Introduction
The name retinitis pigmentosa (RP), described initially as a clinical entity in 1853, was not attached to the disease until 1857.[1] Considered by most to be a misnomer, the term retinitis persists today, even though inflammation plays only a small role in the natural progression of the disease. RP is not a single entity but rather a group of disorders that produce a gradual loss of vision. Also known as hereditary retinal dystrophy, it affects 1 in every 4000 people in the United States and approximately one in 5000 worldwide, making RP the most common inherited disease of the retina.[2] RP is usually bilateral; however, there have been reports of unilateral eye involvement with this condition.[3] While it may present and progress with a variety of clinical manifestations, the first symptom of RP is generally nyctalopia, or loss of night vision, followed by a gradual narrowing of the visual fields. Over time, depending on the severity and rate of progression of the disease, tunnel vision or complete vision loss can result. As the disease progresses, other features may also develop, including loss of accurate color discrimination and eventual loss of visual acuity. Most patients will maintain some light perception even with late-stage RP as the macula continues functioning. Perhaps the most unsettling late effect of RP is the development of photopsia (perceived flashes of light), likely due to sensory deprivation [4] that can progress to the point of visual hallucinations.[5]
The disease may involve vision loss alone, referred to as "nonsyndromic" RP. About 70% to 80% of RP cases fall into the nonsyndromic category.[1] When RP occurs in conjunction with systemic disease, it is termed "syndromic" RP. The most common form of syndromic RP is Usher syndrome, which involves neurosensory hearing loss associated with vision loss.[6][7]
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
Genetic mutations responsible for RP produce biochemical dysfunction that specifically affects rod photoreceptors in the retina.[8] Defects may be associated with multiple injury pathways, including apoptosis, light damage, ciliary transport dysfunction, and endoplasmic reticulum stress. The typical result of all the possible pathways is the death of the rod photoreceptors.[9] Since the rods are responsible for low-light vision, the ever-increasing loss of these cells produces the characteristic night blindness and a gradual diminution of peripheral vision. Eventually, the destruction of large numbers of rods has a deleterious effect on the retinal pigment epithelium (RPE) and affects cone photoreceptors. As cones succumb to the toxic environment created by progressive cell death in the retina, dyschromatopsia, or disturbance of color perception, may develop (rod-cone dystrophy).
RP is genetically heterogeneous, meaning multiple genetic mutations are associated with the condition. The affected genes participate in critical retinal functions, such as phototransduction, the visual cycle, ciliary transport, and photoreceptor structure.[10] To date, over 3100 different mutations have been identified as causing RP [8] and there is phenotypic variability with the same mutation even within the same family.[11] This genetic diversity contributes to the variation in the severity and progression of the disease. Research findings have discovered more than 80 different genes that cause multiple patterns of inheritance and expression for nonsyndromic RP.[12][1] Approximately 5% to 20% of RP cases are autosomal recessive (arRP) [12] and around 15% to 25% of RP is autosomal dominant (adRP), whereas around 5% to 15% of cases are X-linked recessive (xlRP).[12] The remaining cases (around 40%-50%) are termed sporadic or simplex (with a single reported case in the family); no family history or known molecular basis is found.[12] Digenic RP is very rare.[13]
The most commonly mutated genes in arRP include USH2A, ABCA4, CERKL, CRB1, EYS, PDE6A, PDE6B, RPE65, RP1, and SAG.[12] The common genes involved in adRP include RHO, RP1, CRX, GUCA1B, IMPDH1, KLHL7, NR2E3, PRPF8, PRPF3, PRPF31, PRPH2, SEMA4A, SNRNP200, and TOPORS; mutations in genes RPGR, RP2, and OFD1 cause xlRP.[12]
Epidemiology
Nonsyndromic RP has a worldwide prevalence of about 1 in 5000 individuals.[1] RP constitutes around 50% of the cases of inherited retinal diseases, and [8] affects more than 1.5 million people worldwide [14] with varying prevalence. Estimates in prevalence range from 1 in 3026 in Denmark [15] to 1 in 4869 in Birmingham, UK,[16] and as high as 1 in 372 in rural India.[17] Some of this variation may be due to differences in methodology and case definitions across studies; furthermore, the prevalence may be higher in some populations with more consanguineous marriages, as seen in certain Middle Eastern and South Asian countries.[17][18]
Men are affected slightly more often than women due to the X-linked form being expressed more frequently in males. Syndromic RP is much less common, with estimates for Usher syndrome ranging from 4 to 17 cases per 100,000 individuals.[19]
The average age of symptom onset is dependent on the genetic type involved. The autosomal recessive form will develop symptoms in the early adolescent years, but those affected with autosomal dominant RP will likely not have symptoms until well into their 20s. More than three-quarters of individuals with RP will be symptomatic and present for clinical evaluation and diagnosis of the disease by the time they are 30 years.[14] In a study conducted in Japan, the average age of diagnosis was 35.1 years (median age 36.5 years).[20]
Pathophysiology
As previously mentioned, multiple genetically-directed mechanisms for the progress of retinitis pigmentosa exist. Apoptosis is physiologic programming for cell death that a genetic mutation can trigger; cell-to-cell communication between the photoreceptors can also induce apoptosis.[21] Hence, the death of rods can eventually spread to the cone receptors. Other mechanisms of cell death in RP include regulated necrosis and autophagy.[22][23]
Light exposure may exacerbate phototoxic mechanisms. These include mutations in retinol metabolism and acceleration of oxygen consumption in the environment, which can enhance the degeneration of photoreceptors (both rod and cone).[24] Oxidative stress is an essential mechanism for photoreceptor damage in RP as the photoreceptors have high metabolism and oxygen consumption.[25][26] Hyperoxia may cause photoreceptor death due to generating reactive oxygen species (ROS), including superoxide radicals.[27] Other possible mechanisms of RP include metabolic stress and activation of microglia and monocytes.[28][29] Calcium can induce endoplasmic reticulum stress or may even cause apoptosis and nonapoptotic cell death.[30]
The ciliary function is vital to transport nutrients and other substances in the retina. Some genetic mutations, including the one for Usher syndrome, can impair this function and cause cell vulnerability.[31] Stress in the endoplasmic reticulum can release free radicals, subsequently stimulating hypoperfusion of the retina and vascular endothelial cell damage.[32]
Three clinical findings typical of RP are the presence of bone spicule pigmentation, vascular narrowing, and optic nerve head pallor (see Image. Fundus Showing Bony Spicules). The melanin pigment deposits (see Image. Right Eye Retinitis Pigmentosa,) named for their characteristic bone spicule star shape, are due to retinal pigment epithelial cells that detach and migrate to perivascular locations in the retina. The exact cause of this migration is not fully understood, nor is the narrowing of retinal vessels. One suggestion is that this condition results from a decreased metabolic demand due to the death of many photoreceptors. Change in the appearance of the optic disc is probably due to the formation of glial cells (inside the optic disc and on the surface) that produce a "waxy pallor"[1] (see Image. Retinitis Pigmentosa).
Histopathology
Histopathological examination of the retina can reveal characteristic changes in RP, with features including photoreceptor degeneration, outer nuclear layer thinning, retinal pigment clumps, RPE changes, Muller cell gliosis, vascular changes, and optic nerve head changes.[33] One of the hallmark features of RP is the progressive degeneration of photoreceptor cells in the retina, specifically rods and cones.[25] The loss of photoreceptor cell bodies and their outer segments characterize this degeneration histologically. As photoreceptor cells degenerate, the retina's outer nuclear layer (ONL) becomes thinner; areas of the retina that were initially affected by the disease, such as the peripheral regions, have the most pronounced thinning.
The presence of pigment clumps or deposits observed in the retina during histopathological examination gives RP its name. These pigment clumps are often distributed in a characteristic pattern and result from pigment migration from the RPE due to the breakdown of photoreceptor cells. RPE cells may undergo hypertrophy, atrophy, or degeneration in response to the disease. In response to retinal damage, Muller cells, which provide structural and metabolic support to retinal neurons, may undergo gliosis—a reactive process characterized by hypertrophy and increased glial fibrillary acidic protein (GFAP) expression.[34] This gliosis can be observed histologically. In some cases, RP can lead to vascular changes in the retina, including narrowing of blood vessels and vascular attenuation. In advanced stages of RP, the changes in the optic nerve may include optic nerve head pallor and axonal loss. Specific histopathological findings in RP can vary depending on the stage of the disease and the underlying genetic mutation.
History and Physical
The typical presentation of RP involves complaints of visual disturbances beginning at around 20 years. Usually, these disturbances include being unable to see well in low-light situations or situations that require rapid adaptation from light to dim environments. Some individuals note difficulty driving at night, as oncoming headlights and other bright light sources make it difficult for eyes to readjust to the darkness afterward. Further, the narrowing of the visual fields is not initially evident but becomes apparent over time.
A genetic pedigree for the patient's family will be critical for determining the type of inheritance pattern involved and assisting with prognosis. A complete history and review of systems are needed to identify other systems affected and syndromic variants of RP. Additionally, review possible exposure to infectious diseases or toxins that might produce a "mimic" of the disease.
Physical findings include the "classic triad" seen on a fundoscopic exam of bony spicule pigmentation, vascular narrowing, and abnormal waxy pallor of the optic disc.[35] These may not be evident early in the disease, and the degree to which abnormalities occur varies with the severity of the disease. Arteriolar attenuation appears very early. The retina may appear normal (retinitis pigmentosa sine pigmento) in some cases despite the electroretinogram showing rod dysfunction. Other associated physical findings may include posterior subcapsular cataracts (up to 72% of patients) and macular edema.
While the external eye examination is usually routine, patients with RP are at higher risk for keratoconus than the general population. Still, the development of keratoconus is quite rare.[36] Refractive errors (usually high myopia with or without astigmatism) also occur more commonly in patients with RP than in the general population.[1] Other features of RP include dust-like particles in the vitreous cavity, optic disc drusen, white dot-like appearance at the level of RPE (also called retinitis punctata albescence), and exudative vasculopathy similar to Coats disease. Bilaterally symmetrical pigmentary changes with bony spicules at a focal area of the fundus characterize sector RP.[37] Around 50% of the carriers of xlRP may show a golden reflex at the posterior pole. Mild hearing loss may be present in some cases of non-syndromic RP (not associated with Usher syndrome). Juvenile RP is a heterogeneous and severe form of arRP. The most aggressive form is Leber congenital amaurosis, which manifests in early childhood and leads to blindness in most of these children.
Evaluation
A complete ophthalmologic evaluation for any individual suspected of having this condition is needed, including an expert fundoscopic examination and a retinal function evaluation. Test visual acuity and contrast sensitivity to denote the quality of vision. Assess color vision to determine the presence and progression of dyschromatopsia, indicating the degree of cone photoreceptor involvement. These tests will establish a baseline and assist in determining the rate of progression and formulating a prognosis for the disease.
Color fundus and pseudocolor wide-field fundus images can help document the disease and monitor progression.
Fundus autofluorescence (FAF) imaging is a noninvasive technique that provides insight into retinal health by detecting lipofuscin, a byproduct of photoreceptor cell death, in the RPE. In RP, FAF reveals distinct patterns correlating with disease severity and progression. Findings from many studies found that patients with RP exhibit a ring of increased autofluorescence surrounding the fovea, indicating lipofuscin accumulation and RPE stress. This “AF ring” (Robson-Holder ring) corresponds to the area between the functional and nonfunctional retina border.[38][39][40] As RP progresses, the AF ring contracts over time, coinciding with worsening rod and cone function in the parafoveal region within the ring.[41] Once the ring reaches a critical minimum size, central vision becomes impaired.[41] The AF ring diameter also correlates with visual field sensitivity and electroretinogram amplitudes, suggesting it indicates overall retinal health.[38][42] Beyond the AF ring, FAF reveals other patterns in RP. Atrophic patches of decreased AF, indicating RPE and photoreceptor cell death, emerge in the mid-periphery and spread centripetally.[43] The remaining areas of normal or increased FAF within atrophic zones represent “islands” of viable retina.[44] As RP progresses, atrophic patches coalesce while islands disappear. The location and spread of atrophic changes do not correlate well with visual function, unlike that seen in the AF ring.[38] Thus, FAF is valuable for monitoring RP progression and treatment efficacy. FAF provides insight into the complex mosaic of viable, stressed, and nonfunctional retinas seen in this condition.
Visual field assessment with kinetic perimetry is considered the most effective way to evaluate the loss of peripheral vision. Humphrey visual fields can be done to note the remaining visual field to determine the degree of visual disability in various countries. Electrophysiologic evaluation of the entire retina is also helpful for the early detection of electroretinographic abnormalities, even before nyctalopia and fundoscopic abnormalities are present.[1]
The electroretinogram (ERG) has been instrumental in understanding retinal function in RP. Early study results found reduced ERG responses in RP, indicating impaired rod and cone function. Rod responses were detectable but reduced in amplitude and delayed in latency, while cone responses remained normal. Later research confirmed these findings, showing that ERG responses correlated with visual function and age but not dark adaptation and were similar across genetic types of RP.[45] The ERG could detect RP even before ophthalmoscopic fundus changes and assisted in diagnosing and monitoring RP progression and potential treatments. Electroretinography (ERG) not only helps diagnose RP but also quantifies the severity and tracks the progression of the disease. Over time, a-waves decrease in amplitude to a point where the ERG signal is no longer detected.[1]
Fluorescein angiography is instrumental in characterizing the breakdown of the blood-retinal barrier, abnormalities in retinal and choroidal blood flow, cystoid macular edema, and even subtle changes in carriers of RP. Typically, RP is associated with non-leaking macular edema.
Optical coherence tomography (OCT) has been instrumental in evaluating retinal changes in patients with RP. Findings from multiple studies have found significant thinning of the retina in areas affected by RP, while the macula often remains relatively preserved. The photoreceptor layer, including the inner segment/outer segment junction and external limiting membrane, is frequently disrupted or absent, sparing the fovea. Epiretinal membranes are common in RP and identifiable in up to 94% of patients.[46][47] These membranes can sometimes cause cystoid macular edema, evident in 12% to 19% of eyes.[47] Furthermore, 18% of eyes had observable macular pseudocysts.[47] OCT can characterize the spectrum of retinal changes that occur in RP, from photoreceptor loss to epiretinal membrane formation to microvascular abnormalities. By tracking these changes over time, OCT can serve as an essential outcome measure in clinical trials for RP and help clinicians monitor disease progression and treatment response in their patients.
Optical coherence tomography angiography (OCTA) is an emerging noninvasive imaging technique that provides detailed visualization of the retinal microvasculature in patients with RP. Several study results have found that OCTA helps detect vascular changes in RP that correlate with disease severity and progression. Takagi et al [48] and Koyanagi et al [49] found reduced vessel density and flow areas in the superficial and deep capillary plexuses of patients with RP with preserved visual acuity, indicating early vascular changes.
Adaptive optics scanning laser ophthalmoscopy (AOSLO) enables high-resolution evaluation of the retina to detect photoreceptor damage early in the disease. This process is likely to be used in monitoring the disease progression and for evaluating the effectiveness of treatment efficacy.[1] Further, perform a systemic evaluation to rule out the possibility of syndromic RP.
Treatment / Management
There are no standard treatments for patients with RP. Therapies explored in retinitis pigmentosa include vitamin A supplements, gene therapy, stem cell therapy, neuroprotective approaches, and retinal implants. Provide refraction and glasses to improve the central visual acuity. The non-leaking macular edema (foveoschisis) may respond to topical or systemic carbonic anhydrase inhibitors. Leaking CME responds well to sub-Tenon, intravitreal, or systemic steroids. Consider surgery with intraocular lens in patients with significant cataracts. Low vision aids help in mobility and daily activity. Counseling and vocational rehabilitation with a collaborative interprofessional approach help improve patient outcomes.
The research on vitamin A supplementation for retinitis pigmentosa has yielded mixed and inconclusive results. The most widely recommended treatment for many years has been supplementation with vitamin A as some study results have shown vitamin A to slow the rate of retinal deterioration.[36] Chatzinoff and colleagues found no benefit of 11-cis vitamin A for RP.[50] Massoud and colleagues found no difference in plasma vitamin A levels between patients with RP and controls.[51] However, other studies suggest vitamin A may slow vision loss or provide other benefits for some patients with RP. Gouras et al found that massive doses of vitamin A reversed vision abnormalities in patients with RP with abetalipoproteinemia.[52] Berson et al found that 15,000 IU/day of vitamin A slowed vision loss over 5 years in some patients with RP, as measured by electroretinography.[53] Rayapudi and colleagues reviewed multiple studies and found no clear evidence that vitamin A or fish oil benefits most patients with RP regarding vision loss over 1 to 5 years.[54] Thus, while results from some studies have found benefits of vitamin A for slowing vision loss or improving vision in certain patients with RP, especially those with more severe disease or specific genotypes, the evidence does not currently support vitamin A supplementation for most patients with RP. More extensive and targeted studies are still needed to determine if and which subgroups of patients with RP may benefit from vitamin A treatment. It is uncertain if treatment with vitamin A or DHA (docosahexaenoic acid) or both benefits people with RP.[55] When supplementing individual patients with high-dose vitamin A, test liver function.(A1)
Gene therapy shows promising potential for treating RP. Findings from multiple studies have demonstrated that gene therapy can rescue photoreceptor cells and restore vision in animal models of RP.[56][57] The eye is an ideal target for gene therapy due to its accessibility and immunological isolation. Viral vectors like adeno-associated virus (AAV) and lentivirus can deliver therapeutic genes to the retina.[58] In recent clinical trials, subretinal injection of AAV carrying the RPE65 gene led to visual improvements in patients with Leber congenital amaurosis (LCA), a severe early-onset form of RP.[59] The Food and Drug Administration (FDA) approved voretigene neparvovec-rzyl for treating children and adults with LCA due to a confirmed biallelic RPE65 mutation in December 2017. These exciting results provide hope that gene therapy may soon become a viable treatment option for patients with other forms of RP.[60] Long-term safety and efficacy data are necessary before gene therapy is in standard clinical practice. The genetic causes of RP are complex, with over 80 genes involved. Genetic diagnosis is critical for devising customized treatment plans and will become more accessible with advancing genetic screening techniques. A better understanding of the pathways leading from gene mutations to photoreceptor cell death can also reveal new therapeutic targets.[61](B3)
Stem cell therapy shows promising potential for restoring vision in patients with RP. Multiple study findings demonstrated that transplanting stem cells into the eyes of RP animal models can differentiate into photoreceptor cells and integrate into the retina, improving visual function.[62][63] Stem cells explored for this purpose include retinal progenitor, embryonic, induced pluripotent, and mesenchymal stem cells.[62][63] Limoli and colleagues placed mesenchymal stem cells, adipose stem cells, and platelet-rich plasma into the suprachoroidal space of 21 patients with RP, and found minor improvements in threshold sensitivity in microperimetry for patients with a foveal thickness of more than 190 μm.[64] Transplanting bone marrow stem cells into RP mouse models may prevent cone loss.[65] As cones mediate central and color vision, preserving them could maintain critical visual abilities for patients with RP with severe vision loss. While promising, more research is still needed to optimize stem cell therapy for RP. Cell transplantation seeks to replace degenerating photoreceptors. Transplantation of rod precursor cells or retinal pigment epithelial cells has shown promise in animal models.[66] However, significant technical hurdles must be overcome before widespread clinical use, including immune rejection and limited cell integration.(B3)
Neuroprotective approaches aim to slow photoreceptor cell death by providing growth factors, antioxidants, or anti-apoptotic agents.[67] Neuroprotective agents evaluated in managing RP include ciliary neurotrophic factor (CNTF) and fibroblast growth factor (FGF).[68] (B3)
Retinal implants show promise in restoring vision for patients with RP. Findings from several studies showed visual improvements in patients after the implantation of retinal prostheses. Chow et al found that an artificial silicon retina (ASR) chip implanted in the subretinal space of RP patients led to improvements in visual acuity, contrast sensitivity, color perception, and visual field over more than 7 years.[69] Similarly, Gekeler et al reported increased visual function and durability of the RETINA IMPLANT Alpha AMS chip.[70] However, the research limitation is a lack of high-quality evidence and control groups. Hallum et al reviewed findings on retinal implant studies and found all were at high risk of bias from study design and conduct.[71] None had control groups or masking; outcome measures were not always comparable before and after implantation. The most common comparison, device on versus off, is prone to bias. More rigorous studies are needed to quantify visual benefits properly.[71] In February 2013, the FDA approved Argus II retinal prosthesis for adults with advanced RP. A type of technology that has been of interest lately uses auditory information to substitute for visual sensory input.[72] While these avenues are promising for vision restoration and preservation, there are complicated issues for rehabilitating patients with this condition and device-specific challenges, such as functional longevity.[1](A1)
In recent years, genetic causes of RP have been better understood, and novel treatments are under development to combat the disease. Gene-specific or mutation-specific investigations suggest gene augmentation therapy might restore regular gene expression in photoreceptors. Other research involves cell replacement therapy, which includes transplanting retinal progenitor cells (or non-ocular stem cells) into the eye to repopulate the retina with functional photoreceptors.
Treatable causes of RP include Bassen-Kornzweig disease (treated with supplementation of fat-soluble vitamins), Refsum disease (managed by reduction of dietary intake of phytanic acid), and ataxia with vitamin E deficiency (AVED, managed by supplementation of vitamin E).[73] In all patients with recent onset acquired night blindness, especially with normal appearing retina, vitamin A deficiency must be ruled out.
Differential Diagnosis
The differential diagnosis for progressive vision loss is complex. RP is a clinical diagnosis confirmed by bilateral eye involvement with night vision disturbance and gradual loss of peripheral vision. Physical findings on fundoscopic examination reveal bone spicule pigmentation, with vascular narrowing and optic disc pallor. Note that there are variants of RP with significant differences in physical findings.
Retinitis punctata albescens (RPA, also called progressive RPA) is an autosomal recessive variant of RP that frequently presents as night blindness in childhood. Mutations in RLBP1 usually cause this condition.[74] On direct fundoscopic examination, findings include less prominent (or even absent) disc pallor, less pronounced vascular narrowing, and bony spicules are rare or absent altogether. Instead, small white spots cover the majority of the fundus in RPA. Awareness of such variants is necessary to avoid excluding RP and its subclasses from the differential.[74] RPA is progressive with a progressive decline of visual fields and other visual parameters compared to fundus albipunctatus (congenital stationary night blindness), which is a stationary (nonprogressive) disease. However, a mutation in the RLBP1 is known to be associated with Bothnia retinal dystrophy, fundus albipunctatus, and Newfoundland rod-cone dystrophy. Thus, there is a phenotypic variability.[75]
Other diseases may mimic the early findings of this condition; careful evaluation is necessary to make the correct diagnosis. One possible mimic is cone-rod dystrophy. This progressive retinopathy can be differentiated from RP by the onset of dyschromatopsia before nyctalopia, as the cone photoreceptors are affected rather than the rods. Another consideration for nyctalopia in childhood is congenital stationary night blindness (CSNB).[76][77] CSN presents with 2 types and nyctalopia in childhood. The first type is CSNB (both Riggs and Schubert-Bornstein type), which presents without abnormalities on the fundoscopic exam. The second type has visual fundoscopic abnormalities, including Oguchi disease and fundus albipunctatus. Oguchi disease is associated with the Mizuo-Nakamura phenomenon in which the fundus has a golden sheen on exposure to light, and the typical color of the fundus reappears after prolonged dark adaptation.[78] Fundus albipunctata is characterized by small yellowish-white round dots sparing the fovea and extending to or beyond the mid-periphery. As previously mentioned, these conditions are nonprogressive.[77]
Chorioretinal infections, such as syphilis, cytomegalovirus, or even Lyme disease, may produce symptoms suggestive of RP. Take a careful history to help identify these possibilities and perform laboratory testing for confirmation.
Vision loss can result from multiple other diseases, including sarcoidosis and systemic lupus erythematosus. Systemic involvement, in addition to vision loss, should prompt a thorough assessment of these diseases and other inflammatory processes. Also, consider trauma and Vitamin A deficiency, especially in children or adults with recent onset acquired nyctalopia. In older patients with recent onset of nyctalopia and retinal changes similar to RP, cancer-associated retinopathy and possible occult malignancy should be ruled out.[79]
Unilateral retinitis pigmentosa (URP) is a rare retinitis pigmentosa that affects only one eye. According to results from multiple studies, URP diagnosis occurs only after ruling out all possible infective causes, and the affected eye shows all the clinical signs of retinitis pigmentosa. In contrast, the unaffected eye remains completely normal.[80] However, the exact cause of most cases of this condition remains a mystery. Electroretinography and electrooculography help diagnose URP, as the affected eye will show greatly diminished rod function and nonrecordable or severely decreased electroretinogram responses, while the unaffected eye remains normal. Patients with URP often do not notice symptoms in the eye, though the condition can be present from birth. Some cases of URP appear to be genetic. Mukhopadhyay and colleagues described a patient with a germline mutation in the RP1 gene who developed URP. However, the cause remains unclear of this patient's unilateral expression of the disease.[81] Whether URP represents a distinct clinical entity or an abortive form of bilateral retinitis pigmentosa is still debated.
Diseases that can cause features similar to URP (called unilateral pseudo RP) or unilateral pigmentary retinopathy include
- Inflammation: diffuse unilateral subacute neuroretinitis (DUSN), acute zonal occult outer retinopathy (AZOOR), severe posterior uveitis, retinal vasculitis
- Trauma: blunt trauma (after resolution of commotion retinae or spontaneously settled retinal detachment), retained intraocular foreign body (iron foreign body-siderosis), forceps-associated birth trauma
- Infection: syphilis, cytomegalovirus, measles, rubella, toxoplasmosis, tuberculosis
- Autoimmune disorders: autoimmune retinopathy (AIR)
- Malignancy: cancer-associated retinopathy (CAR, paraneoplastic syndrome),[79] choroidal melanoma
- Drug toxicity: chloroquine, thioridazine, phenothiazine, hydroxychloroquine, oral contraceptives, cephaloridine
- Other retinal disorders: self-settled retinal detachment [82]
- Vascular diseases: ophthalmic artery occlusion [83]
Syndromic RP
RP usually occurs as an isolated condition (nonsyndromic RP) but may be associated with several syndromes and genetic disorders. These syndromes often involve RP as one of their clinical features, along with other systemic manifestations. Important syndromes associated with RP include the following:
- Usher syndrome: Usher syndrome is the most common syndrome associated with RP. Around 18% of all patients with RP may have Usher syndrome.[84] Sensorineural hearing loss and, in some cases, vestibular (balance) issues characterize this condition. There are 3 main types of Usher syndrome, each with different genetic causes and degrees of severity.
- Bardet-Biedl syndrome: Bardet-Biedl syndrome (BBS) is a rare genetic disorder characterized by RP, obesity, kidney dysfunction, extra fingers or toes (polydactyly), and other features such as intellectual disability and developmental delays.[85] Multiple genes can be involved in BBS, leading to significant variability in clinical presentation.
- Joubert Syndrome: Joubert syndrome is a rare genetic disorder characterized by aplasia of the cerebellar vermis, causing the "molar tooth sign" on brain imaging and a range of neurological and systemic features. Some individuals with Joubert syndrome may also develop RP.
- Senior-Løken Syndrome: Senior-Løken syndrome combines the features of nephronophthisis and RP. This condition is a rare autosomal recessive disorder characterized by early-onset kidney disease and progressive vision loss.
- Refsum Disease: Refsum disease is a metabolic disorder characterized by the accumulation of phytanic acid in the body and can lead to RP, as well as other symptoms such as ataxia (difficulty with coordination), peripheral neuropathy, and cardiac issues.
- Kearns-Sayre syndrome: Kearns-Sayre syndrome is a rare mitochondrial disorder characterized by external ophthalmoplegia (paralysis of eye muscles), cardiac conduction defects, and pigmentary retinopathy resembling RP.
- Abetalipoproteinemia (Bassen-Kornzweig disease): This is an autosomal recessive disorder characterized by low or absent plasma cholesterol, low-density lipoproteins (LDL) and very low-density lipoproteins (VLDL). Other features include RP, acanthocytosis, spinocerebellar degeneration, and fat malabsorption.[86]
- Neurodegeneration with brain iron accumulation (NBIA): Some forms of NBIA, a group of rare genetic disorders characterized by abnormal iron accumulation in the brain, may present with retinal abnormalities resembling RP. The most typical variant (pantothenate kinase-associated neurodegeneration, PKAN) shows the "eye of the tiger" sign on a T2-weighted magnetic resonance image due to a hyperintense signal at the center of the globus pallidus.
- Other systemic diseases associated with RP include familial isolated vitamin E deficiency and Alström syndrome.
These syndromes often have underlying genetic mutations that affect multiple organ systems, including the retina. It is important to note that the severity and specific clinical manifestations can vary widely among individuals with the same syndrome due to genetic heterogeneity and the influence of other genetic and environmental factors. Genetic testing and a multidisciplinary approach involving ophthalmologists, geneticists, and other specialists are typically necessary for accurate diagnosis and management of this condition.
Prognosis
The prognosis for patients with retinitis pigmentosa depends on the age of onset and pattern of inheritance. Expect early-onset symptoms, severe vision loss, and night blindness with the autosomal recessive form of RP. The autosomal dominant expression is the least severe and is associated with the more gradual onset of symptoms later in adulthood. The most severe vision loss occurs with X-linked recessive RP.[87] Tunnel vision is expected late in all forms of RP, and almost all patients with RP will be legally blind at some point in the progression of their disease. Fortunately, total vision loss is uncommon, as the macular function will generally allow light perception, even after acuity is lost. Many patients retain good central vision into their 40s or 50s.[88] Genetic testing is now available to determine the specific gene mutation causing a patient's RP, which may help predict disease severity and progression.[89]
Complications
Functional difficulties associated with RP include night blindness, progressive loss of vision, and a gradual reduction in peripheral and central vision. In advanced stages, patients with RP may become legally blind or have minimal vision. Severely impaired night vision may make navigating in low-light conditions challenging. RP typically leads to gradually losing peripheral vision (tunnel vision). This narrowing of the visual field can affect daily activities such as mobility and driving.
In some cases, RP can progress to affect central vision, which is essential for tasks like reading and recognizing faces. Some individuals with RP may experience color vision abnormalities, including difficulty distinguishing between specific colors or a reduced ability to perceive colors accurately. Increased sensitivity to light (photophobia) can complicate RP.[90] Bright lights can cause discomfort and glare, making it difficult to tolerate well-lit environments. RP can reduce contrast sensitivity, making distinguishing objects or text from their background difficult. Reduced peripheral vision and contrast sensitivity can lead to difficulties with depth perception, making it harder to judge distances accurately. Reduced vision, especially in low-light conditions, can increase the risk of accidents and injuries, both indoors and outdoors. Individuals with RP may have depression, anxiety, and reduced quality of life.[91] As vision deteriorates, individuals with RP may become increasingly dependent on others for daily tasks and mobility, impacting their independence. Due to vision-related limitations, children and adults with RP may face difficulties in educational and occupational settings.
Ocular complications associated with RP include cataracts, especially posterior subcapsular cataracts. The lens may have weak zonules resulting in anterior capsular phimosis, subluxation, or dislocation of capsular bag-intraocular lens complex after cataract surgery.[92] Patients with RP may have retrolental or vitreous cells. Some cases are associated with intermediate uveitis and leaking cystoid macular edema on fluorescein angiography.[93][94] Such patients may respond favorably with posterior subtenon triamcinolone acetonide, intravitreal, or systemic steroids.[95][96] Macular complications associated with RP include foveoschisis (non-leaking on fluorescein angiogram), CME (petaloid leak on fluorescein angiogram), macular holes, epiretinal membrane, vitreomacular traction, and choroidal neovascular membrane.[97] Myopia and astigmatism may be expected in patients with RP.[98] Both open-angle and closed-angle glaucoma may be associated with RP.[99][100] The prevalence of angle-closure glaucoma has been reported as 1% to 2.3% in populations of Canada and China, respectively.[101][102] Around 2% to 12% of patients with RP have primary open-angle glaucoma.[99] Around 68% of retinal detachments associated with RP are rhegmatogenous.[103] There may be an absence of complete posterior vitreous detachment and a predominance of round holes in younger patients. Proliferative vitreoretinopathy is common, and titration of laser burns is necessary during surgery for rhegmatogenous retinal detachment.[103] Coats disease-like exudative vitreoretinopathy may be noted in up to 5% of patients with RP.[104] Such manifestation is usually bilateral and may cause exudative retinal detachment.[104][105] Patients with RP may develop retinal vascular abnormalities like microaneurysms, telangiectasia, and neovascularization.[106] These abnormalities can cause recurrent vitreous hemorrhage.[106]
Deterrence and Patient Education
Upon diagnosis of RP, patient and family education becomes a primary goal. Understanding the type of disease which affects the patient is critical. In a study involving vision-related quality of life and coping mechanisms for managing RP, results indicated that most individuals with RP were unaware of their subtype. With this understanding, it is evident that these patients can fully understand their disease progression and implications for their future. [107]
While no treatment options guarantee to arrest the progression of vision loss, patients should be aware of the possible benefits of several interventions. Nutritional supplementation, especially with vitamin A, is still an area of interest, but there has yet to be a definite identification of benefits. Avoidance of excessively bright light exposure has been a recommendation for many years, as it may decrease phototoxic effects on the retina. Good quality sunglasses are, therefore, a must, and patients should avoid smoking.[108]
Patient education should also include preparation for the anticipated progression of RP. Genetic analysis helps in prognosticating. After diagnosis and the prognosis becomes relatively predictable, the patient and family must look toward adaptive changes to enable the patient to function with maximal independence for as long as possible (ie, things as simple as not rearranging the furniture in the home and installing additional lighting in work areas). As vision impairs the ability to drive and function outside the home without assistance, consider referral to community support agencies. As the disease affects independence and the patient begins to feel isolated, depression can be a severe complication.[109] Study results found that patients with RP have 5- to 6-times increased odds of anxiety or depression.[109] Educate patients and family members about the warning signs of depression, and clinicians should maintain vigilance in assessing the mental health of their patients.
Promote awareness of ongoing research and the development of new therapies. This knowledge will enhance the patient's ability to make an informed decision about treatment options when they become available. The National Eye Institute of the National Institutes of Health, Retinitis Pigmentosa Foundation Fighting Blindness, and the American Foundation for the Blind are excellent sources of information. These organizations can place patients and their families in touch with support groups and other sources for assistance and provide information about the disease.
Enhancing Healthcare Team Outcomes
The evaluation and treatment of retinitis pigmentosa require educating the patient and family. While the ophthalmologist primarily manages the disorder, an ancillary team consisting of a geneticist and pharmacist may also help. Consider genetic testing and counseling for those affected by RP. In many cases, the long-term prognosis can be better predicted, and the possibility of gene transmission to descendants will be an essential consideration for some patients.
While no treatment options guarantee to arrest the progression of vision loss, patients should be aware of the possible benefits of several interventions. Nutritional supplementation, especially with vitamin A, is still an area of interest, but the pharmacist should warn the patient that benefits are unidentified. In addition, monitor vitamin A intake for the long term because of its potential toxicity to the liver. The ophthalmology nurse should educate the patient on avoiding excessively bright light exposure, which may decrease phototoxic effects on the retina. Good sunglasses are a must. Understanding the type of disease that affects the patient is also critical. With this understanding, patients can fully appreciate and learn to adapt to their disease progression and its implications. An interprofessional approach involving a specialty-trained nurse and clinician assisting in patient management and education will provide the best outcome.
Media
(Click Image to Enlarge)
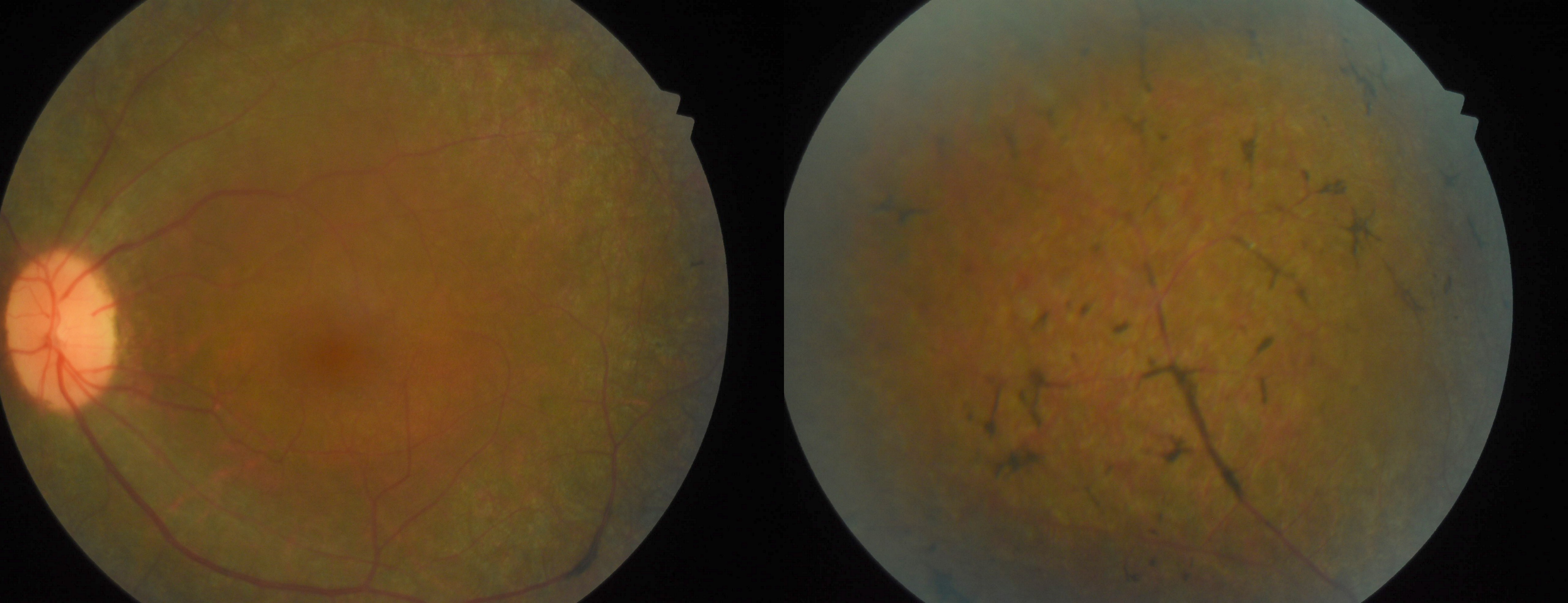
Fundus Showing Bony Spicules
Courtesy of Koushik Tripathy, MD
(Click Image to Enlarge)
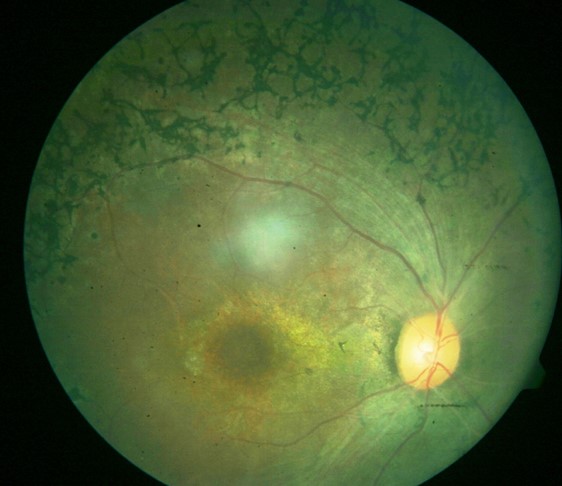
Right Eye Retinitis Pigmentosa
Courtesy of Ogugua Okonkwo, MD FRCS
(Click Image to Enlarge)
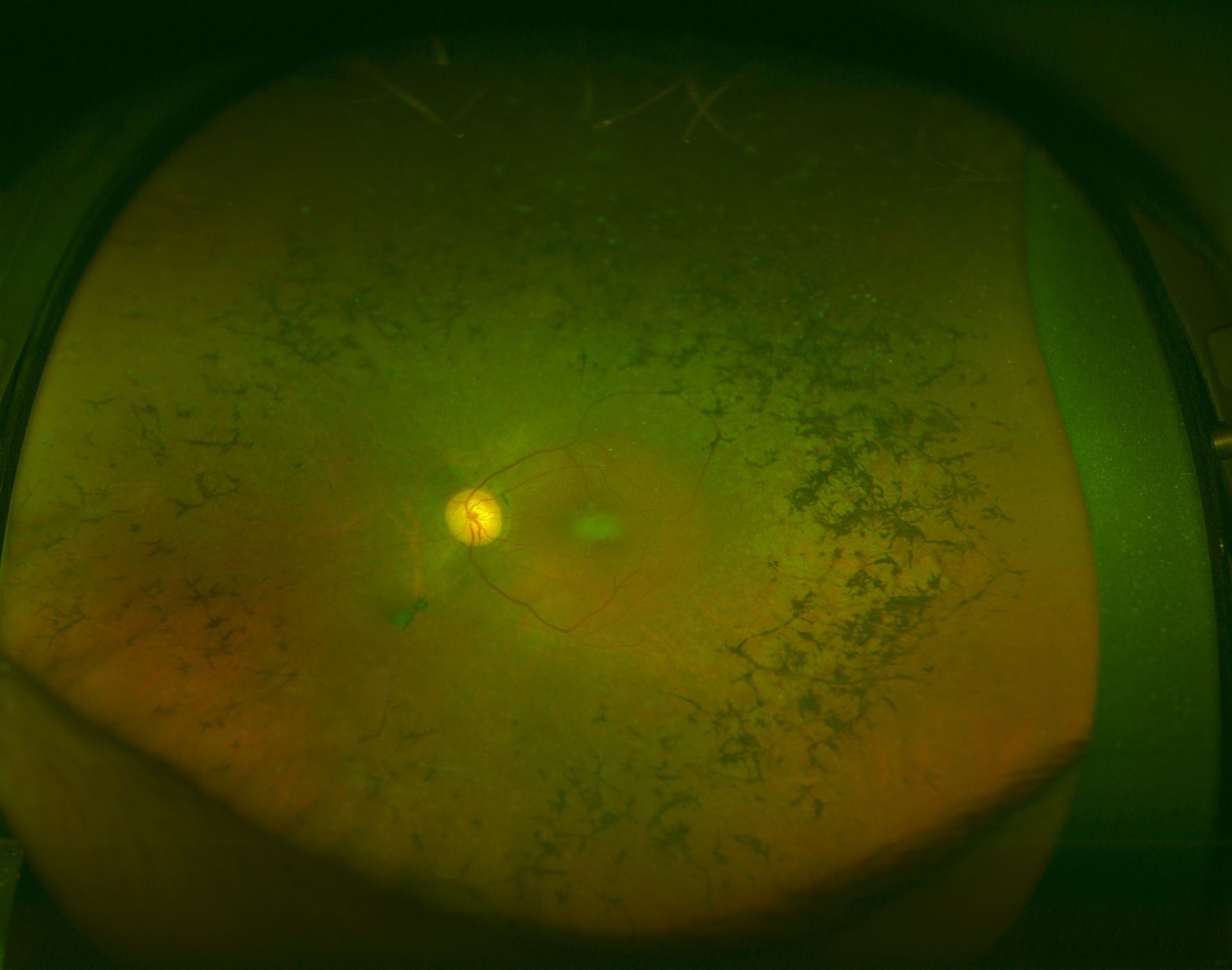
Retinitis Pigmentosa: The fundus appearance of RP classically includes a triad of retinal vessel attenuation, waxy pallor of the optic disc, and bone spicule intraretinal pigmentation.
Courtesy of K Tripathy, MD
References
Verbakel SK, van Huet RAC, Boon CJF, den Hollander AI, Collin RWJ, Klaver CCW, Hoyng CB, Roepman R, Klevering BJ. Non-syndromic retinitis pigmentosa. Progress in retinal and eye research. 2018 Sep:66():157-186. doi: 10.1016/j.preteyeres.2018.03.005. Epub 2018 Mar 27 [PubMed PMID: 29597005]
Boughman JA, Conneally PM, Nance WE. Population genetic studies of retinitis pigmentosa. American journal of human genetics. 1980 Mar:32(2):223-35 [PubMed PMID: 7386458]
Weller JM, Michelson G, Juenemann AG. Unilateral retinitis pigmentosa: 30 years follow-up. BMJ case reports. 2014 Feb 10:2014():. doi: 10.1136/bcr-2013-202236. Epub 2014 Feb 10 [PubMed PMID: 24515232]
Level 3 (low-level) evidenceBittner AK, Diener-West M, Dagnelie G. Characteristics and possible visual consequences of photopsias as vision measures are reduced in retinitis pigmentosa. Investigative ophthalmology & visual science. 2011 Aug 11:52(9):6370-6. doi: 10.1167/iovs.11-7195. Epub 2011 Aug 11 [PubMed PMID: 21693605]
Level 2 (mid-level) evidenceO'Hare F, Bentley SA, Wu Z, Guymer RH, Luu CD, Ayton LN. Charles Bonnet Syndrome in Advanced Retinitis Pigmentosa. Ophthalmology. 2015 Sep:122(9):1951-3. doi: 10.1016/j.ophtha.2015.03.006. Epub 2015 Apr 11 [PubMed PMID: 25870080]
Wolfrum U, Nagel-Wolfrum K. [The Usher Syndrome, a Human Ciliopathy]. Klinische Monatsblatter fur Augenheilkunde. 2018 Mar:235(3):273-280. doi: 10.1055/a-0573-9431. Epub 2018 Mar 13 [PubMed PMID: 29534264]
Mathur P, Yang J. Usher syndrome: Hearing loss, retinal degeneration and associated abnormalities. Biochimica et biophysica acta. 2015 Mar:1852(3):406-20. doi: 10.1016/j.bbadis.2014.11.020. Epub 2014 Dec 4 [PubMed PMID: 25481835]
Daiger SP, Sullivan LS, Bowne SJ. Genes and mutations causing retinitis pigmentosa. Clinical genetics. 2013 Aug:84(2):132-41. doi: 10.1111/cge.12203. Epub 2013 Jun 19 [PubMed PMID: 23701314]
Yang YJ, Peng J, Ying D, Peng QH. A Brief Review on the Pathological Role of Decreased Blood Flow Affected in Retinitis Pigmentosa. Journal of ophthalmology. 2018:2018():3249064. doi: 10.1155/2018/3249064. Epub 2018 Feb 25 [PubMed PMID: 29682340]
Phelan JK, Bok D. A brief review of retinitis pigmentosa and the identified retinitis pigmentosa genes. Molecular vision. 2000 Jul 8:6():116-24 [PubMed PMID: 10889272]
Level 3 (low-level) evidenceChang S, Vaccarella L, Olatunji S, Cebulla C, Christoforidis J. Diagnostic challenges in retinitis pigmentosa: genotypic multiplicity and phenotypic variability. Current genomics. 2011 Jun:12(4):267-75. doi: 10.2174/138920211795860116. Epub [PubMed PMID: 22131872]
Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, Fahim AT, Daiger SP, Weleber RG. Nonsyndromic Retinitis Pigmentosa Overview. GeneReviews(®). 1993:(): [PubMed PMID: 20301590]
Level 3 (low-level) evidenceKajiwara K, Berson EL, Dryja TP. Digenic retinitis pigmentosa due to mutations at the unlinked peripherin/RDS and ROM1 loci. Science (New York, N.Y.). 1994 Jun 10:264(5165):1604-8 [PubMed PMID: 8202715]
Cross N, van Steen C, Zegaoui Y, Satherley A, Angelillo L. Retinitis Pigmentosa: Burden of Disease and Current Unmet Needs. Clinical ophthalmology (Auckland, N.Z.). 2022:16():1993-2010. doi: 10.2147/OPTH.S365486. Epub 2022 Jun 20 [PubMed PMID: 35757022]
Haim M, Holm NV, Rosenberg T. Prevalence of retinitis pigmentosa and allied disorders in Denmark. I Main results. Acta ophthalmologica. 1992 Apr:70(2):178-86 [PubMed PMID: 1609565]
Level 2 (mid-level) evidenceBundey S, Crews SJ. A study of retinitis pigmentosa in the City of Birmingham. I Prevalence. Journal of medical genetics. 1984 Dec:21(6):417-20 [PubMed PMID: 6512829]
Level 2 (mid-level) evidenceSen P, Bhargava A, George R, Ve Ramesh S, Hemamalini A, Prema R, Kumaramanickavel G, Vijaya L. Prevalence of retinitis pigmentosa in South Indian population aged above 40 years. Ophthalmic epidemiology. 2008 Jul-Aug:15(4):279-81. doi: 10.1080/09286580802105814. Epub [PubMed PMID: 18780262]
Level 2 (mid-level) evidenceGupta KK, Gurung G, Tulsyan N. Prevalence of Retinitis Pigmentosa in a Tertiary Eye Hospital of Nepal. Nepalese journal of ophthalmology : a biannual peer-reviewed academic journal of the Nepal Ophthalmic Society : NEPJOPH. 2022 Jan:14(27):31-38. doi: 10.3126/nepjoph.v14i1.38977. Epub [PubMed PMID: 35996901]
Delmaghani S, El-Amraoui A. The genetic and phenotypic landscapes of Usher syndrome: from disease mechanisms to a new classification. Human genetics. 2022 Apr:141(3-4):709-735. doi: 10.1007/s00439-022-02448-7. Epub 2022 Mar 30 [PubMed PMID: 35353227]
Tsujikawa M, Wada Y, Sukegawa M, Sawa M, Gomi F, Nishida K, Tano Y. Age at onset curves of retinitis pigmentosa. Archives of ophthalmology (Chicago, Ill. : 1960). 2008 Mar:126(3):337-40. doi: 10.1001/archopht.126.3.337. Epub [PubMed PMID: 18332312]
Xu H, Chen M, Forrester JV. Para-inflammation in the aging retina. Progress in retinal and eye research. 2009 Sep:28(5):348-68. doi: 10.1016/j.preteyeres.2009.06.001. Epub 2009 Jun 26 [PubMed PMID: 19560552]
Level 3 (low-level) evidenceArango-Gonzalez B, Trifunović D, Sahaboglu A, Kranz K, Michalakis S, Farinelli P, Koch S, Koch F, Cottet S, Janssen-Bienhold U, Dedek K, Biel M, Zrenner E, Euler T, Ekström P, Ueffing M, Paquet-Durand F. Identification of a common non-apoptotic cell death mechanism in hereditary retinal degeneration. PloS one. 2014:9(11):e112142. doi: 10.1371/journal.pone.0112142. Epub 2014 Nov 13 [PubMed PMID: 25392995]
Level 3 (low-level) evidenceViringipurampeer IA, Gregory-Evans CY, Metcalfe AL, Bashar E, Moritz OL, Gregory-Evans K. Cell Death Pathways in Mutant Rhodopsin Rat Models Identifies Genotype-Specific Targets Controlling Retinal Degeneration. Molecular neurobiology. 2019 Mar:56(3):1637-1652. doi: 10.1007/s12035-018-1192-8. Epub 2018 Jun 18 [PubMed PMID: 29911255]
Cideciyan AV, Jacobson SG, Aleman TS, Gu D, Pearce-Kelling SE, Sumaroka A, Acland GM, Aguirre GD. In vivo dynamics of retinal injury and repair in the rhodopsin mutant dog model of human retinitis pigmentosa. Proceedings of the National Academy of Sciences of the United States of America. 2005 Apr 5:102(14):5233-8 [PubMed PMID: 15784735]
Level 3 (low-level) evidenceNewton F, Megaw R. Mechanisms of Photoreceptor Death in Retinitis Pigmentosa. Genes. 2020 Sep 24:11(10):. doi: 10.3390/genes11101120. Epub 2020 Sep 24 [PubMed PMID: 32987769]
Wellard J, Lee D, Valter K, Stone J. Photoreceptors in the rat retina are specifically vulnerable to both hypoxia and hyperoxia. Visual neuroscience. 2005 Jul-Aug:22(4):501-7 [PubMed PMID: 16212707]
Level 3 (low-level) evidenceB Domènech E, Marfany G. The Relevance of Oxidative Stress in the Pathogenesis and Therapy of Retinal Dystrophies. Antioxidants (Basel, Switzerland). 2020 Apr 23:9(4):. doi: 10.3390/antiox9040347. Epub 2020 Apr 23 [PubMed PMID: 32340220]
Zhang L, Justus S, Xu Y, Pluchenik T, Hsu CW, Yang J, Duong JK, Lin CS, Jia Y, Bassuk AG, Mahajan VB, Tsang SH. Reprogramming towards anabolism impedes degeneration in a preclinical model of retinitis pigmentosa. Human molecular genetics. 2016 Oct 1:25(19):4244-4255. doi: 10.1093/hmg/ddw256. Epub 2016 Aug 11 [PubMed PMID: 27516389]
Gallenga CE, Lonardi M, Pacetti S, Violanti SS, Tassinari P, Di Virgilio F, Tognon M, Perri P. Molecular Mechanisms Related to Oxidative Stress in Retinitis Pigmentosa. Antioxidants (Basel, Switzerland). 2021 May 26:10(6):. doi: 10.3390/antiox10060848. Epub 2021 May 26 [PubMed PMID: 34073310]
Groenendyk J, Agellon LB, Michalak M. Calcium signaling and endoplasmic reticulum stress. International review of cell and molecular biology. 2021:363():1-20. doi: 10.1016/bs.ircmb.2021.03.003. Epub 2021 May 19 [PubMed PMID: 34392927]
Badano JL, Mitsuma N, Beales PL, Katsanis N. The ciliopathies: an emerging class of human genetic disorders. Annual review of genomics and human genetics. 2006:7():125-48 [PubMed PMID: 16722803]
Andresen H M, Regueira H T, Leighton F. [Oxidative stress in critically ill patients]. Revista medica de Chile. 2006 May:134(5):649-56 [PubMed PMID: 16802059]
Milam AH, Li ZY, Fariss RN. Histopathology of the human retina in retinitis pigmentosa. Progress in retinal and eye research. 1998 Apr:17(2):175-205 [PubMed PMID: 9695792]
Zhao TT, Tian CY, Yin ZQ. Activation of Müller cells occurs during retinal degeneration in RCS rats. Advances in experimental medicine and biology. 2010:664():575-83. doi: 10.1007/978-1-4419-1399-9_66. Epub [PubMed PMID: 20238061]
Level 3 (low-level) evidenceTripathy K, Sharma YR, Chawla R, Basu K, Vohra R, Venkatesh P. Triads in Ophthalmology: A Comprehensive Review. Seminars in ophthalmology. 2017:32(2):237-250. doi: 10.3109/08820538.2015.1045150. Epub 2015 Jul 6 [PubMed PMID: 26148300]
Shintani K, Shechtman DL, Gurwood AS. Review and update: current treatment trends for patients with retinitis pigmentosa. Optometry (St. Louis, Mo.). 2009 Jul:80(7):384-401. doi: 10.1016/j.optm.2008.01.026. Epub [PubMed PMID: 19545852]
Level 3 (low-level) evidenceCoussa RG, Basali D, Maeda A, DeBenedictis M, Traboulsi EI. Sector retinitis pigmentosa: Report of ten cases and a review of the literature. Molecular vision. 2019:25():869-889 [PubMed PMID: 31908405]
Level 3 (low-level) evidencePopović P, Jarc-Vidmar M, Hawlina M. Abnormal fundus autofluorescence in relation to retinal function in patients with retinitis pigmentosa. Graefe's archive for clinical and experimental ophthalmology = Albrecht von Graefes Archiv fur klinische und experimentelle Ophthalmologie. 2005 Oct:243(10):1018-27 [PubMed PMID: 15906064]
Robson AG, Egan C, Holder GE, Bird AC, Fitzke FW. Comparing rod and cone function with fundus autofluorescence images in retinitis pigmentosa. Advances in experimental medicine and biology. 2003:533():41-7 [PubMed PMID: 15180246]
Level 2 (mid-level) evidencePichi F, Abboud EB, Ghazi NG, Khan AO. Fundus autofluorescence imaging in hereditary retinal diseases. Acta ophthalmologica. 2018 Aug:96(5):e549-e561. doi: 10.1111/aos.13602. Epub 2017 Nov 2 [PubMed PMID: 29098804]
Robson AG, Lenassi E, Saihan Z, Luong VA, Fitzke FW, Holder GE, Webster AR. Comparison of fundus autofluorescence with photopic and scotopic fine matrix mapping in patients with retinitis pigmentosa: 4- to 8-year follow-up. Investigative ophthalmology & visual science. 2012 Sep 14:53(10):6187-95. doi: 10.1167/iovs.12-10195. Epub 2012 Sep 14 [PubMed PMID: 22899761]
Lee J, Asano S, Inoue T, Fujino Y, Matsuura M, Kitamoto K, Hashimoto Y, Ogawa A, Yanagisawa M, Azuma K, Murata H, Obata R, Asaoka R. Investigating the Usefulness of Fundus Autofluorescence in Retinitis Pigmentosa. Ophthalmology. Retina. 2018 Oct:2(10):1062-1070. doi: 10.1016/j.oret.2018.03.007. Epub 2018 May 18 [PubMed PMID: 31047495]
Yung M, Klufas MA, Sarraf D. Clinical applications of fundus autofluorescence in retinal disease. International journal of retina and vitreous. 2016:2():12. doi: 10.1186/s40942-016-0035-x. Epub 2016 Apr 8 [PubMed PMID: 27847630]
Trichonas G, Traboulsi EI, Ehlers JP. Ultra-widefield fundus autofluorescence patterns in retinitis pigmentosa and other retinal dystrophies. Ophthalmic genetics. 2017 Jan-Feb:38(1):98-100. doi: 10.3109/13816810.2015.1137328. Epub 2016 Apr 6 [PubMed PMID: 27049178]
Marmor MF. The electroretinogram in retinitis pigmentosa. Archives of ophthalmology (Chicago, Ill. : 1960). 1979 Jul:97(7):1300-4 [PubMed PMID: 454267]
Grigoropoulos VG, Emfietzoglou J, Nikolaidis P, Chatzistefanou K, Vergados J, Theodossiadis GP, Theodossiadis PG. Optical coherence tomography findings in patients with retinitis pigmentosa and low visual acuity. Ophthalmic surgery, lasers & imaging : the official journal of the International Society for Imaging in the Eye. 2010 Jan-Feb:41(1):35-9. doi: 10.3928/15428877-20091230-07. Epub [PubMed PMID: 20128568]
Level 2 (mid-level) evidenceTriolo G, Pierro L, Parodi MB, De Benedetto U, Gagliardi M, Manitto MP, Bandello F. Spectral domain optical coherence tomography findings in patients with retinitis pigmentosa. Ophthalmic research. 2013:50(3):160-4. doi: 10.1159/000351681. Epub 2013 Aug 28 [PubMed PMID: 23989166]
Level 2 (mid-level) evidenceTakagi S, Hirami Y, Takahashi M, Fujihara M, Mandai M, Miyakoshi C, Tomita G, Kurimoto Y. Optical coherence tomography angiography in patients with retinitis pigmentosa who have normal visual acuity. Acta ophthalmologica. 2018 Aug:96(5):e636-e642. doi: 10.1111/aos.13680. Epub 2018 Mar 1 [PubMed PMID: 29498230]
Koyanagi Y, Murakami Y, Funatsu J, Akiyama M, Nakatake S, Fujiwara K, Tachibana T, Nakao S, Hisatomi T, Yoshida S, Ishibashi T, Sonoda KH, Ikeda Y. Optical coherence tomography angiography of the macular microvasculature changes in retinitis pigmentosa. Acta ophthalmologica. 2018 Feb:96(1):e59-e67. doi: 10.1111/aos.13475. Epub 2017 May 31 [PubMed PMID: 28561452]
Chatzinoff A, Nelson E, Stahl N, Clahane A. Eleven-CIS vitamin A in the treatment of retinitis pigmentosa. A negative study. Archives of ophthalmology (Chicago, Ill. : 1960). 1968 Oct:80(4):417-9 [PubMed PMID: 4877320]
Massoud WH, Bird AC, Perkins ES. Plasma vitamin A and beta-carotene in retinitis pigmentosa. The British journal of ophthalmology. 1975 Apr:59(4):200-4 [PubMed PMID: 1138843]
Gouras P, Carr RE, Gunkel RD. Retinitis pigmentosa in abetalipoproteinemia: Effects of vitamin A. Investigative ophthalmology. 1971 Oct:10(10):784-93 [PubMed PMID: 5124019]
Berson EL, Rosner B, Sandberg MA, Hayes KC, Nicholson BW, Weigel-DiFrano C, Willett W. Vitamin A supplementation for retinitis pigmentosa. Archives of ophthalmology (Chicago, Ill. : 1960). 1993 Nov:111(11):1456-9 [PubMed PMID: 8240091]
Level 3 (low-level) evidenceRayapudi S, Schwartz SG, Wang X, Chavis P. Vitamin A and fish oils for retinitis pigmentosa. The Cochrane database of systematic reviews. 2013 Dec 19:2013(12):CD008428. doi: 10.1002/14651858.CD008428.pub2. Epub 2013 Dec 19 [PubMed PMID: 24357340]
Level 1 (high-level) evidenceSchwartz SG, Wang X, Chavis P, Kuriyan AE, Abariga SA. Vitamin A and fish oils for preventing the progression of retinitis pigmentosa. The Cochrane database of systematic reviews. 2020 Jun 18:6(6):CD008428. doi: 10.1002/14651858.CD008428.pub3. Epub 2020 Jun 18 [PubMed PMID: 32573764]
Level 1 (high-level) evidencePiri N, Grodsky JD, Kaplan HJ. Gene therapy for retinitis pigmentosa. Taiwan journal of ophthalmology. 2021 Oct-Dec:11(4):348-351. doi: 10.4103/tjo.tjo_47_21. Epub 2021 Nov 19 [PubMed PMID: 35070662]
Xi Z, Vats A, Sahel JA, Chen Y, Byrne LC. Gene augmentation prevents retinal degeneration in a CRISPR/Cas9-based mouse model of PRPF31 retinitis pigmentosa. Nature communications. 2022 Dec 13:13(1):7695. doi: 10.1038/s41467-022-35361-8. Epub 2022 Dec 13 [PubMed PMID: 36509783]
Arsenijevic Y, Berger A, Udry F, Kostic C. Lentiviral Vectors for Ocular Gene Therapy. Pharmaceutics. 2022 Jul 31:14(8):. doi: 10.3390/pharmaceutics14081605. Epub 2022 Jul 31 [PubMed PMID: 36015231]
Maguire AM, Simonelli F, Pierce EA, Pugh EN Jr, Mingozzi F, Bennicelli J, Banfi S, Marshall KA, Testa F, Surace EM, Rossi S, Lyubarsky A, Arruda VR, Konkle B, Stone E, Sun J, Jacobs J, Dell'Osso L, Hertle R, Ma JX, Redmond TM, Zhu X, Hauck B, Zelenaia O, Shindler KS, Maguire MG, Wright JF, Volpe NJ, McDonnell JW, Auricchio A, High KA, Bennett J. Safety and efficacy of gene transfer for Leber's congenital amaurosis. The New England journal of medicine. 2008 May 22:358(21):2240-8. doi: 10.1056/NEJMoa0802315. Epub 2008 Apr 27 [PubMed PMID: 18441370]
Dias MF, Joo K, Kemp JA, Fialho SL, da Silva Cunha A Jr, Woo SJ, Kwon YJ. Molecular genetics and emerging therapies for retinitis pigmentosa: Basic research and clinical perspectives. Progress in retinal and eye research. 2018 Mar:63():107-131. doi: 10.1016/j.preteyeres.2017.10.004. Epub 2017 Oct 31 [PubMed PMID: 29097191]
Level 3 (low-level) evidenceCross N, van Steen C, Zegaoui Y, Satherley A, Angelillo L. Current and Future Treatment of Retinitis Pigmentosa. Clinical ophthalmology (Auckland, N.Z.). 2022:16():2909-2921. doi: 10.2147/OPTH.S370032. Epub 2022 Aug 31 [PubMed PMID: 36071725]
Sharma A, Jaganathan BG. Stem Cell Therapy for Retinal Degeneration: The Evidence to Date. Biologics : targets & therapy. 2021:15():299-306. doi: 10.2147/BTT.S290331. Epub 2021 Jul 27 [PubMed PMID: 34349498]
He Y, Zhang Y, Liu X, Ghazaryan E, Li Y, Xie J, Su G. Recent advances of stem cell therapy for retinitis pigmentosa. International journal of molecular sciences. 2014 Aug 20:15(8):14456-74. doi: 10.3390/ijms150814456. Epub 2014 Aug 20 [PubMed PMID: 25141102]
Level 3 (low-level) evidenceLimoli PG, Vingolo EM, Limoli C, Nebbioso M. Stem Cell Surgery and Growth Factors in Retinitis Pigmentosa Patients: Pilot Study after Literature Review. Biomedicines. 2019 Nov 30:7(4):. doi: 10.3390/biomedicines7040094. Epub 2019 Nov 30 [PubMed PMID: 31801246]
Level 3 (low-level) evidenceSmith LE. Bone marrow-derived stem cells preserve cone vision in retinitis pigmentosa. The Journal of clinical investigation. 2004 Sep:114(6):755-7 [PubMed PMID: 15372096]
Level 3 (low-level) evidenceSurendran H, Nandakumar S, Reddy K VB, Stoddard J, Mohan K V, Upadhyay PK, McGill TJ, Pal R. Transplantation of retinal pigment epithelium and photoreceptors generated concomitantly via small molecule-mediated differentiation rescues visual function in rodent models of retinal degeneration. Stem cell research & therapy. 2021 Jan 19:12(1):70. doi: 10.1186/s13287-021-02134-x. Epub 2021 Jan 19 [PubMed PMID: 33468244]
Hill D, Compagnoni C, Cordeiro MF. Investigational neuroprotective compounds in clinical trials for retinal disease. Expert opinion on investigational drugs. 2021 May:30(5):571-577. doi: 10.1080/13543784.2021.1896701. Epub 2021 Apr 1 [PubMed PMID: 33641585]
Level 3 (low-level) evidenceBirch DG, Bennett LD, Duncan JL, Weleber RG, Pennesi ME. Long-term Follow-up of Patients With Retinitis Pigmentosa Receiving Intraocular Ciliary Neurotrophic Factor Implants. American journal of ophthalmology. 2016 Oct:170():10-14. doi: 10.1016/j.ajo.2016.07.013. Epub 2016 Jul 25 [PubMed PMID: 27457255]
Chow AY, Bittner AK, Pardue MT. The artificial silicon retina in retinitis pigmentosa patients (an American Ophthalmological Association thesis). Transactions of the American Ophthalmological Society. 2010 Dec:108():120-54 [PubMed PMID: 21212852]
Level 3 (low-level) evidenceGekeler K, Bartz-Schmidt KU, Sachs H, MacLaren RE, Stingl K, Zrenner E, Gekeler F. Implantation, removal and replacement of subretinal electronic implants for restoration of vision in patients with retinitis pigmentosa. Current opinion in ophthalmology. 2018 May:29(3):239-247. doi: 10.1097/ICU.0000000000000467. Epub [PubMed PMID: 29528862]
Level 3 (low-level) evidenceHallum LE, Dakin SC. Retinal Implantation of Electronic Vision Prostheses to Treat Retinitis Pigmentosa: A Systematic Review. Translational vision science & technology. 2021 Aug 12:10(10):8. doi: 10.1167/tvst.10.10.8. Epub [PubMed PMID: 34383874]
Level 1 (high-level) evidenceWard J, Meijer P. Visual experiences in the blind induced by an auditory sensory substitution device. Consciousness and cognition. 2010 Mar:19(1):492-500. doi: 10.1016/j.concog.2009.10.006. Epub 2009 Dec 1 [PubMed PMID: 19955003]
Level 3 (low-level) evidenceGrant CA, Berson EL. Treatable forms of retinitis pigmentosa associated with systemic neurological disorders. International ophthalmology clinics. 2001 Winter:41(1):103-10 [PubMed PMID: 11198137]
Dessalces E, Bocquet B, Bourien J, Zanlonghi X, Verdet R, Meunier I, Hamel CP. Early-onset foveal involvement in retinitis punctata albescens with mutations in RLBP1. JAMA ophthalmology. 2013 Oct:131(10):1314-23. doi: 10.1001/jamaophthalmol.2013.4476. Epub [PubMed PMID: 23929416]
Hipp S, Zobor G, Glöckle N, Mohr J, Kohl S, Zrenner E, Weisschuh N, Zobor D. Phenotype variations of retinal dystrophies caused by mutations in the RLBP1 gene. Acta ophthalmologica. 2015 Jun:93(4):e281-6. doi: 10.1111/aos.12573. Epub 2014 Nov 27 [PubMed PMID: 25429852]
Kurata K, Hosono K, Hotta Y. Long-Term Clinical Course in a Patient with Complete Congenital Stationary Night Blindness. Case reports in ophthalmology. 2017 Jan-Apr:8(1):237-244. doi: 10.1159/000462961. Epub 2017 Apr 10 [PubMed PMID: 28512427]
Level 3 (low-level) evidenceZeitz C, Friedburg C, Preising MN, Lorenz B. [Overview of Congenital Stationary Night Blindness with Predominantly Normal Fundus Appearance]. Klinische Monatsblatter fur Augenheilkunde. 2018 Mar:235(3):281-289. doi: 10.1055/s-0043-123072. Epub 2018 Feb 1 [PubMed PMID: 29390235]
Level 3 (low-level) evidenceAgarwal R, Tripathy K, Bandyopadhyay G, Basu K. Mizuo-Nakamura phenomenon in an Indian male. Clinical case reports. 2019 Feb:7(2):401-403. doi: 10.1002/ccr3.1990. Epub 2019 Jan 13 [PubMed PMID: 30847219]
Level 3 (low-level) evidenceSingh D, Tripathy K. Cancer-Associated Retinopathy. StatPearls. 2024 Jan:(): [PubMed PMID: 35201711]
Thakur A, Puri L. Unilateral retinitis pigmentosa. Clinical & experimental optometry. 2010 Mar:93(2):102-4. doi: 10.1111/j.1444-0938.2009.00435.x. Epub [PubMed PMID: 20406260]
Level 3 (low-level) evidenceMukhopadhyay R, Holder GE, Moore AT, Webster AR. Unilateral retinitis pigmentosa occurring in an individual with a germline mutation in the RP1 gene. Archives of ophthalmology (Chicago, Ill. : 1960). 2011 Jul:129(7):954-6. doi: 10.1001/archophthalmol.2011.171. Epub [PubMed PMID: 21746989]
Level 3 (low-level) evidenceTripathy K, Chawla R, Meena S, Agarwal P. Unilateral giant peripapillary drusen and retinal drusenoid deposits in a case of X-linked retinoschisis. BMJ case reports. 2016 Feb 23:2016():. doi: 10.1136/bcr-2016-214558. Epub 2016 Feb 23 [PubMed PMID: 26907824]
Level 3 (low-level) evidenceCarr RE, Siegel IM. Unilateral retinitis pigmentosa. Archives of ophthalmology (Chicago, Ill. : 1960). 1973 Jul:90(1):21-6 [PubMed PMID: 4714794]
Toms M, Pagarkar W, Moosajee M. Usher syndrome: clinical features, molecular genetics and advancing therapeutics. Therapeutic advances in ophthalmology. 2020 Jan-Dec:12():2515841420952194. doi: 10.1177/2515841420952194. Epub 2020 Sep 17 [PubMed PMID: 32995707]
Level 3 (low-level) evidenceTripathy K, Chawla R, Sarkar S. Girl with polydactyly and pigmentary retinopathy. Journal of paediatrics and child health. 2017 Apr:53(4):424. doi: 10.1111/jpc.1_13323. Epub [PubMed PMID: 28370859]
Junaid SZS, Patel K. Abetalipoproteinemia. StatPearls. 2023 Jan:(): [PubMed PMID: 30020727]
Berson EL. Long-term visual prognoses in patients with retinitis pigmentosa: the Ludwig von Sallmann lecture. Experimental eye research. 2007 Jul:85(1):7-14 [PubMed PMID: 17531222]
Menghini M, Cehajic-Kapetanovic J, MacLaren RE. Monitoring progression of retinitis pigmentosa: current recommendations and recent advances. Expert opinion on orphan drugs. 2020:8(2-3):67-78. doi: 10.1080/21678707.2020.1735352. Epub 2020 Mar 2 [PubMed PMID: 32231889]
Level 3 (low-level) evidenceFahim A. Retinitis pigmentosa: recent advances and future directions in diagnosis and management. Current opinion in pediatrics. 2018 Dec:30(6):725-733. doi: 10.1097/MOP.0000000000000690. Epub [PubMed PMID: 30234647]
Level 3 (low-level) evidenceOtsuka Y, Oishi A, Miyata M, Oishi M, Hasegawa T, Numa S, Ikeda HO, Tsujikawa A. Wavelength of light and photophobia in inherited retinal dystrophy. Scientific reports. 2020 Sep 9:10(1):14798. doi: 10.1038/s41598-020-71707-2. Epub 2020 Sep 9 [PubMed PMID: 32908200]
Watanabe K, Aouadj C, Hiratsuka Y, Yamamoto S, Murakami A. Quality of Life and Economic Impacts of Retinitis Pigmentosa on Japanese Patients: A Non-interventional Cross-sectional Study. Advances in therapy. 2023 May:40(5):2375-2393. doi: 10.1007/s12325-023-02446-9. Epub 2023 Mar 22 [PubMed PMID: 36947329]
Level 2 (mid-level) evidenceNajjar DM, Igbre AO, Tsai FF. Late capsular bag contraction and intraocular lens subluxation in retinitis pigmentosa: a case report. Journal of medical case reports. 2011 Feb 14:5():65. doi: 10.1186/1752-1947-5-65. Epub 2011 Feb 14 [PubMed PMID: 21320335]
Level 3 (low-level) evidenceTripathy K, Chawla R, Venkatesh P, Vohra R, Sharma YR, Gogia V, Jain S, Behera A. Ultra-wide Field Fluorescein Angiography in Retinitis Pigmentosa with Intermediate Uveitis. Journal of ophthalmic & vision research. 2016 Apr-Jun:11(2):237-9. doi: 10.4103/2008-322X.183929. Epub [PubMed PMID: 27413510]
Chauhan K, Tripathy K. Pars Planitis. StatPearls. 2024 Jan:(): [PubMed PMID: 28613790]
Tripathy K. Commentary: Posterior subtenon triamcinolone - The unsung hero for managing various ocular disorders. Indian journal of ophthalmology. 2023 Jan:71(1):181-182. doi: 10.4103/ijo.IJO_1963_22. Epub [PubMed PMID: 36588232]
Level 3 (low-level) evidenceTripathy K. Cystoid Macular Edema in Retinitis Pigmentosa with Intermediate Uveitis Responded Well to Oral and Posterior Subtenon Steroid. Seminars in ophthalmology. 2018:33(4):492-493. doi: 10.1080/08820538.2017.1303521. Epub 2017 Mar 29 [PubMed PMID: 28353369]
Malik A, Sood S, Narang S. Successful treatment of choroidal neovascular membrane in retinitis pigmentosa with intravitreal bevacizumab. International ophthalmology. 2010 Aug:30(4):425-8. doi: 10.1007/s10792-009-9337-4. Epub 2010 Jan 5 [PubMed PMID: 20049508]
Level 3 (low-level) evidenceSieving PA, Fishman GA. Refractive errors of retinitis pigmentosa patients. The British journal of ophthalmology. 1978 Mar:62(3):163-7 [PubMed PMID: 638108]
Pradhan C, Khadka S, Joshi P. Angle Closure Glaucoma in Retinitis Pigmentosa. Case reports in ophthalmological medicine. 2020:2020():6023586. doi: 10.1155/2020/6023586. Epub 2020 May 29 [PubMed PMID: 32551144]
Level 3 (low-level) evidenceHung MC, Chen YY. Patients with Retinitis Pigmentosa May Have a Higher Risk of Developing Open-Angle Glaucoma. Journal of ophthalmology. 2022:2022():9719095. doi: 10.1155/2022/9719095. Epub 2022 Jun 22 [PubMed PMID: 35783342]
Badeeb O, Trope G, Musarella M. Primary angle closure glaucoma and retinitis pigmentosa. Acta ophthalmologica. 1993 Dec:71(6):727-32 [PubMed PMID: 8154244]
Level 3 (low-level) evidencePeng DW. [Retinitis pigmentosa associated with glaucoma]. [Zhonghua yan ke za zhi] Chinese journal of ophthalmology. 1991 Sep:27(5):262-4 [PubMed PMID: 1815915]
Chan WO, Brennan N, Webster AR, Michaelides M, Muqit MMK. Retinal detachment in retinitis pigmentosa. BMJ open ophthalmology. 2020:5(1):e000454. doi: 10.1136/bmjophth-2020-000454. Epub 2020 Jul 9 [PubMed PMID: 32671228]
Level 2 (mid-level) evidenceMoinuddin O, Sathrasala S, Jayasundera KT, Branham KH, Chang EY, Qian CX, Recchia FM, Fahim AT, Besirli CG. Coats-like Exudative Vitreoretinopathy in Retinitis Pigmentosa: Ocular Manifestations and Treatment Outcomes. Ophthalmology. Retina. 2021 Jan:5(1):86-96. doi: 10.1016/j.oret.2020.03.026. Epub 2020 Apr 9 [PubMed PMID: 32507488]
Level 2 (mid-level) evidenceGupta A, Paulbuddhe VS, Shukla UV, Tripathy K. Exudative Retinitis (Coats Disease). StatPearls. 2024 Jan:(): [PubMed PMID: 32809517]
Nao-i N, Fukiyama J, Sawada A. Retinitis pigmentosa with recurrent vitreous hemorrhage. Acta ophthalmologica Scandinavica. 1996 Oct:74(5):509-12 [PubMed PMID: 8950405]
Level 3 (low-level) evidenceAnil K, Garip G. Coping strategies, vision-related quality of life, and emotional health in managing retinitis pigmentosa: a survey study. BMC ophthalmology. 2018 Jan 30:18(1):21. doi: 10.1186/s12886-018-0689-2. Epub 2018 Jan 30 [PubMed PMID: 29378559]
Level 2 (mid-level) evidenceOishi A, Noda K, Birtel J, Miyake M, Sato A, Hasegawa T, Miyata M, Numa S, Charbel Issa P, Tsujikawa A. Effect of smoking on macular function and retinal structure in retinitis pigmentosa. Brain communications. 2020:2(2):fcaa117. doi: 10.1093/braincomms/fcaa117. Epub 2020 Jul 23 [PubMed PMID: 33134916]
Le P, Nguyen M, Vu T, Dao DP, Olson D, Zhang AY. Anxiety and Depression in Patients With Retinitis Pigmentosa. Journal of vitreoretinal diseases. 2021 Mar-Apr:5(2):114-120. doi: 10.1177/2474126420936455. Epub 2020 Aug 18 [PubMed PMID: 37009075]