Introduction
Sturge-Weber syndrome (SWS) is a neurocutaneous syndrome characterized by angiomas involving the face, choroid, and leptomeninges. The facial capillary vascular malformation is also known as "port-wine stain" or "nevus flammeus" and usually is seen in the territory of the trigeminal nerve. Sturge-Weber syndrome is also called encephalotrigeminal angiomatosis. It is the third most common neurocutaneous syndrome after neurofibromatosis and tuberous sclerosis.[1] The neurologic manifestations of SWS include atonic, tonic, or myoclonic seizures.
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
Sturge-Weber syndrome is a sporadic developmental disorder caused by somatic mosaic mutations in the GNAQ gene which is located on the long arm of chromosome 9.[2] It has been postulated that the presence of facial and pial angiomas suggests the persistence of primordial sinusoidal vascular channels. Another possibility is that the superficial venous drainage of the brain never fully develops and the capillaries and small venous channels become dilated to compensate.
Epidemiology
The incidence of Sturge-Weber syndrome is not well known and estimated to be 1 in 20,000-50,000 live births. SWS affects males and females equally and there is no race predilection.[1]
Pathophysiology
The facial nevus is composed of multiple thin-walled vessels that resemble capillaries. The neuropathological finding is an angioma that consists of multiple capillaries and small venous channels and is usually confined to the pia mater. There is a relative lack of superficial cortical veins, and the blood is shunted to the deep venous system by the enlarged medullary veins which result in stasis and ischemic changes.
History and Physical
A seizure is usually the first neurological manifestation of Sturge-Weber syndrome. Infantile spasms are seen in approximately 90% of affected patients in the first year of life followed by atonic, tonic, or myoclonic seizures. Increasing hemiparesis commonly accompanies seizures. Seizures progressively become refractory to medication.[3] Facial nevus (port-wine stain) is another common finding and is typically seen along the ophthalmic or maxillary segment of the trigeminal nerve (forehead, cheeks). It is typically unilateral, present at birth, and does not change with the age of the patient.[4] This appearance is in sharp contrast to the infantile hemangioma which is more common than a port-wine stain; however, it is not present at birth. It slowly grows and is followed by involution. A child with a facial port-wine stain has a 10% to 35% risk of brain involvement. If there is involvement of both upper and lower eyelids, then the risk of glaucoma increases up to 50%. Glaucoma is almost always ipsilateral to the facial port-wine stain. Not all patients with port-wine stains have Sturge-Weber syndrome. Patients with Sturge-Weber syndrome may present with cerebral symptoms without facial findings. Ocular involvement in infancy may present with increased vascularity of the conjunctiva, eye enlargement, strabismus, and increased tearing. Other symptoms are intellectual disability, early handedness, and gaze preferences. Diffuse choroidal hemangioma is seen in about 20% of patients with Sturge-Weber syndrome and is usually on the same side as a facial port-wine stain.[5] Choroidal hemangiomas grow slowly and usually do not cause any symptoms. Retina overlying the choroidal hemangioma may be normal; however, it may also show pathological changes such as epithelial atrophy/proliferation, drusen formation, or detachment.
Evaluation
Diagnosis of Sturge-Weber syndrome is based on typical clinical symptoms, facial appearance, and brain magnetic resonance imaging (MRI) findings.[6] An ophthalmic examination is required to rule out glaucoma. Ocular ultrasound can demonstrate diffuse choroidal thickening which suggests choroidal hemangioma. Gyriform calcifications can be seen on the skull radiographs and are classically described as "tram-track sign." Computed tomogram (CT) is the best modality to detect calcifications and also show the other changes such as cortical atrophy and leptomeningeal enhancement on the post-contrast studies. However, CT uses ionizing radiation, and the routine use of CT in children is not recommended. Therefore, MRI of the brain with contrast is the recommended imaging modality of choice. The most common locations are occipital and posterior parietal/temporal lobes. The MRI findings depend on the stage of the disease. In the early phase, there is transient hyperperfusion with accelerated myelin maturation, leptomeningeal enhancement (seen as serpiginous enhancement along the sulci), and restricted diffusion if there is an associated acute ischemic event. In the late phase, an increased T2 signal is seen in the region of gliosis with decreased pial enhancement and cortical atrophy. There is a lack of superficial cortical veins with prominent deep medullary/subependymal veins and enlarged choroid plexus. Gyriform calcifications are best seen on T2* or SWI (susceptibility-weighted images) and appear as areas of signal loss along the gyri in a serpentine pattern. Patients with cutaneous and ocular manifestations with a normal brain MRI at one year of age are unlikely to have brain involvement in the future. Choroidal angiomas can be seen on MRI as increased enhancement along the posterior choroid layer of the globe. Fluorodeoxyglucose-positron emission tomography (FDG-PET) may be a useful modality to study cerebral metabolism in patients with Sturge-Weber syndrome. The affected area is usually hypermetabolic in the early stages with hypometabolism in the late stage.[7] PET may be useful in surgical planning when cortical resection is required for the treatment of intractable seizures.
Treatment / Management
There is no specific treatment for Sturge-Weber syndrome. The primary aim is to minimize seizure activity with anticonvulsive medications. Surgery may be considered in patients who fail medical management and continue to have refractory seizures. The surgical procedures for Sturge-Weber syndrome include hemispherectomy or focal resection of the seizure focus. Patients with bilateral involvement are typically not good candidates for surgery.[8] Low-dose aspirin has also been shown to be effective in decreasing the frequency of seizures and stroke-like episodes.[9] An annual ophthalmologic examination is recommended even if the early evaluation does not detect glaucoma. The goal of glaucoma treatment is to reduce the intraocular fluid which decreases the intraocular pressure with subsequently decreased the risk of vision loss. Topical medication is considered first for late-onset glaucoma. The surgery is considered for patients with early-onset glaucoma and associated angle abnormalities and includes goniotomy or trabeculotomy. Further surgeries such as trabeculectomy or glaucoma drainage device may be considered for resistant patients.[10] The port-wine stain can be treated with laser photocoagulation which results in irreversible damage to the blood vessels without damage to the other skin components.(B3)
Differential Diagnosis
- Blue rubber bleb nevus syndrome
- Klippel-Trenaunay-Weber syndrome
- PHACES (posterior fossa abnormalities, hemangiomas, arterial anomalies, cardiac, eye, and sternal anomalies)
- Wyburg-Mason syndrome
Sturge-Weber syndrome can easily be differentiated from these conditions based on clinical history, physical examination, and brain MRI findings.
Prognosis
The prognosis for these patients is guarded and depends on the severity of associated anomalies.[11] [Level 5]
Complications
- Seizures
- Stroke-like episodes
- Glaucoma
Deterrence and Patient Education
The patient and parents should be made to understand that there is no cure for this disease and the maximum we can offer is to give symptomatic treatment and control the neurologic and ocular manifestations.
Enhancing Healthcare Team Outcomes
The management of Sturge-Weber syndrome is best accomplished with an interprofessional team approach. The team may include a geneticist, pediatricians, pediatric ophthalmologists, pediatric radiologists, pediatric neurologists, nurse practitioners, and pediatric neurosurgeons. There is no cure for the syndrome and all treatments are symptom-based. In addition to preventing seizures, patients need ophthalmology follow-up due to the risk of developing glaucoma. The skin lesions can be managed by a dermatologist. Nurses play an important role in patient and parent education.
Media
(Click Image to Enlarge)
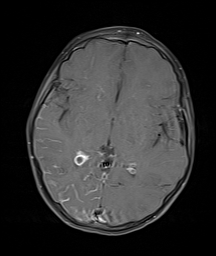
Right occipital and temporal leptomeningeal enhancement with enlarged choroid plexus in a patient with Sturge-Weber syndrome. Contributed by Achint Singh
References
Comi AM. Sturge-Weber syndrome. Handbook of clinical neurology. 2015:132():157-68. doi: 10.1016/B978-0-444-62702-5.00011-1. Epub [PubMed PMID: 26564078]
Shirley MD, Tang H, Gallione CJ, Baugher JD, Frelin LP, Cohen B, North PE, Marchuk DA, Comi AM, Pevsner J. Sturge-Weber syndrome and port-wine stains caused by somatic mutation in GNAQ. The New England journal of medicine. 2013 May 23:368(21):1971-9. doi: 10.1056/NEJMoa1213507. Epub 2013 May 8 [PubMed PMID: 23656586]
Pinto A, Sahin M, Pearl PL. Epileptogenesis in neurocutaneous disorders with focus in Sturge Weber syndrome. F1000Research. 2016:5():. pii: F1000 Faculty Rev-370. doi: 10.12688/f1000research.7605.1. Epub 2016 Mar 18 [PubMed PMID: 27019697]
Enjolras O, Riche MC, Merland JJ. Facial port-wine stains and Sturge-Weber syndrome. Pediatrics. 1985 Jul:76(1):48-51 [PubMed PMID: 4011357]
Level 2 (mid-level) evidenceMaraña Pérez AI, Ruiz-Falcó Rojas ML, Puertas Martín V, Domínguez Carral J, Carreras Sáez I, Duat Rodríguez A, Sánchez González V. Analysis of Sturge-Weber syndrome: A retrospective study of multiple associated variables. Neurologia (Barcelona, Spain). 2017 Jul-Aug:32(6):363-370. doi: 10.1016/j.nrl.2015.12.012. Epub 2016 Mar 8 [PubMed PMID: 26964511]
Level 2 (mid-level) evidenceComi AM. Update on Sturge-Weber syndrome: diagnosis, treatment, quantitative measures, and controversies. Lymphatic research and biology. 2007:5(4):257-64. doi: 10.1089/lrb.2007.1016. Epub [PubMed PMID: 18370916]
Chugani HT, Mazziotta JC, Phelps ME. Sturge-Weber syndrome: a study of cerebral glucose utilization with positron emission tomography. The Journal of pediatrics. 1989 Feb:114(2):244-53 [PubMed PMID: 2783735]
Arzimanoglou AA, Andermann F, Aicardi J, Sainte-Rose C, Beaulieu MA, Villemure JG, Olivier A, Rasmussen T. Sturge-Weber syndrome: indications and results of surgery in 20 patients. Neurology. 2000 Nov 28:55(10):1472-9 [PubMed PMID: 11094100]
Bay MJ, Kossoff EH, Lehmann CU, Zabel TA, Comi AM. Survey of aspirin use in Sturge-Weber syndrome. Journal of child neurology. 2011 Jun:26(6):692-702. doi: 10.1177/0883073810388646. Epub 2011 Mar 22 [PubMed PMID: 21427442]
Level 3 (low-level) evidenceThavikulwat AT, Edward DP, AlDarrab A, Vajaranant TS. Pathophysiology and management of glaucoma associated with phakomatoses. Journal of neuroscience research. 2019 Jan:97(1):57-69. doi: 10.1002/jnr.24241. Epub 2018 Apr 1 [PubMed PMID: 29607552]
Higueros E, Roe E, Granell E, Baselga E. Sturge-Weber Syndrome: A Review. Actas dermo-sifiliograficas. 2017 Jun:108(5):407-417. doi: 10.1016/j.ad.2016.09.022. Epub 2017 Jan 23 [PubMed PMID: 28126187]