Introduction
Stroke is a significant cause of mortality and disability worldwide, which affects about 2.5% of the global population.[1] The ischemic core, the most proximal part of the arterio-vascular occlusion, sustains maximal damage during a stroke. Between the ischemic core and healthy brain tissue resides the "penumbra," characterized by mild-to-moderate hypoxia. This area risks irreversible damage without restoring blood flow to normal levels within a critical period. Without therapeutic interventions and continued ischemia, the loss of brain tissue can be staggering, with an estimated decline of 1.9 million neurons, 14 billion synapses, and 12 km of myelinated fibers every minute.[2][3][4][5]
Specifically, 1 hour of ischemic brain damage equals 3.6 years of normal brain aging. Acute stroke therapeutics target the tissue damage occurring at the penumbra level and restore the penumbra's functionality. Alteplase, a tissue plasminogen activator (tPA), is the only clot-busting medication approved by the United States Food and Drug Administration (FDA) and recanalizes thrombosed/occluded vasculature in ischemic stroke cases.[6][7] Many studies consistently demonstrate improved outcomes in acute ischemic stroke (AIS) patients treated with tPA. However, interventions such as newer proven endovascular techniques such as mechanical thrombectomy aimed at recanalizing thrombosed vessels, when combined with tPA, may paradoxically result in detrimental effects on ischemic tissue due to complex biochemical and pathological events. In a subacute context, procedures such as carotid endarterectomy and stenting may also lead to reperfusion injury.[8][9][10] Such functional, microscopic, and sometimes macroscopic injury consequential to blood flow restoration is termed an ischemia-reperfusion injury (IRI).
IRI presents a clinical challenge characterized by the exacerbation of cellular dysfunction and death upon restoration of blood flow to previously ischemic tissues. Despite the necessity of restoring blood flow to salvage ischemic tissues, reperfusion paradoxically induces further damage, thereby impeding the function and viability of the affected organ. This phenomenon is observed across various organs, including the heart, lungs, kidneys, gut, skeletal muscles, and brain. Furthermore, IRI may extend beyond the primary ischemic site, triggering systemic damage that culminates in multisystem organ failure. The multifaceted nature of reperfusion injury involves intricate molecular and cellular mechanisms, culminating in extensive tissue destruction.
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
Prolonged cerebral ischemia deprives the brain cells of energy, leading to physiological dysfunction and eventual cell death. Numerous biochemical, physiological, and morphological changes in the cell and its environment precede its eventual death, such as energy failure, lactic acidosis, increases in oxygen extraction fraction and glucose utilization, protein synthesis inhibition, chromatin condensation, cell organelle disruption, and cell shrinkage.[11]
The cascade of events initiated by the ischemic insult contributes to the molecular, microscopic, functional, and macroscopic derangements observed in IRI (see Image. Ischemic Injury and its Molecular Consequences). Among the notable effects are oxidative stress, recruitment of leukocytes, and disruption of the blood-brain barrier (BBB).
Epidemiology
Due to the lack of worldwide assessment data on AIS reperfusion injury after thrombolysis and mechanical thrombectomy, predicting the epidemiology of IRI proves challenging. However, the term "hemorrhagic transformation" can be considered an expression of reperfusion injury. In the historic National Institute of Neurological Disorders and Stroke (NINDS) trial, hemorrhagic transformation occurred in 6.4% of recombinant tPA-treated patients compared to 0.3% of patients treated with a placebo. Notably, this represents symptomatic hemorrhagic transformation (sHT).[7][12]
In the large observational Safe Implementation of Thrombolysis in Stroke-Monitoring Study (SITS-MOST), sHT was observed in 7.3% of ischemic stroke patients undergoing thrombolysis.[13] A comprehensive insight into the epidemiology of thrombectomy-based reperfusion injury can be sourced from the Highly Effective Reperfusion evaluated in Multiple Endovascular Stroke Trials (HERMES) meta-analyses by Goyal et al, where sHT was reported at 4.4%.[14] In the recently concluded extended window endovascular trials, DEFUSE 3 and DAWN, sHT rates were 7% and 6%, respectively.[8][15]
Pathophysiology
Most treatment strategies for ischemic stroke focus on reopening thrombosed vessels, underscoring the importance of time. However, beyond a critical window, rather than preserving brain tissues, restoring oxygen exacerbates the damage to an already compromised neurovascular and brain parenchymal environment. This activity has piqued scientific interest long before the advent of thrombolytic therapies.
Prolonged ischemia and hypoxia following thrombosis of a cerebral vessel induce a shift to anaerobic metabolism, leading to insufficient energy balances and ion dysregulation due to pump failures, including Na+/K+ ATPase, among many others. This cascade leads to an overload of Na+ and Ca+2, causing neuronal swelling and subsequent morphological and functional disruption of cellular organelles, particularly the mitochondria. Damage and swelling of the mitochondria further exacerbate the energy dysfunction in brain cells.[16][17]
Continued ischemia of the brain cells leads to the activation of microglial cells, which initiate post-ischemic inflammation by producing pro-inflammatory chemokines and cytokines such as tumor necrosis factor (TNF)-α or interleukin (IL)-1β. Additionally, activated neutrophils produce reactive oxygen species (ROS) post-ischemia. Beyond a critical period, restoration of oxygen exacerbates an already compromised neurovascular and brain parenchymal milieu, setting the stage for an imminent IRI (see Image. Ischemia-Reperfusion Injury, Inflammation, and the Complement System).[11]
The most critical consequence of introducing oxygen by restoring blood flow to oxygen-deprived tissues is the aggravated generation of ROS. The unchecked generation of ROS directly damages the neurons and indirectly triggers an overactive immune response.
- Formation of ROS: Ischemic tissue is a region where ROS forms. Hypoxanthine, accumulated during ischemia, undergoes sudden metabolism (due to oxygen in reperfusion) by hypoxanthine oxidase, facilitating the production of reperfusion mediators (such as O-, HOCl-, and HO). Mitochondria also contribute to the formation of ROS, paving the way for oxidative stress.
- ROS in reperfusion injury: ROS initiates peroxidation of cell membranes and directly damages the cells. Unchecked ROS intensifies and abets the ongoing pro-inflammatory molecular cascades, along with the recruitment and activation of more leukocytes (see Image. Ischemia-Reperfusion Injury, Reactive Oxygen Species).[18][19]
Inflammatory responses can also have a significantly deleterious impact during reperfusion, as mentioned below.
- Many animal studies have established a temporal relationship between the activation of inflammatory cascades and ischemia. Within the ischemic penumbra, adhesion molecules such as P-selectins and ICAM-1 are upregulated, facilitating leucocyte-endothelial interactions. The unchecked production of ROS contributes to the enhanced P-selectin–mediated rolling and ICAM-1–mediated adhesion of leukocytes, ultimately resulting in platelet-endothelial cell adhesion molecule 1 (PECAM-1)-assisted transmigration or diapedesis of leukocytes into the affected tissues.[20][21][22]
- Oxidative stress also leads to complement activation, involving both C3a and the potent anaphylatoxin C5a. C5a promotes the production of pro-inflammatory cytokines such as TNF-α, IL-1, and IL-6. This cytokine production facilitates leukocyte aggregation due to increased upregulation of leukocyte adhesion molecules. The formation of C5b-9, alongside other activated complement components, forms a membrane attack complex (MAC), further activating leukocytes and lysing cell membranes. Additionally, immunoglobulin M (IgM) antibodies have been observed to deposit in ischemic tissues. When the flow is resumed, complement proteins adhere to these antibodies, activating the complement pathway and exacerbating the injury.[23][24]
- Ischemia-reperfusion significantly contributes to the activation of platelets. P-selectins, upregulated during this process, facilitate the adhesion of activated platelets and leukocytes. Aggregating activated platelets and neutrophils can lead to clogging or microvascular obstruction in the brain, a phenomenon known as "no-reflow," which may occur following reperfusion. Additionally, activated platelets form many biochemical molecules, further enhancing the leukotaxis, promoting leukocyte extravasation, and worsening tissue injury.
The BBB, comprising the vascular endothelium, basement membrane, pericytes, and astrocyte foot processes, creates a unique environment that shields the brain from plasma fluctuations. This barrier's uniqueness lies in the non-fenestrated basement membrane, minimal pinocytic transport, and tight junctions. However, under certain physiological conditions, there may be a minimal opening of the BBB (see Image. Blood Brain Barrier).[25]
A complex interplay of ROS-mediated lipid peroxidation, leukocyte-endothelial adhesion, activated complement, aggregation of activated platelets, and leukocytes (primarily neutrophils) culminates in interrupting the tight junctions and breaching the BBB. This breach allows access to leukocytes, particularly activated neutrophils and their toxins, which derange the physiological milieu of the brain tissue. Additionally, reperfusion triggers the activation of proteases such as matrix metalloproteinases (MMPs), compromising the integrity of the capillary basal lamina, increasing capillary permeability, and ultimately leading to BBB opening.[26]
Apoptotic Cell Death During Ischemia-Reperfusion Injury
Tissue damage resulting from IRI may occur through necrotic or apoptotic pathways. Apoptosis, also known as programmed cell death, is an orchestrated process involving gene-directed events leading to characteristic cellular changes, controlled DNA fragmentation, and eventual cell death. The involvement of apoptosis in reperfusion-induced tissue damage has garnered significant attention in recent research. Oxidative stress and ROS production can trigger apoptosis, particularly in cerebral ischemia-reperfusion, where distinctive apoptotic features are observed. Similarly, renal and cardiac ischemia-reperfusion exhibit measurable levels of apoptosis in affected tissues. Apoptosis thus emerges as a pivotal mechanism contributing to cellular damage in various tissues during reperfusion.
Nevertheless, the role of apoptosis in skeletal muscle ischemic reperfusion remains contentious. Studies have not identified apoptosis in rat skeletal myocytes following ischemic reperfusion, indicating a potentially tissue-specific mode of cell death in this context. Nonetheless, targeting the apoptotic pathway with specific inhibitors against pro-apoptotic caspase enzymes has demonstrated partial effectiveness in animal models, mitigating the severity and extent of injury following hepatic and cardiac ischemia-reperfusion.
Proteases and Metalloproteinases
MMPs represent a family of zinc-dependent enzymes pivotal in extracellular matrix degradation. Coupled with their inhibitors, tissue inhibitors of metalloproteinases, MMPs intricately regulate extracellular matrix turnover. Their involvement spans critical physiological processes such as wound healing, periodontal disease, cancer metastasis, and, notably, vascular pathologies, including aneurysm formation, atherosclerotic plaque development, and reperfusion injury.
Elevated MMP-2 and MMP-9 levels have been observed post-pulmonary, hepatic, and cardiac IRI. Similarly, cerebral IRI elicits upregulation of MMPs, correlating with BBB disruption, basal lamina degradation, heightened capillary permeability, and cerebral edema.[27] Notably, studies utilizing selective MMP-9 inhibitors and MMP-9 knockout mice underscore MMP-9's pivotal role in cerebral IRI pathophysiology, significantly reducing infarct size.[28]
Conversely, the involvement of MMPs in renal IRI remains less elucidated.[29] Although MMP-2 elevation is observed in late-stage renal IRI, MMP inhibition does not effectively mitigate the severity of renal dysfunction, suggesting a nuanced role for MMPs in this context. Investigations correlating systemic plasma MMP-9 levels with MRI-detected hyperintense acute reperfusion injury markers (HARM) in AIS patients underscore MMP-9's association with BBB disruption post-stroke, hinting at MMP-9 inhibition as a potential therapeutic avenue.[30] Importantly, even in the absence of reperfusion, permanent ischemia triggers MMP-2 and MMP-9 elevation, which aligns with the destruction of basement membrane components, notably type IV collagen and laminin.
No Re-flow Phenomenon
"No-reflow" refers to the impairment of microvascular perfusion after restoring flow to previously ischemic tissue. While the precise etiology of this phenomenon remains incompletely understood, it is undoubtedly multifactorial. Factors contributing to "no-reflow" include leukocyte recruitment, platelet activation, and increased blood viscosity due to water movement from plasma to the perivascular space in ischemic tissue, which further clogs the microvasculature of the downstream tissue.[31] This results in increased interstitial pressure alongside a decrease in net intravascular pressure. Furthermore, leukocyte plugging mediated by CD18 exacerbates the obstruction of post-capillary venules, exacerbating the no-reflow phenomenon.[32][33]
The clogged microvasculature proves detrimental to an already oxygen- and nutrient-starved tissue. Despite recanalization and blood flow resumption, the clogged microvasculature leads to continued ischemia and possibly ischemic expansion.[34][16] Studies have shown that depletion of neutrophils effectively mitigates no-reflow in various tissues, including the myocardium, brain, and skeletal muscle, highlighting the pivotal role of neutrophils in this process.
History and Physical
The phrase "time is brain" underscores the rapid loss of healthy brain tissue as a stroke progresses, highlighting the critical importance of prompt care, whether through thrombolysis or endovascular therapy, for patients with AIS. Conducting a prompt, focused history and physical examination in patients presenting with acute-onset neurological symptoms suspicious of AIS is crucial.
During patient assessment, it is essential to gather the patient's past information about when the patient was last known to be well, the onset of symptoms, any relevant risk factors, medications, and other pertinent details suggestive of an underlying condition. Assessing vital signs is crucial, alongside evaluating the level of consciousness, head or gaze deviation, and the laterality of voluntary movements. Ensuring cardiovascular stability is paramount before proceeding with imaging evaluations. In addition, conducting a targeted neurological examination using the National Institutes of Health Stroke Scale (NIHSS) is imperative. The neurological examination findings may be categorized using the Hunt-Hess scale or the World Federation of Neurological Surgeons scale. Please see StatPearls' companion resources, "Acute Stroke" and "Acute Ischemic Stroke," for further information.
Evaluation
Following recanalization using recombinant tPA, the time from symptom onset to disruption of the BBB has been observed to be approximately 12.9 hours.[35] The time for BBB disruption is also associated with age. Usually, imaging of penumbra is not indicated for regular recombinant tPA usage if the patient meets all the criteria. However, penumbral imaging becomes crucial after prolonged ischemia (beyond 6 hours). To pursue further treatment options, including thrombectomy, and to prevent any iatrogenic breach of BBB or hemorrhagic transformation, it is necessary to know the lesion's age. Computed tomography (CT) and magnetic resonance imaging (MRI)-based perfusion studies are utilized to identify the ischemic core and penumbra, assessing the risk-benefit aspects of reperfusion therapies while mitigating unintended consequences of recanalizing a thrombosed cerebral vessel.[36][37][38]
The HARM is a radiological phenomenon characterized by a hyperintense signal within the cerebrospinal fluid spaces observed on post-contrast fluid-attenuated inversion recovery (FLAIR) sequences. This finding is indicative of permeability changes in the BBB during acute stroke. Additionally, single-photon emission computed tomography (SPECT) imaging with 99mTc-duramycin has demonstrated utility in detecting apoptotic neuronal cell death in a rat model of IRI.[39][40][41]
Treatment / Management
Based on the pathophysiology, there are many potential IRI treatment targets. The therapeutic targets include complement depletion, excess ROS reduction, inflammatory cascade mitigation, and leukocyte activation and platelet recruitment inhibition. Given the close association between prolonged ischemia and subsequent IRI with pro-inflammatory conditions, there is a theoretical possibility of addressing this pro-inflammatory state with glucocorticoids.[22] Although dexamethasone showed promise in rat models of ischemic stroke, human clinical trials did not yield beneficial results.[42](B3)
ROS, implicated in much of the oxidative stress associated with ischemia, escalates significantly during reperfusion injury, surpassing the cellular milieu's capacity to contain this excess production. Studies indicate that inhaling hydrogen gas can mitigate mitochondrial pore formation, eventual mitochondrial cell death, and apoptosis. Inhalation of hydrogen gas has been proposed to mop up these free ROS. The underlying principle of hydrogen gas supplementation as a treatment possibility lies in its ability to react with ROS, forming free water and thus reducing ROS toxicity.[43][44] This theory has been successful in animal models but has not yet been tested in human trials.[43][45] (B3)
A clinical trial investigated the potential of infusing superoxide dismutase to prevent reperfusion injury in patients with hemorrhagic shock, showing promise but requiring further exploration on a larger scale.[46] A Rho-kinase inhibitor aimed at inhibiting the NADH oxidase and further limiting ROS production, as well as apocynin, has demonstrated promise in rat models of IRI.[47](A1)
In animal models of cerebral ischemia, hyperbaric oxygen treatment has demonstrated effectiveness in mitigating MMP-2-led injury and inhibiting apoptosis. Additionally, hypothermia, inhalation of isoflurane, and noninvasive vagal nerve stimulation have all shown positive effects in mitigating the damage caused by activated MMP in ischemic tissue.[48][49] A review by Yongchang Li et al provides detailed insight into the emerging possibilities for mitigating IRI.[50][51][52][53]
The failure of the aforementioned substances in clinical trials versus their apparent successes in animal studies raises a fundamental question about the validity of animal ischemia models and their ability to simulate human cerebral ischemia accurately. However, ongoing research in targeted treatments and advancements in modern, functional, and temporal neuroimaging offer promising avenues for future promise.
Preventing IRI requires a comprehensive assessment, including perfusion studies, to evaluate the risk-benefit profile of reperfusion therapies and mitigate potential adverse effects of recanalizing thrombosed cerebral vessels. Identifying HARM correlates with BBB permeability changes in acute stroke, with disruption potentially leading to hemorrhagic transformation and neurological instability. Prompt admission to a specialized neuroscience intensive care unit is associated with favorable outcomes.[54]
Supportive management of an IRI may include aiming for normovolemia and normotension, preventing hypoglycemia, considering antiepileptic drugs if seizures occur, and ensuring cerebral perfusion pressure remains above 70 mmHg. In cases of significant edema leading to clinical decompensation, osmotic therapies such as mannitol or hypertonic saline may be considered. Notably, while mannitol can help reduce edema, it also carries the risk of increasing hemorrhagic risk due to vessel damage and its osmotic effect.[55] (B3)
In certain clinical scenarios where imminent herniation or decompensation due to elevated intracranial pressures is observed, decompressive hemicraniectomy is used and has demonstrated beneficial outcomes. In addition, it is important to highlight that the data concerning the acute application of osmotic therapies in cerebral edema originates from studies encompassing stroke in general rather than specifically focusing on reperfusion injury. Additionally, steroids are contraindicated in such presentations and have been associated with unfavorable clinical outcomes.
Differential Diagnosis
In the context of recent admission, deteriorating signs associated with reperfusion typically present fewer differentials. However, differential diagnoses such as bleeding from a tumor, arteriovenous malformations, infections, subarachnoid hemorrhage, subdural hematomas, and meningitis should be considered. Although they are less likely in reperfusion, these possibilities should be meticulously ruled out based on the patient's history, physical examination findings, and evaluation.
Prognosis
The prognosis of patients with stroke reperfusion injury can vary significantly based on various factors, including the age of the patient, size of the ischemic core, reperfusion strategies used, and extent of the reperfusion injury, and other prognostic markers. While reperfusion therapies such as thrombolysis and endovascular clot retrieval have revolutionized acute stroke care, they also carry the risk of exacerbating tissue damage through reperfusion injury.
Individuals who endure severe reperfusion injury may encounter an increased risk of neurological deficits, cognitive impairment, and functional disability. Nonetheless, timely implementation of targeted management strategies, such as neuroprotective interventions and rehabilitation therapies, can facilitate substantial recovery and improve long-term outcomes for some patients. Optimizing prognosis and enhancing the quality of life for those affected by stroke reperfusion injury necessitates close monitoring, personalized care plans, and interdisciplinary collaboration.
Complications
Complications arising from IRI in AIS patients can significantly impact clinical outcomes and long-term prognosis. While reperfusion therapies are vital for restoring blood flow to ischemic brain tissue, they can paradoxically exacerbate tissue damage through mechanisms such as oxidative stress, inflammation, and excitotoxicity. These complications may manifest as penumbral damage, ischemia expansion, hemorrhagic transformation, malignant cerebral edema and herniation, seizures, and disruption of the BBB, leading to secondary brain injury and neurological deterioration. Moreover, reperfusion injury can contribute to post-stroke complications such as recurrent ischemic events, vasospasm, and systemic inflammatory response syndrome. Managing these complications requires a multifaceted approach, including close monitoring for early detection, tailored interventions to mitigate further damage, and comprehensive rehabilitation strategies to optimize recovery and minimize long-term disability. Healthcare professionals can improve patient outcomes and enhance the quality of care for affected patients by addressing the complexities of IRI and its associated complications.
Deterrence and Patient Education
Deterrence and patient education are critical in mitigating stroke reperfusion injury and optimizing outcomes. Educating patients about modifiable risk factors, such as hypertension, diabetes, and smoking, empowers them to make lifestyle modifications that can reduce their susceptibility to ischemic stroke and subsequent reperfusion injury. Moreover, raising awareness about the importance of seeking immediate medical attention at the onset of stroke symptoms facilitates timely intervention, minimizing ischemic damage and mitigating the need for extensive reperfusion therapy.
Patient education should also emphasize the risks associated with reperfusion therapies, including the possibility of reperfusion injury, hemorrhagic transformation, and other complications, to facilitate informed decision-making and enhance treatment adherence. By fostering a collaborative partnership between clinicians and patients, deterrence strategies and patient education initiatives can promote optimal stroke care outcomes and prevent stroke reperfusion injury.
Enhancing Healthcare Team Outcomes
Time sensitivity is the most limiting factor in treating stroke and IRI patients. Achieving optimal outcomes and enhancing patient care requires a dedicated interprofessional team effort involving neurologists, neurosurgeons, emergency physicians, critical care physicians, anesthetists, advanced care practitioners, nurses, and other healthcare professionals.[56]
Each healthcare professional must possess specialized skills relevant to their role in managing stroke reperfusion injury. Physicians and advanced practitioners require expertise in diagnosing and treating stroke, including assessing reperfusion injury risk and delivering timely interventions. Clinicians excel in patient assessment, monitoring vital signs, and providing comprehensive, compassionate care. Pharmacists contribute by ensuring accurate medication management, dosing, and monitoring for potential drug interactions or adverse effects.
Collaboratively developing and implementing evidence-based protocols and care pathways is essential for optimizing IRI management. This involves establishing standardized protocols for administering reperfusion therapies, monitoring patients for complications, and initiating neuroprotective strategies. Regularly reviewing and refining protocols based on emerging research findings and clinical outcomes are critical for ensuring the most effective patient care.
Effective communication among healthcare team members is essential for coordinating care. This involves clear and concise communication of patient information, treatment plans, and updates on patient status. Regular interdisciplinary team meetings facilitate collaborative decision-making, problem-solving, and addressing any concerns or challenges in stroke reperfusion injury management. This includes facilitating rapid transfer to stroke centers equipped to deliver reperfusion therapies, ensuring timely treatment administration, and coordinating post-acute care and rehabilitation services. Care coordination also involves liaising with community resources and support services to address patients' psychosocial and long-term care needs.
The effective interprofessional team management of IRI in patients, characterized by collaboration, clear communication, and coordinated care, is paramount in optimizing patient outcomes and enhancing the quality of stroke care delivery. By leveraging each team member's diverse expertise and resources, healthcare professionals can mitigate reperfusion injury risks, improve patient safety, and ultimately contribute to improving the long-term prognosis for stroke patients.
Media
(Click Image to Enlarge)

Ischemic Injury and its Molecular Consequences. Abbreviation: ROS, reactive oxygen species.
Contributed by D Sikri, MD
(Click Image to Enlarge)

Ischemia-Reperfusion Injury, Reactive Oxygen Species. The image explores the involvement of reactive oxygen species in ischemia-reperfusion injury.
Contributed by D Sikri, MD
(Click Image to Enlarge)
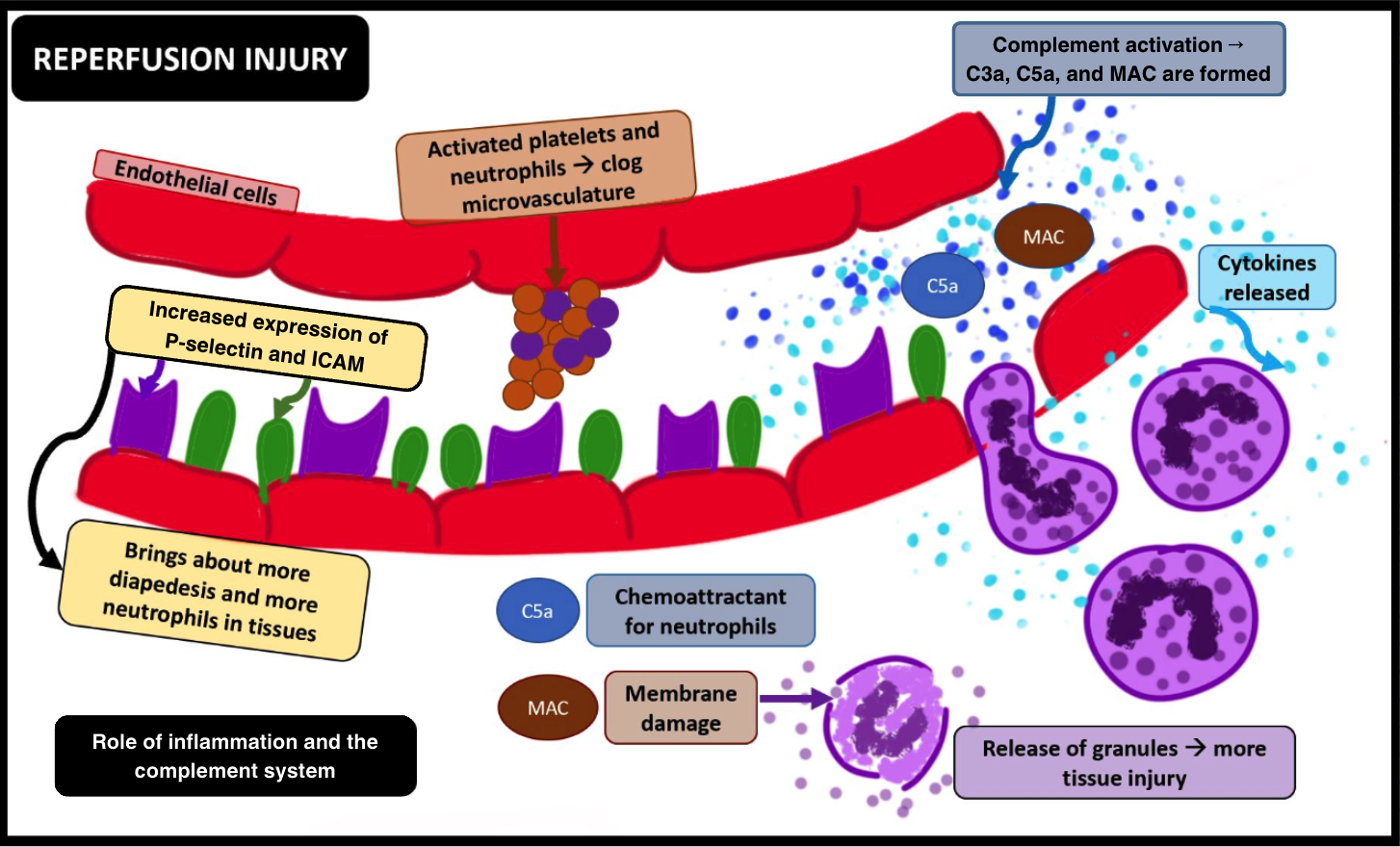
Ischemia-Reperfusion Injury, Inflammation and the Complement System. This image elucidates the involvement of inflammation and the complement system in ischemia-reperfusion injury. Abbreviations: ICAM, intercellular adhesion molecule; MAC, membrane attack complex.
Contributed by D Sikri, MD
(Click Image to Enlarge)

Blood Brain Barrier. The image illustrates the significance of astrocyte foot processes in maintaining the integrity and function of the blood-brain barrier.
Ben Brahim Mohammed, Public Domain, via Wikimedia Commons
References
Virani SS, Alonso A, Benjamin EJ, Bittencourt MS, Callaway CW, Carson AP, Chamberlain AM, Chang AR, Cheng S, Delling FN, Djousse L, Elkind MSV, Ferguson JF, Fornage M, Khan SS, Kissela BM, Knutson KL, Kwan TW, Lackland DT, Lewis TT, Lichtman JH, Longenecker CT, Loop MS, Lutsey PL, Martin SS, Matsushita K, Moran AE, Mussolino ME, Perak AM, Rosamond WD, Roth GA, Sampson UKA, Satou GM, Schroeder EB, Shah SH, Shay CM, Spartano NL, Stokes A, Tirschwell DL, VanWagner LB, Tsao CW, American Heart Association Council on Epidemiology and Prevention Statistics Committee and Stroke Statistics Subcommittee. Heart Disease and Stroke Statistics-2020 Update: A Report From the American Heart Association. Circulation. 2020 Mar 3:141(9):e139-e596. doi: 10.1161/CIR.0000000000000757. Epub 2020 Jan 29 [PubMed PMID: 31992061]
Saver JL. Time is brain--quantified. Stroke. 2006 Jan:37(1):263-6 [PubMed PMID: 16339467]
Butcher K, Parsons M, Baird T, Barber A, Donnan G, Desmond P, Tress B, Davis S. Perfusion thresholds in acute stroke thrombolysis. Stroke. 2003 Sep:34(9):2159-64 [PubMed PMID: 12893953]
Level 1 (high-level) evidenceGomez CR. Time Is Brain: The Stroke Theory of Relativity. Journal of stroke and cerebrovascular diseases : the official journal of National Stroke Association. 2018 Aug:27(8):2214-2227. doi: 10.1016/j.jstrokecerebrovasdis.2018.04.001. Epub 2018 Apr 25 [PubMed PMID: 29705088]
Heiss WD. The ischemic penumbra: correlates in imaging and implications for treatment of ischemic stroke. The Johann Jacob Wepfer award 2011. Cerebrovascular diseases (Basel, Switzerland). 2011:32(4):307-20. doi: 10.1159/000330462. Epub 2011 Sep 15 [PubMed PMID: 21921593]
Hacke W, Kaste M, Fieschi C, Toni D, Lesaffre E, von Kummer R, Boysen G, Bluhmki E, Höxter G, Mahagne MH. Intravenous thrombolysis with recombinant tissue plasminogen activator for acute hemispheric stroke. The European Cooperative Acute Stroke Study (ECASS). JAMA. 1995 Oct 4:274(13):1017-25 [PubMed PMID: 7563451]
Level 1 (high-level) evidenceNational Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group. Tissue plasminogen activator for acute ischemic stroke. The New England journal of medicine. 1995 Dec 14:333(24):1581-7 [PubMed PMID: 7477192]
Level 1 (high-level) evidenceNogueira RG, Jadhav AP, Haussen DC, Bonafe A, Budzik RF, Bhuva P, Yavagal DR, Ribo M, Cognard C, Hanel RA, Sila CA, Hassan AE, Millan M, Levy EI, Mitchell P, Chen M, English JD, Shah QA, Silver FL, Pereira VM, Mehta BP, Baxter BW, Abraham MG, Cardona P, Veznedaroglu E, Hellinger FR, Feng L, Kirmani JF, Lopes DK, Jankowitz BT, Frankel MR, Costalat V, Vora NA, Yoo AJ, Malik AM, Furlan AJ, Rubiera M, Aghaebrahim A, Olivot JM, Tekle WG, Shields R, Graves T, Lewis RJ, Smith WS, Liebeskind DS, Saver JL, Jovin TG, DAWN Trial Investigators. Thrombectomy 6 to 24 Hours after Stroke with a Mismatch between Deficit and Infarct. The New England journal of medicine. 2018 Jan 4:378(1):11-21. doi: 10.1056/NEJMoa1706442. Epub 2017 Nov 11 [PubMed PMID: 29129157]
Lieb M, Shah U, Hines GL. Cerebral hyperperfusion syndrome after carotid intervention: a review. Cardiology in review. 2012 Mar-Apr:20(2):84-9. doi: 10.1097/CRD.0b013e318237eef8. Epub [PubMed PMID: 22183061]
Farooq MU, Goshgarian C, Min J, Gorelick PB. Pathophysiology and management of reperfusion injury and hyperperfusion syndrome after carotid endarterectomy and carotid artery stenting. Experimental & translational stroke medicine. 2016:8(1):7. doi: 10.1186/s13231-016-0021-2. Epub 2016 Sep 6 [PubMed PMID: 27602202]
Eltzschig HK, Eckle T. Ischemia and reperfusion--from mechanism to translation. Nature medicine. 2011 Nov 7:17(11):1391-401. doi: 10.1038/nm.2507. Epub 2011 Nov 7 [PubMed PMID: 22064429]
Miller DJ, Simpson JR, Silver B. Safety of thrombolysis in acute ischemic stroke: a review of complications, risk factors, and newer technologies. The Neurohospitalist. 2011 Jul:1(3):138-47. doi: 10.1177/1941875211408731. Epub [PubMed PMID: 23983849]
Wahlgren N, Ahmed N, Dávalos A, Ford GA, Grond M, Hacke W, Hennerici MG, Kaste M, Kuelkens S, Larrue V, Lees KR, Roine RO, Soinne L, Toni D, Vanhooren G, SITS-MOST investigators. Thrombolysis with alteplase for acute ischaemic stroke in the Safe Implementation of Thrombolysis in Stroke-Monitoring Study (SITS-MOST): an observational study. Lancet (London, England). 2007 Jan 27:369(9558):275-82 [PubMed PMID: 17258667]
Goyal M, Menon BK, van Zwam WH, Dippel DW, Mitchell PJ, Demchuk AM, Dávalos A, Majoie CB, van der Lugt A, de Miquel MA, Donnan GA, Roos YB, Bonafe A, Jahan R, Diener HC, van den Berg LA, Levy EI, Berkhemer OA, Pereira VM, Rempel J, Millán M, Davis SM, Roy D, Thornton J, Román LS, Ribó M, Beumer D, Stouch B, Brown S, Campbell BC, van Oostenbrugge RJ, Saver JL, Hill MD, Jovin TG, HERMES collaborators. Endovascular thrombectomy after large-vessel ischaemic stroke: a meta-analysis of individual patient data from five randomised trials. Lancet (London, England). 2016 Apr 23:387(10029):1723-31. doi: 10.1016/S0140-6736(16)00163-X. Epub 2016 Feb 18 [PubMed PMID: 26898852]
Level 1 (high-level) evidenceAlbers GW, Marks MP, Kemp S, Christensen S, Tsai JP, Ortega-Gutierrez S, McTaggart RA, Torbey MT, Kim-Tenser M, Leslie-Mazwi T, Sarraj A, Kasner SE, Ansari SA, Yeatts SD, Hamilton S, Mlynash M, Heit JJ, Zaharchuk G, Kim S, Carrozzella J, Palesch YY, Demchuk AM, Bammer R, Lavori PW, Broderick JP, Lansberg MG, DEFUSE 3 Investigators. Thrombectomy for Stroke at 6 to 16 Hours with Selection by Perfusion Imaging. The New England journal of medicine. 2018 Feb 22:378(8):708-718. doi: 10.1056/NEJMoa1713973. Epub 2018 Jan 24 [PubMed PMID: 29364767]
Kalogeris T, Baines CP, Krenz M, Korthuis RJ. Cell biology of ischemia/reperfusion injury. International review of cell and molecular biology. 2012:298():229-317. doi: 10.1016/B978-0-12-394309-5.00006-7. Epub [PubMed PMID: 22878108]
Level 3 (low-level) evidenceHalestrap AP. A pore way to die: the role of mitochondria in reperfusion injury and cardioprotection. Biochemical Society transactions. 2010 Aug:38(4):841-60. doi: 10.1042/BST0380841. Epub [PubMed PMID: 20658967]
Level 3 (low-level) evidenceBolli R, Jeroudi MO, Patel BS, Aruoma OI, Halliwell B, Lai EK, McCay PB. Marked reduction of free radical generation and contractile dysfunction by antioxidant therapy begun at the time of reperfusion. Evidence that myocardial "stunning" is a manifestation of reperfusion injury. Circulation research. 1989 Sep:65(3):607-22 [PubMed PMID: 2548761]
Level 3 (low-level) evidenceJennings RB. Historical perspective on the pathology of myocardial ischemia/reperfusion injury. Circulation research. 2013 Aug 2:113(4):428-38. doi: 10.1161/CIRCRESAHA.113.300987. Epub [PubMed PMID: 23908330]
Level 3 (low-level) evidenceYang C, Hawkins KE, Doré S, Candelario-Jalil E. Neuroinflammatory mechanisms of blood-brain barrier damage in ischemic stroke. American journal of physiology. Cell physiology. 2019 Feb 1:316(2):C135-C153. doi: 10.1152/ajpcell.00136.2018. Epub 2018 Oct 31 [PubMed PMID: 30379577]
Eltzschig HK, Carmeliet P. Hypoxia and inflammation. The New England journal of medicine. 2011 Feb 17:364(7):656-65. doi: 10.1056/NEJMra0910283. Epub [PubMed PMID: 21323543]
Level 3 (low-level) evidencePanés J, Perry M, Granger DN. Leukocyte-endothelial cell adhesion: avenues for therapeutic intervention. British journal of pharmacology. 1999 Feb:126(3):537-50 [PubMed PMID: 10188959]
Level 3 (low-level) evidenceAlawieh A, Elvington A, Tomlinson S. Complement in the Homeostatic and Ischemic Brain. Frontiers in immunology. 2015:6():417. doi: 10.3389/fimmu.2015.00417. Epub 2015 Aug 12 [PubMed PMID: 26322048]
Zhang M, Austen WG Jr, Chiu I, Alicot EM, Hung R, Ma M, Verna N, Xu M, Hechtman HB, Moore FD Jr, Carroll MC. Identification of a specific self-reactive IgM antibody that initiates intestinal ischemia/reperfusion injury. Proceedings of the National Academy of Sciences of the United States of America. 2004 Mar 16:101(11):3886-91 [PubMed PMID: 14999103]
Level 3 (low-level) evidenceEngelhardt B, Coisne C. Fluids and barriers of the CNS establish immune privilege by confining immune surveillance to a two-walled castle moat surrounding the CNS castle. Fluids and barriers of the CNS. 2011 Jan 18:8(1):4. doi: 10.1186/2045-8118-8-4. Epub 2011 Jan 18 [PubMed PMID: 21349152]
Mun-Bryce S, Rosenberg GA. Matrix metalloproteinases in cerebrovascular disease. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 1998 Nov:18(11):1163-72 [PubMed PMID: 9809504]
Level 3 (low-level) evidenceFujimura M, Gasche Y, Morita-Fujimura Y, Massengale J, Kawase M, Chan PH. Early appearance of activated matrix metalloproteinase-9 and blood-brain barrier disruption in mice after focal cerebral ischemia and reperfusion. Brain research. 1999 Sep 18:842(1):92-100 [PubMed PMID: 10526099]
Asahi M, Asahi K, Jung JC, del Zoppo GJ, Fini ME, Lo EH. Role for matrix metalloproteinase 9 after focal cerebral ischemia: effects of gene knockout and enzyme inhibition with BB-94. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2000 Dec:20(12):1681-9 [PubMed PMID: 11129784]
Jain S, Bicknell GR, Nicholson ML. Molecular changes in extracellular matrix turnover after renal ischaemia-reperfusion injury. The British journal of surgery. 2000 Sep:87(9):1188-92 [PubMed PMID: 10971426]
Roach DM, Fitridge RA, Laws PE, Millard SH, Varelias A, Cowled PA. Up-regulation of MMP-2 and MMP-9 leads to degradation of type IV collagen during skeletal muscle reperfusion injury; protection by the MMP inhibitor, doxycycline. European journal of vascular and endovascular surgery : the official journal of the European Society for Vascular Surgery. 2002 Mar:23(3):260-9 [PubMed PMID: 11914015]
Niccoli G, Marino M, Spaziani C, Crea F. Prevention and treatment of no-reflow. Acute cardiac care. 2010 Sep:12(3):81-91. doi: 10.3109/17482941.2010.498919. Epub [PubMed PMID: 20698732]
Ames A 3rd, Wright RL, Kowada M, Thurston JM, Majno G. Cerebral ischemia. II. The no-reflow phenomenon. The American journal of pathology. 1968 Feb:52(2):437-53 [PubMed PMID: 5635861]
Level 3 (low-level) evidencedel Zoppo GJ, Schmid-Schönbein GW, Mori E, Copeland BR, Chang CM. Polymorphonuclear leukocytes occlude capillaries following middle cerebral artery occlusion and reperfusion in baboons. Stroke. 1991 Oct:22(10):1276-83 [PubMed PMID: 1926239]
Level 3 (low-level) evidenceKloner RA, Ganote CE, Jennings RB. The "no-reflow" phenomenon after temporary coronary occlusion in the dog. The Journal of clinical investigation. 1974 Dec:54(6):1496-508 [PubMed PMID: 4140198]
Level 3 (low-level) evidenceWarach S, Latour LL. Evidence of reperfusion injury, exacerbated by thrombolytic therapy, in human focal brain ischemia using a novel imaging marker of early blood-brain barrier disruption. Stroke. 2004 Nov:35(11 Suppl 1):2659-61 [PubMed PMID: 15472105]
Campbell BCV, Ma H, Parsons MW, Churilov L, Yassi N, Kleinig TJ, Hsu CY, Dewey HM, Butcher KS, Yan B, Desmond PM, Wijeratne T, Curtze S, Barber PA, De Silva DA, Thijs V, Levi CR, Bladin CF, Sharma G, Bivard A, Donnan GA, Davis SM. Association of Reperfusion After Thrombolysis With Clinical Outcome Across the 4.5- to 9-Hours and Wake-up Stroke Time Window: A Meta-Analysis of the EXTEND and EPITHET Randomized Clinical Trials. JAMA neurology. 2021 Feb 1:78(2):236-240. doi: 10.1001/jamaneurol.2020.4123. Epub [PubMed PMID: 33137171]
Baird AE, Benfield A, Schlaug G, Siewert B, Lövblad KO, Edelman RR, Warach S. Enlargement of human cerebral ischemic lesion volumes measured by diffusion-weighted magnetic resonance imaging. Annals of neurology. 1997 May:41(5):581-9 [PubMed PMID: 9153519]
Barber PA, Darby DG, Desmond PM, Yang Q, Gerraty RP, Jolley D, Donnan GA, Tress BM, Davis SM. Prediction of stroke outcome with echoplanar perfusion- and diffusion-weighted MRI. Neurology. 1998 Aug:51(2):418-26 [PubMed PMID: 9710013]
Level 1 (high-level) evidenceZhang Y, Stevenson GD, Barber C, Furenlid LR, Barrett HH, Woolfenden JM, Zhao M, Liu Z. Imaging of rat cerebral ischemia-reperfusion injury using(99m)Tc-labeled duramycin. Nuclear medicine and biology. 2013 Jan:40(1):80-8. doi: 10.1016/j.nucmedbio.2012.09.004. Epub 2012 Nov 2 [PubMed PMID: 23123139]
Level 3 (low-level) evidenceNour M, Scalzo F, Liebeskind DS. Ischemia-reperfusion injury in stroke. Interventional neurology. 2013 Sep:1(3-4):185-99. doi: 10.1159/000353125. Epub [PubMed PMID: 25187778]
Choi HY, Lee KM, Kim HG, Kim EJ, Choi WS, Kim BJ, Heo SH, Chang DI. Role of Hyperintense Acute Reperfusion Marker for Classifying the Stroke Etiology. Frontiers in neurology. 2017:8():630. doi: 10.3389/fneur.2017.00630. Epub 2017 Nov 29 [PubMed PMID: 29276498]
Sun WH, He F, Zhang NN, Zhao ZA, Chen HS. Time dependent neuroprotection of dexamethasone in experimental focal cerebral ischemia: The involvement of NF-κB pathways. Brain research. 2018 Dec 15:1701():237-245. doi: 10.1016/j.brainres.2018.09.029. Epub 2018 Sep 21 [PubMed PMID: 30248332]
Ohsawa I, Ishikawa M, Takahashi K, Watanabe M, Nishimaki K, Yamagata K, Katsura K, Katayama Y, Asoh S, Ohta S. Hydrogen acts as a therapeutic antioxidant by selectively reducing cytotoxic oxygen radicals. Nature medicine. 2007 Jun:13(6):688-94 [PubMed PMID: 17486089]
Level 3 (low-level) evidenceWood KC, Gladwin MT. The hydrogen highway to reperfusion therapy. Nature medicine. 2007 Jun:13(6):673-4 [PubMed PMID: 17554332]
Level 3 (low-level) evidenceOharazawa H, Igarashi T, Yokota T, Fujii H, Suzuki H, Machide M, Takahashi H, Ohta S, Ohsawa I. Protection of the retina by rapid diffusion of hydrogen: administration of hydrogen-loaded eye drops in retinal ischemia-reperfusion injury. Investigative ophthalmology & visual science. 2010 Jan:51(1):487-92. doi: 10.1167/iovs.09-4089. Epub 2009 Oct 15 [PubMed PMID: 19834032]
Level 3 (low-level) evidenceMarzi I, Bühren V, Schüttler A, Trentz O. Value of superoxide dismutase for prevention of multiple organ failure after multiple trauma. The Journal of trauma. 1993 Jul:35(1):110-9; discussion 119-20 [PubMed PMID: 8331700]
Level 1 (high-level) evidenceWang Q, Tompkins KD, Simonyi A, Korthuis RJ, Sun AY, Sun GY. Apocynin protects against global cerebral ischemia-reperfusion-induced oxidative stress and injury in the gerbil hippocampus. Brain research. 2006 May 23:1090(1):182-9 [PubMed PMID: 16650838]
Level 3 (low-level) evidenceHu Q, Manaenko A, Bian H, Guo Z, Huang JL, Guo ZN, Yang P, Tang J, Zhang JH. Hyperbaric Oxygen Reduces Infarction Volume and Hemorrhagic Transformation Through ATP/NAD(+)/Sirt1 Pathway in Hyperglycemic Middle Cerebral Artery Occlusion Rats. Stroke. 2017 Jun:48(6):1655-1664. doi: 10.1161/STROKEAHA.116.015753. Epub 2017 May 11 [PubMed PMID: 28495827]
Lyden P, Hemmen T, Grotta J, Rapp K, Ernstrom K, Rzesiewicz T, Parker S, Concha M, Hussain S, Agarwal S, Meyer B, Jurf J, Altafullah I, Raman R, Collaborators. Results of the ICTuS 2 Trial (Intravascular Cooling in the Treatment of Stroke 2). Stroke. 2016 Dec:47(12):2888-2895 [PubMed PMID: 27834742]
Li Y, Zhong W, Jiang Z, Tang X. New progress in the approaches for blood-brain barrier protection in acute ischemic stroke. Brain research bulletin. 2019 Jan:144():46-57. doi: 10.1016/j.brainresbull.2018.11.006. Epub 2018 Nov 15 [PubMed PMID: 30448453]
Michalski D, Pelz J, Weise C, Kacza J, Boltze J, Grosche J, Kamprad M, Schneider D, Hobohm C, Härtig W. Early outcome and blood-brain barrier integrity after co-administered thrombolysis and hyperbaric oxygenation in experimental stroke. Experimental & translational stroke medicine. 2011 Jun 16:3(1):5. doi: 10.1186/2040-7378-3-5. Epub 2011 Jun 16 [PubMed PMID: 21679435]
Kim SY, Cheon SY, Kim EJ, Lee JH, Kam EH, Kim JM, Park M, Koo BN. Isoflurane Postconditioning Inhibits tPA-Induced Matrix Metalloproteinases Activation After Hypoxic Injury via Low-Density Lipoprotein Receptor-Related Protein and Extracellular Signal-Regulated Kinase Pathway. Neurochemical research. 2017 May:42(5):1533-1542. doi: 10.1007/s11064-017-2211-2. Epub 2017 Mar 16 [PubMed PMID: 28303501]
Yang Y, Yang LY, Orban L, Cuylear D, Thompson J, Simon B, Yang Y. Non-invasive vagus nerve stimulation reduces blood-brain barrier disruption in a rat model of ischemic stroke. Brain stimulation. 2018 Jul-Aug:11(4):689-698. doi: 10.1016/j.brs.2018.01.034. Epub 2018 Feb 15 [PubMed PMID: 29496430]
Diringer MN, Edwards DF. Admission to a neurologic/neurosurgical intensive care unit is associated with reduced mortality rate after intracerebral hemorrhage. Critical care medicine. 2001 Mar:29(3):635-40 [PubMed PMID: 11373434]
Rapoport SI. Osmotic opening of the blood-brain barrier: principles, mechanism, and therapeutic applications. Cellular and molecular neurobiology. 2000 Apr:20(2):217-30 [PubMed PMID: 10696511]
Level 3 (low-level) evidenceHacke W, Donnan G, Fieschi C, Kaste M, von Kummer R, Broderick JP, Brott T, Frankel M, Grotta JC, Haley EC Jr, Kwiatkowski T, Levine SR, Lewandowski C, Lu M, Lyden P, Marler JR, Patel S, Tilley BC, Albers G, Bluhmki E, Wilhelm M, Hamilton S, ATLANTIS Trials Investigators, ECASS Trials Investigators, NINDS rt-PA Study Group Investigators. Association of outcome with early stroke treatment: pooled analysis of ATLANTIS, ECASS, and NINDS rt-PA stroke trials. Lancet (London, England). 2004 Mar 6:363(9411):768-74 [PubMed PMID: 15016487]
Level 1 (high-level) evidence