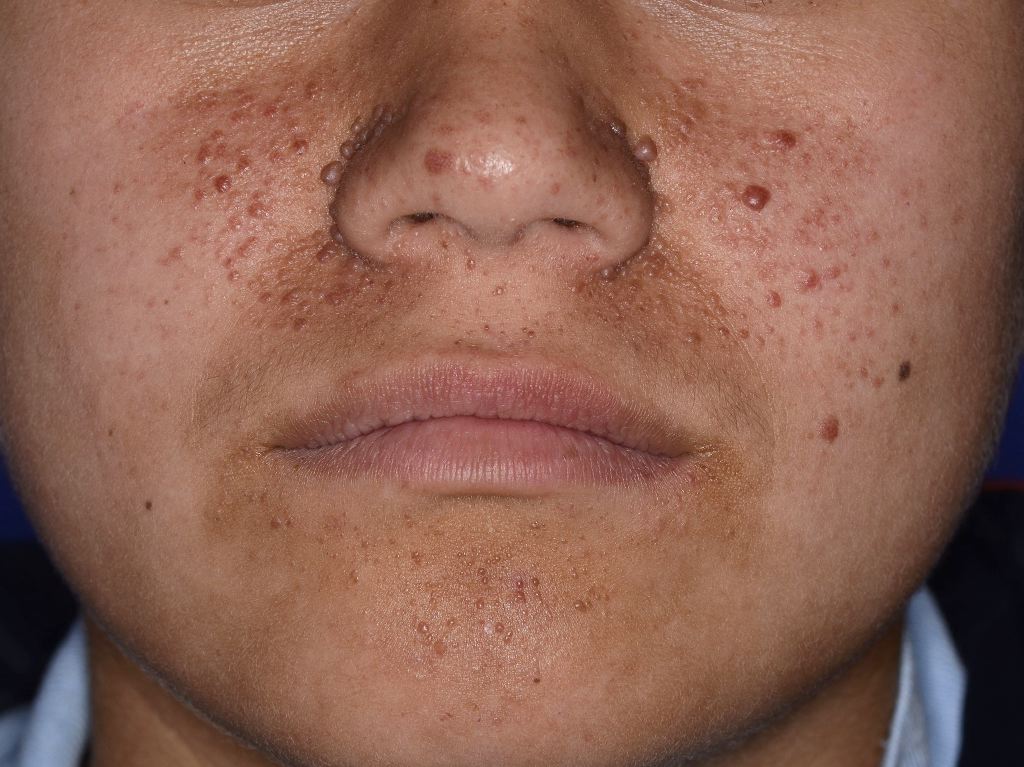
Facial Angiofibromas Observed in Tuberous Sclerosis. Facial angiofibromas are considered one of the most apparent clinical presentations of tuberous sclerosis, an autosomal dominant hamartomatous disorder that affects the skin, kidneys, heart, brain, and lungs.
(Click Image to Enlarge)
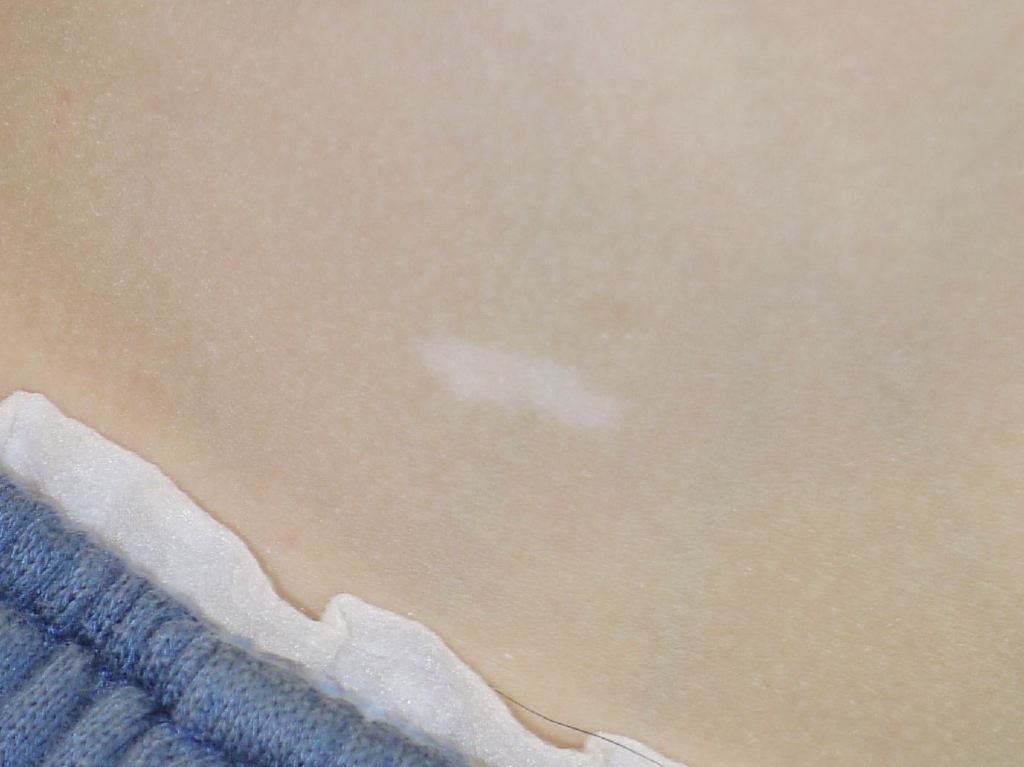
Tuberous Sclerosis Ash Leaf. The ash-leaf spot is the earliest skin lesion in patients with tuberous sclerosis.
(Click Image to Enlarge)
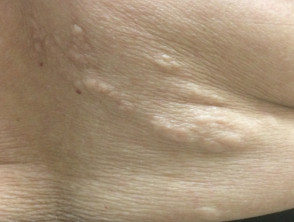
Shagreen Patch of Tuberous Sclerosis. These patches are specific for tuberous sclerosis and present as large plaques on the lower back, with a bumpy or orange-peel surface. They appear commonly in children younger than 10.
(Click Image to Enlarge)
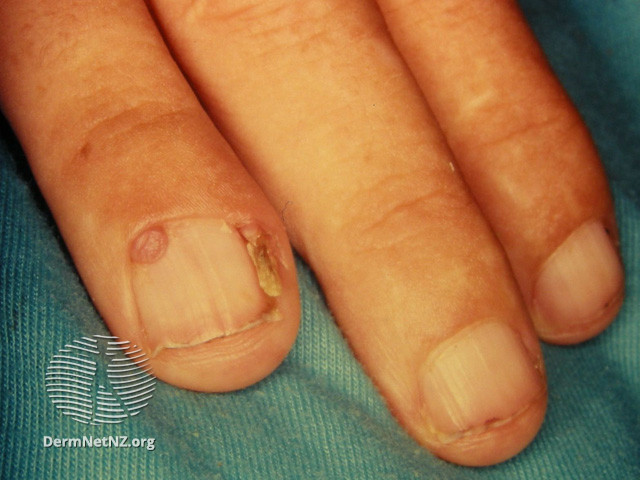
Periungual Fibroma of Tuberous Sclerosis. The pathology of this fibroma is similar to angiofibromas, but with increased vascularity and dense collagen that can extend to the hypodermis.
References
De Waele L, Lagae L, Mekahli D. Tuberous sclerosis complex: the past and the future. Pediatric nephrology (Berlin, Germany). 2015 Oct:30(10):1771-80. doi: 10.1007/s00467-014-3027-9. Epub 2014 Dec 23 [PubMed PMID: 25533384]
Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. The New England journal of medicine. 2006 Sep 28:355(13):1345-56 [PubMed PMID: 17005952]
Manoukian SB, Kowal DJ. Comprehensive imaging manifestations of tuberous sclerosis. AJR. American journal of roentgenology. 2015 May:204(5):933-43. doi: 10.2214/AJR.13.12235. Epub [PubMed PMID: 25905927]
Ng KH, Ng SM, Parker A. Annual review of children with tuberous sclerosis. Archives of disease in childhood. Education and practice edition. 2015 Jun:100(3):114-21. doi: 10.1136/archdischild-2013-304948. Epub 2014 Aug 11 [PubMed PMID: 25112285]
Northrup H, Krueger DA, International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 Iinternational Tuberous Sclerosis Complex Consensus Conference. Pediatric neurology. 2013 Oct:49(4):243-54. doi: 10.1016/j.pediatrneurol.2013.08.001. Epub [PubMed PMID: 24053982]
Level 3 (low-level) evidenceDabora SL, Jozwiak S, Franz DN, Roberts PS, Nieto A, Chung J, Choy YS, Reeve MP, Thiele E, Egelhoff JC, Kasprzyk-Obara J, Domanska-Pakiela D, Kwiatkowski DJ. Mutational analysis in a cohort of 224 tuberous sclerosis patients indicates increased severity of TSC2, compared with TSC1, disease in multiple organs. American journal of human genetics. 2001 Jan:68(1):64-80 [PubMed PMID: 11112665]
Level 2 (mid-level) evidenceHenske EP, McCormack FX. Lymphangioleiomyomatosis - a wolf in sheep's clothing. The Journal of clinical investigation. 2012 Nov:122(11):3807-16. doi: 10.1172/JCI58709. Epub 2012 Nov 1 [PubMed PMID: 23114603]
Saxton RA, Sabatini DM. mTOR Signaling in Growth, Metabolism, and Disease. Cell. 2017 Mar 9:168(6):960-976. doi: 10.1016/j.cell.2017.02.004. Epub [PubMed PMID: 28283069]
Yates JR, Maclean C, Higgins JN, Humphrey A, le Maréchal K, Clifford M, Carcani-Rathwell I, Sampson JR, Bolton PF, Tuberous Sclerosis 2000 Study Group. The Tuberous Sclerosis 2000 Study: presentation, initial assessments and implications for diagnosis and management. Archives of disease in childhood. 2011 Nov:96(11):1020-5. doi: 10.1136/adc.2011.211995. Epub 2011 Aug 3 [PubMed PMID: 21813552]
Doubková M, Štefániková M, Čan V, Merta Z, Svoboda M. Lymphangioleiomyomatosis. Klinicka onkologie : casopis Ceske a Slovenske onkologicke spolecnosti. 2019 Fall:32(5):367-374. doi: 10.14735/amko2019367. Epub [PubMed PMID: 31610670]
Lin S, Zeng JB, Zhao GX, Yang ZZ, Huang HP, Lin MT, Wu ZY, Wang N, Chen WJ, Fang L. Tuberous Sclerosis Complex in Chinese patients: Phenotypic analysis and mutational screening of TSC1/TSC2 genes. Seizure. 2019 Oct:71():322-327. doi: 10.1016/j.seizure.2019.08.010. Epub 2019 Aug 23 [PubMed PMID: 31525612]
Gruber V, Scholl T, Samueli S, Gröppel G, Mühlebner A, Hainfellner JA, Feucht M. Pathophysiology of neurodevelopmental mTOR pathway-associated epileptic conditions: Current status of biomedical research. Clinical neuropathology. 2019 Sep/Oct:38(5):210-224. doi: 10.5414/NP301214. Epub [PubMed PMID: 31347492]
Northrup H, Aronow ME, Bebin EM, Bissler J, Darling TN, de Vries PJ, Frost MD, Fuchs Z, Gosnell ES, Gupta N, Jansen AC, Jóźwiak S, Kingswood JC, Knilans TK, McCormack FX, Pounders A, Roberds SL, Rodriguez-Buritica DF, Roth J, Sampson JR, Sparagana S, Thiele EA, Weiner HL, Wheless JW, Towbin AJ, Krueger DA, International Tuberous Sclerosis Complex Consensus Group. Updated International Tuberous Sclerosis Complex Diagnostic Criteria and Surveillance and Management Recommendations. Pediatric neurology. 2021 Oct:123():50-66. doi: 10.1016/j.pediatrneurol.2021.07.011. Epub 2021 Jul 24 [PubMed PMID: 34399110]
Level 3 (low-level) evidenceLuo C, Ye WR, Shi W, Yin P, Chen C, He YB, Chen MF, Zu XB, Cai Y. Perfect match: mTOR inhibitors and tuberous sclerosis complex. Orphanet journal of rare diseases. 2022 Mar 4:17(1):106. doi: 10.1186/s13023-022-02266-0. Epub 2022 Mar 4 [PubMed PMID: 35246210]
Cascarino M, Leclerc-Mercier S. Histological Patterns of Skin Lesions in Tuberous Sclerosis Complex: A Panorama. Dermatopathology (Basel, Switzerland). 2021 Jul 4:8(3):236-252. doi: 10.3390/dermatopathology8030029. Epub 2021 Jul 4 [PubMed PMID: 34287284]
Altman NR, Purser RK, Post MJ. Tuberous sclerosis: characteristics at CT and MR imaging. Radiology. 1988 May:167(2):527-32 [PubMed PMID: 3357966]
Kassiri J, Snyder TJ, Bhargava R, Wheatley BM, Sinclair DB. Cortical tubers, cognition, and epilepsy in tuberous sclerosis. Pediatric neurology. 2011 May:44(5):328-32. doi: 10.1016/j.pediatrneurol.2011.01.001. Epub [PubMed PMID: 21481739]
Level 2 (mid-level) evidenceGoodman M, Lamm SH, Engel A, Shepherd CW, Houser OW, Gomez MR. Cortical tuber count: a biomarker indicating neurologic severity of tuberous sclerosis complex. Journal of child neurology. 1997 Feb:12(2):85-90 [PubMed PMID: 9075016]
Level 1 (high-level) evidenceRebaine Y, Nasser M, Girerd B, Leroux C, Cottin V. Tuberous sclerosis complex for the pulmonologist. European respiratory review : an official journal of the European Respiratory Society. 2021 Sep 30:30(161):. doi: 10.1183/16000617.0348-2020. Epub 2021 Aug 3 [PubMed PMID: 34348978]
Gupta N, Henske EP. Pulmonary manifestations in tuberous sclerosis complex. American journal of medical genetics. Part C, Seminars in medical genetics. 2018 Sep:178(3):326-337. doi: 10.1002/ajmg.c.31638. Epub 2018 Jul 28 [PubMed PMID: 30055039]
Uysal SP, Şahin M. Tuberous sclerosis: a review of the past, present, and future. Turkish journal of medical sciences. 2020 Nov 3:50(SI-2):1665-1676. doi: 10.3906/sag-2002-133. Epub 2020 Nov 3 [PubMed PMID: 32222129]
Capal JK, Franz DN. Profile of everolimus in the treatment of tuberous sclerosis complex: an evidence-based review of its place in therapy. Neuropsychiatric disease and treatment. 2016:12():2165-72. doi: 10.2147/NDT.S91248. Epub 2016 Aug 25 [PubMed PMID: 27601910]
Oyerinde O, Buccine D, Treichel A, Hong C, Lee CR, Moss J, Darling TN. Fibrous cephalic plaques in tuberous sclerosis complex. Journal of the American Academy of Dermatology. 2018 Apr:78(4):717-724. doi: 10.1016/j.jaad.2017.12.027. Epub 2017 Dec 16 [PubMed PMID: 29258863]
Jóźwiak S, Schwartz RA, Janniger CK, Michałowicz R, Chmielik J. Skin lesions in children with tuberous sclerosis complex: their prevalence, natural course, and diagnostic significance. International journal of dermatology. 1998 Dec:37(12):911-7 [PubMed PMID: 9888331]
Hoogeveen-Westerveld M, Ekong R, Povey S, Mayer K, Lannoy N, Elmslie F, Bebin M, Dies K, Thompson C, Sparagana SP, Davies P, van Eeghen AM, Thiele EA, van den Ouweland A, Halley D, Nellist M. Functional assessment of TSC2 variants identified in individuals with tuberous sclerosis complex. Human mutation. 2013 Jan:34(1):167-75. doi: 10.1002/humu.22202. Epub 2012 Oct 11 [PubMed PMID: 22903760]
Caban C, Khan N, Hasbani DM, Crino PB. Genetics of tuberous sclerosis complex: implications for clinical practice. The application of clinical genetics. 2017:10():1-8. doi: 10.2147/TACG.S90262. Epub 2016 Dec 21 [PubMed PMID: 28053551]
Krueger DA, Northrup H, International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex surveillance and management: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatric neurology. 2013 Oct:49(4):255-65. doi: 10.1016/j.pediatrneurol.2013.08.002. Epub [PubMed PMID: 24053983]
Level 3 (low-level) evidenceJóźwiak S, Nabbout R, Curatolo P, participants of the TSC Consensus Meeting for SEGA and Epilepsy Management. Management of subependymal giant cell astrocytoma (SEGA) associated with tuberous sclerosis complex (TSC): Clinical recommendations. European journal of paediatric neurology : EJPN : official journal of the European Paediatric Neurology Society. 2013 Jul:17(4):348-52. doi: 10.1016/j.ejpn.2012.12.008. Epub 2013 Feb 5 [PubMed PMID: 23391693]
Ebrahimi-Fakhari D, Franz DN. Pharmacological treatment strategies for subependymal giant cell astrocytoma (SEGA). Expert opinion on pharmacotherapy. 2020 Aug:21(11):1329-1336. doi: 10.1080/14656566.2020.1751124. Epub 2020 Apr 27 [PubMed PMID: 32338549]
Level 3 (low-level) evidenceCuratolo P, Bombardieri R, Jozwiak S. Tuberous sclerosis. Lancet (London, England). 2008 Aug 23:372(9639):657-68. doi: 10.1016/S0140-6736(08)61279-9. Epub [PubMed PMID: 18722871]
Sooriakumaran P, Gibbs P, Coughlin G, Attard V, Elmslie F, Kingswood C, Taylor J, Corbishley C, Patel U, Anderson C. Angiomyolipomata: challenges, solutions, and future prospects based on over 100 cases treated. BJU international. 2010 Jan:105(1):101-6. doi: 10.1111/j.1464-410X.2009.08649.x. Epub 2009 Jun 2 [PubMed PMID: 19493268]
Level 2 (mid-level) evidenceHatano T, Inaba H, Endo K, Egawa S. Intermittent everolimus administration for renal angiomyolipoma associated with tuberous sclerosis complex. International journal of urology : official journal of the Japanese Urological Association. 2017 Nov:24(11):780-785. doi: 10.1111/iju.13428. Epub 2017 Sep 14 [PubMed PMID: 28905429]
Siroky BJ, Towbin AJ, Trout AT, Schäfer H, Thamann AR, Agricola KD, Tudor C, Capal J, Dixon BP, Krueger DA, Franz DN. Improvement in Renal Cystic Disease of Tuberous Sclerosis Complex After Treatment with Mammalian Target of Rapamycin Inhibitor. The Journal of pediatrics. 2017 Aug:187():318-322.e2. doi: 10.1016/j.jpeds.2017.05.015. Epub 2017 Jun 7 [PubMed PMID: 28600153]
Bissler JJ, Kingswood JC, Radzikowska E, Zonnenberg BA, Frost M, Belousova E, Sauter M, Nonomura N, Brakemeier S, de Vries PJ, Berkowitz N, Miao S, Segal S, Peyrard S, Budde K. Everolimus for renal angiomyolipoma in patients with tuberous sclerosis complex or sporadic lymphangioleiomyomatosis: extension of a randomized controlled trial. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 2016 Jan:31(1):111-9. doi: 10.1093/ndt/gfv249. Epub 2015 Jul 8 [PubMed PMID: 26156073]
Level 1 (high-level) evidenceJohnson SR, Cordier JF, Lazor R, Cottin V, Costabel U, Harari S, Reynaud-Gaubert M, Boehler A, Brauner M, Popper H, Bonetti F, Kingswood C, Review Panel of the ERS LAM Task Force. European Respiratory Society guidelines for the diagnosis and management of lymphangioleiomyomatosis. The European respiratory journal. 2010 Jan:35(1):14-26. doi: 10.1183/09031936.00076209. Epub [PubMed PMID: 20044458]
Xu KF, Tian X, Ryu JH. Recent advances in the management of lymphangioleiomyomatosis. F1000Research. 2018:7():. pii: F1000 Faculty Rev-758. doi: 10.12688/f1000research.14564.1. Epub 2018 Jun 18 [PubMed PMID: 29946430]
Level 3 (low-level) evidenceZöllner JP, Franz DN, Hertzberg C, Nabbout R, Rosenow F, Sauter M, Schubert-Bast S, Wiemer-Kruel A, Strzelczyk A. A systematic review on the burden of illness in individuals with tuberous sclerosis complex (TSC). Orphanet journal of rare diseases. 2020 Jan 21:15(1):23. doi: 10.1186/s13023-019-1258-3. Epub 2020 Jan 21 [PubMed PMID: 31964424]
Level 1 (high-level) evidenceSuzuki E, Kurihara M, Tsuboshima K, Watanabe K, Okamoto S, Seyama K. The effects of total pleural covering on pneumothorax recurrence and pulmonary function in lymphangioleiomyomatosis patients without history of pleurodesis or thoracic surgeries for pneumothorax. Journal of thoracic disease. 2021 Jan:13(1):113-124. doi: 10.21037/jtd-20-2286. Epub [PubMed PMID: 33569191]
Goh S, Butler W, Thiele EA. Subependymal giant cell tumors in tuberous sclerosis complex. Neurology. 2004 Oct 26:63(8):1457-61 [PubMed PMID: 15505165]
Level 2 (mid-level) evidenceClarke MJ, Foy AB, Wetjen N, Raffel C. Imaging characteristics and growth of subependymal giant cell astrocytomas. Neurosurgical focus. 2006 Jan 15:20(1):E5 [PubMed PMID: 16459995]
Level 2 (mid-level) evidenceKingswood JC, Belousova E, Benedik MP, Budde K, Carter T, Cottin V, Curatolo P, Dahlin M, D'Amato L, d'Augères GB, de Vries PJ, Ferreira JC, Feucht M, Fladrowski C, Hertzberg C, Jozwiak S, Lawson JA, Macaya A, Marques R, Nabbout R, O'Callaghan F, Qin J, Sander V, Sauter M, Shah S, Takahashi Y, Touraine R, Youroukos S, Zonnenberg B, Jansen AC, TOSCA Consortium and TOSCA Investigators. TuberOus SClerosis registry to increAse disease awareness (TOSCA) Post-Authorisation Safety Study of Everolimus in Patients With Tuberous Sclerosis Complex. Frontiers in neurology. 2021:12():630378. doi: 10.3389/fneur.2021.630378. Epub 2021 Mar 23 [PubMed PMID: 33833726]
Level 2 (mid-level) evidenceVerhoef S, Bakker L, Tempelaars AM, Hesseling-Janssen AL, Mazurczak T, Jozwiak S, Fois A, Bartalini G, Zonnenberg BA, van Essen AJ, Lindhout D, Halley DJ, van den Ouweland AM. High rate of mosaicism in tuberous sclerosis complex. American journal of human genetics. 1999 Jun:64(6):1632-7 [PubMed PMID: 10330349]