Introduction
Pulmonary hypertension encompasses a diverse group of conditions characterized by high pulmonary pressures. The World Health Organization classifies pulmonary hypertension into 5 clinical groups based on pathophysiology, hemodynamic characteristics, clinical features, and management (see Table 1. Clinical Classification of Pulmonary Hypertension).[1]
TABLE 1: Clinical Classification of Pulmonary Hypertension
|
Group 1: Pulmonary Arterial Hypertension (PAH) |
Group 2: Pulmonary Hypertension due to left heart disease (PH-LHD) |
Group 3: Pulmonary Hypertension due to Lung Diseases, Hypoxia, or Both |
Group 4: Pulmonary Hypertension Due to Pulmonary Artery Obstructions |
Group 5: Pulmonary Hypertension with Unclear or Multifactorial Mechanisms |
|
1.1 Idiopathic PAH (IPAH) 1.1.1 Non-responders to vasoreactivity testing 1.1.2 Acute responders to vasoreactivity testing |
2.1 Heart failure 2.1.1 With preserved ejection fraction 2.1.2 With reduced ejection fraction |
3.1 Obstructive lung disease |
4.1 Chronic thromboembolic pulmonary hypertension (CTEPH) |
5.1 Hematological disorders |
|
1.2 Heritable PAH (HPAH) |
2.2 Valvular heart disease |
3.2 Restrictive lung disease |
4.2 Other pulmonary artery obstructions |
5.2 Systemic disorders |
|
1.3 Drug and Toxin induced PAH |
2.3 Congenital and acquired cardiovascular conditions leading to postcapillary pulmonary hypertension |
3.3 Lung diseases with mixed restrictive and obstructive patterns |
|
5.3 Metabolic disorders |
|
1.4 PAH associated with 1.4.1 Connective tissue disease (CTD) 1.4.2 HIV infection 1.4.3 Portal hypertension 1.4.4 Congenital heart disease (CHD) 1.4.5 Schistosomiasis |
|
3.4 Hypoventilation syndromes |
|
5.4 Chronic renal failure with and without hemodialysis |
|
1.5 PAH with overt features of venous or capillary involvement, ie, pulmonary venoocclusive disease and pulmonary capillary hemangiomatosis |
|
3.5 Hypoxia without lung disease |
|
5.5 Pulmonary tumor thrombotic microangiopathy |
|
1.6 Persistent pulmonary hypertension of the newborn |
|
3.6 Developmental lung disorders |
|
5.6 Fibrosing mediastinitis |
Besides the above clinical distinctions, pulmonary hypertension has a hemodynamic classification scheme that can aid diagnosis. A mean pulmonary artery pressure (mPAP) greater than 20 mm Hg is above the upper normal limit.[2][3][4][5] However, a mere mPAP elevation is insufficient to define pulmonary hypertension, as this elevation could be due to a cardiac output or pulmonary artery wedge pressure (PAWP) increase. Thus, the 6th World Symposium on Pulmonary Hypertension and the European Society of Cardiology/European Respiratory Society (ESC/ERS) guidelines also classify pulmonary hypertension based on pulmonary vascular resistance (PVR) and PAWP. (see Table 2. Hemodynamic Classification of Pulmonary Hypertension).[6]
TABLE 2: Hemodynamic Classification of Pulmonary Hypertension
|
Definition |
Hemodynamic Characteristics |
Clinical Groups |
|
Precapillary pulmonary hypertension |
mPAP >20 mm Hg PAWP ≤ 15 mm Hg PVR > 2 Wood units |
1, 3, 4, and 5 |
|
Isolated postcapillary pulmonary hypertension (IpcPH) |
mPAP > 20 mm Hg PAWP > 15 mm Hg PVR ≤ 2 Wood units |
2 and 5 |
|
Combined precapillary and postcapillary pulmonary hypertension (CpcPH) |
mPAP > 20 mm Hg PAWP > 15 mm Hg PVR > 2 Wood units |
2 and 5 |
Pulmonary vessel measurements are obtained during right heart catheterization (RHC) at rest in the supine position. The latest ESC/ERS guidelines also define exercise pulmonary hypertension as a change in mPAP/cardiac output slope between rest and exercise greater than 3 mm Hg/L/min. However, this definition does not differentiate between precapillary and postcapillary pulmonary hypertension. A PAWP/cardiac output slope change greater than 2 mm Hg/L/min between rest, and exercise may offer such differentiation, but measuring PAWP during exercise is challenging.[7][8]
As the above table shows, all pulmonary hypertension groups can have precapillary and postcapillary components. Thus, the condition should be classified based on the presumed predominant underlying cause of the increased pulmonary artery pressures. Patients with PAH typically present with precapillary pulmonary hypertension, excluding conditions seen in groups 3 and 4 that can also cause precapillary hypertension.
Pulmonary Circulation Anatomy and Physiology
The pulmonary vasculature comprises the network of blood vessels transporting blood between the heart and lungs. Deoxygenated blood from the heart’s right side is pumped into the pulmonary arteries, which branch into smaller arterioles and eventually into capillaries within the lungs. Gas exchange occurs in the pulmonary arteries, with carbon dioxide diffusing out of the blood and oxygen entering the blood. Oxygenated blood then travels through the pulmonary venules and veins, returning to the left side of the heart via the pulmonary veins. From there, oxygen-rich blood is pumped into the systemic circulation to supply oxygen to tissues throughout the body.
Physiologically, the pulmonary vasculature is characterized by low resistance, allowing efficient blood flow through the lungs. The pulmonary arteries and arterioles are highly compliant and capable of expanding and contracting to accommodate blood flow and pressure changes. Smooth muscle cells within the vessel walls regulate vascular tone, helping to maintain appropriate blood flow distribution and pulmonary artery pressure. Additionally, the pulmonary circulation operates parallel to the systemic circulation, ensuring that blood is oxygenated adequately before being distributed to the body's tissues. The pulmonary vasculature supports effective gas exchange and oxygenation within the lungs, which is essential for maintaining overall health and function.
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
Genetic mutations are associated with various pulmonary hypertension types, including IPAH, HPAH, and hereditary hemorrhagic telangiectasia (HHT) associated with PAH. Individuals with HPAH have an inheritable genetic mutation. People with IPAH have a genetic predisposition, but the mutations are sporadic. These groups are clinically indistinguishable.[9] The various mutations associated with IPAH/HPAH are BMPR2 (most common), SMAD1, SMAD9, KCNK3, CAV1, and SOX17, while those associated with HHT are ALK1, ENG, and SMAD4.
The drugs and toxins known to cause PAH include aminorex, fenfluramine, dexfenfluramine, benfluorex, methamphetamines, dasatinib, and toxic rapeseed oil. Some substances that may give rise to PAH include cocaine, phenylpropanolamine, L-tryptophan, St. John's wort, amphetamines, interferon -α and -β, alkylating agents, bosutinib, direct-acting hepatitis C antivirals, leflunomide, and indirubin. Various CTDs are also known to cause PAH, including systemic sclerosis, systemic lupus erythematosus, rheumatoid arthritis, Raynaud disease, and mixed connective tissue disease (MCTD). Among these, systemic sclerosis is the most notorious for causing PAH.
Pulmonary hypertension associated with CHD (PH-CHD) is classified into CHD that causes Eisenmenger syndrome, PAH associated with prevalent systemic-to-pulmonary shunts, PAH with small or coincidental defects, and PAH after defect correction.[10] While PH-CHD is predominantly classified under group 1 pulmonary hypertension, some defects are more appropriately classified under group 2. These conditions include mitral valve disease, left ventricular inflow or outflow obstruction, and left ventricular systolic or diastolic dysfunction that may also result in postcapillary pulmonary hypertension.
Group 3 hypoventilation syndromes comprise conditions causing sleep-disordered breathing. Hypoxia without hypoventilation is seen at high altitudes. Group 4 etiologies include chronic thromboembolic disease, sarcoma, other malignant tumors like renal carcinoma, uterine carcinoma, testicular germ cell tumors, and nonmalignant tumors like leiomyoma, arteritis without CTD, congenital pulmonary artery stenoses, and hydatidosis. These conditions can obstruct the pulmonary artery and cause pulmonary hypertension.
Chronic hemolytic anemia and myeloproliferative disorders are hematological disorders that cause group 5 pulmonary hypertension. Systemic diseases that can cause pulmonary hypertension include pulmonary Langerhans cell histiocytosis, neurofibromatosis type 1, and sarcoidosis. Glycogen storage diseases and Gaucher disease are metabolic disorders that can cause pulmonary hypertension.
Epidemiology
Pulmonary hypertension can affect people of any age. Present estimates suggest a prevalence of approximately 1% in the global population. Left heart disease (LHD) is the leading cause, followed by lung disease, particularly chronic obstructive pulmonary disease (COPD).[11] In developing countries, CHD, infectious diseases like schistosomiasis and HIV, and high altitude are significant pulmonary hypertension causes. PAH incidence is approximately 6 cases per million adults, and the prevalence is about 49 to 55 cases per million adults. PAH was initially thought to affect predominantly young women. However, recent data show that the condition is also prevalent in patients aged 65 years and older with cardiovascular comorbidities, leveling the sex distribution.[12][13][14]
At least 50% of patients with heart failure with preserved ejection fraction either have IpcPH or CpcPH.[15][16] The prevalence of pulmonary hypertension increases with disease severity in these patients, with 60 to 70% of patients with severe and symptomatic mitral valve disease and 50% with symptomatic aortic stenosis affected by pulmonary hypertension.[17][18] Mild pulmonary hypertension is common in patients with advanced COPD and interstitial lung disease (ILD). Only 1% to 5% of patients with advanced COPD were found to have severe pulmonary hypertension. The prevalence increases with increasing severity in patients with idiopathic pulmonary fibrosis and is as high as 60% in patients with end-stage disease.[19][20]
Registry data indicate that CTEPH’s incidence and prevalence are 2 to 6 and 26 to 39 cases per million adults, respectively.[21][22] Pulmonary hypertension in individuals with sarcoidosis is frequent and often associated with increased mortality and morbidity.[23][24]
History and Physical
History
The hallmark presenting symptom of pulmonary hypertension is shortness of breath on exertion. Nonspecific symptoms may also be reported, including hemoptysis, fatigue, early exhaustion, palpitations, dizziness, and syncope. As the disease progresses, symptoms of right-sided heart failure manifest, such as weight gain, edema, abdominal distention, and ascites.[25]
Rarely, pulmonary artery enlargement may present with chest pain on exertion from left main coronary artery compression. Hoarseness may manifest from left recurrent laryngeal nerve compression—a condition known as Ortner syndrome. Bronchial compression may present as atelectasis. Pulmonary artery enlargement may also cause wheezing, cough, and frequent lower respiratory tract infections.
Patients may also have symptoms related to their underlying diseases, such as joint pains, skin rashes, cough, daytime somnolence, and a history of blood clots. Family, sexual, and travel history are highly relevant when evaluating a patient with suspected pulmonary hypertension.
Decompensated patients may present with pulselessness, apnea, and unconsciousness. These are signs of cardiorespiratory arrest, warranting immediate resuscitation.
Physical Examination
An increased P2 (pulmonic) component of the second heart sound is usually the initial physical finding. Jugular venous distention with a prominent "a" wave is seen with an eventual prominent "v" wave, signifying tricuspid regurgitation as the disease progresses and right ventricular dysfunction ensues. Tricuspid and pulmonic regurgitation murmurs and right-sided S3 or S4 may be heard. Elevated pulmonary pressures and, eventually, right ventricular failure cause these abnormalities.
Ascites, abdominal distention, hepatomegaly with or without splenomegaly, and dependent edema may also be present. Pallor, delayed capillary refill, and peripheral cyanosis may be seen as right ventricular failure progresses, signifying a low cardiac output state.
Various other physical exam findings may be present in patients with CTD or chronic lung diseases, such as digital clubbing, telangiectasias, Raynaud phenomenon, digital ulceration, gastroesophageal reflux signs like abdominal tenderness, crackles or wheezing on lung auscultation, and joint swelling and erythema.
Evaluation
Basic Concepts in Pulmonary Hypertension Evaluation
Despite technological advances, pulmonary hypertension detection still takes more than 2 years from symptom onset, thus requiring a high index of suspicion.[26][27] Patients with unexplained shortness of breath or symptoms concerning for pulmonary hypertension should be thoroughly assessed. The evaluation process should include a comprehensive medical and family history, physical examination, monitoring of blood pressure, heart rate, oxygen saturation, and blood tests, including brain natriuretic peptide (BNP) or N-terminal pro-BNP (NT-proBNP) and resting electrocardiogram (ECG). These initial steps help raise suspicion of a cardiopulmonary illness. The next step in the workup involves cardiac assessment with echocardiography. Signs indicative of pulmonary dysfunction warrant pulmonary function tests (PFTs), chest imaging modalities like x-ray and computed tomography (CT), and, in some instances, cardiopulmonary exercise testing (CPET).
The echocardiogram can help assess whether the probability of pulmonary hypertension is low, intermediate, or high. Patients with intermediate-to-high pulmonary hypertension probability on echocardiogram and low-probability patients with PAH or CTEPH risk factors but no other identified causes for their symptoms should be referred to a specialized facility for further comprehensive testing and confirmation of pulmonary hypertension.
PAH risk factors include CTD, HIV, family history of PAH, and portal hypertension. CTEPH risk factors include a history of pulmonary embolism, permanent intravascular devices, malignancy, inflammatory bowel diseases, essential thrombocythemia, and high-dose thyroid replacement.
Warning signs warrant a fast-track referral to a pulmonary hypertension center, including rapidly evolving right ventricular failure symptoms, syncope, low cardiac output signs, hemodynamic decompensation, and poorly tolerated arrhythmias.
Laboratory Testing
Routine hematology studies, renal function tests, liver function tests (LFTs), iron profiles, and thyroid studies should be performed in all patients with clinical signs of pulmonary hypertension. Thyroid dysfunction should always be suspected in individuals with abrupt deterioration, as it is common in PAH and may occur during the disease course.
Routine HIV screening, hepatitis virus serologies, and CTD tests should be performed. Patients with scleroderma have a high PAH prevalence and should be routinely screened with antinuclear antibody immunofluorescence. Clinical findings should determine if other CTD tests, such as anticentromere, antitopoisomerase, anti-RNA polymerase III, double-stranded DNA, anti-Ro, anti-La, and U1-RNP, should be obtained. Patients with CTEPH and those with CTD associated with thrombophilic states should also be screened for coagulopathies and thrombophilias, including lupus anticoagulant, anticardiolipin antibodies, and anti-β2-glycoprotein antibodies. BNP and NT-proBNP levels are independent outcome predictors and may also be considered during evaluation.
Patients may have abnormal LFT values due to congestive hepatopathy from either right heart failure, liver disease, or endothelin receptor antagonist (ERA) therapy. Viral hepatitis should still be ruled out in these individuals by obtaining hepatitis serologies.
ECG
A normal ECG does not exclude the diagnosis of pulmonary hypertension. However, an abnormal ECG may indicate severe disease, especially if QRS and QTc are prolonged.[28][29][30][31] ECG abnormalities may include signs of right-sided heart strain and chamber enlargement, including P pulmonale, right axis deviation, right ventricular strain, right ventricular hypertrophy, right bundle branch block, and QTc prolongation. Supraventricular tachycardias, including atrial flutter and fibrillation, can occur in advanced disease, but ventricular arrhythmias are rare.[32]
Chest Radiography
Chest radiography may reveal underlying pulmonary hypertension, such as right atrial enlargement, pulmonary artery enlargement, peripheral vessel pruning, and a water bottle-shaped cardiac silhouette. Signs of left heart disease like Kerley B lines, pleural effusions, and left heart enlargement may also be present. Patients with lung disease may have diaphragmatic flattening, hyperlucency, volume loss, or reticular opacifications on x-ray, depending on their condition. A normal chest X-ray does not rule out pulmonary hypertension.[33][34][35]
Pulmonary Function Tests and Arterial Blood Gases
A complete set of PFTs can give diagnostic and prognostic information. Most patients with PAH have decreased carbon monoxide diffusion capacity (DLCO). A DLCO of less than 45% is associated with poor outcomes.[36][37] Spirometry can help detect obstructive airway disorders. Decreased lung volumes and DLCO may indicate interstitial lung disease.
Patients with PAH usually have a low normal or slightly low partial pressure of oxygen (PaO2). A severely reduced PaO2 should raise suspicion for shunting, as in a patent foramen ovale or hepatic disease. Patients with PAH may also have alveolar hyperventilation, often producing a low or low-to-normal carbon dioxide partial pressure (PaCO2).[38] Unfavorable outcomes have been reported in patients with low PaCO2 at diagnosis and follow-up.[39] An elevated PaCO2 is unusual and should raise suspicion of sleep-disordered breathing or hypoventilation.[40]
Chest CT and Digital Subtraction Angiography
Chest CT is essential for identifying ILDs, helping differentiate between various pulmonary hypertension groups (see Image. Pulmonary Hypertension from Connective Tissue Disease on CT). This modality also provides additional information that may increase suspicion of this condition, such as enlarged pulmonary artery diameter, main pulmonary artery-ascending aorta diameter ratio greater than 0.9, and enlarged right heart chambers. A pulmonary artery diameter of at least 30 mm, right ventricular outflow tract (RVOT) thickness of at least 6 mm, septal deviation greater than 140°, or right-to-left ventricle ratio of at least 1 is highly predictive of pulmonary hypertension.[41]
Contrast CT pulmonary angiography may demonstrate CTEPH signs such as webs, bands, filling defects, enlarged bronchial arteries, and mosaic perfusion. CT pulmonary angiography often has a high diagnostic accuracy for CTEPH when high-quality multi-detector CT images are interpreted by experienced readers.[42][43] Digital subtraction angiography (DSA) with conventional 2- or 3-planar imaging is used in most centers to confirm CTEPH diagnosis and assess operability for endarterectomy or balloon pulmonary angioplasty.
Echocardiogram
Transthoracic echocardiography remains the single most crucial noninvasive assessment tool for pulmonary hypertension. However, echocardiographic assessment should only be used to estimate pulmonary hypertension probability. RHC is used for diagnostic confirmation and therapeutic guidance.
Data obtained from normal adults led to the ESC/ERS guidelines for classifying pulmonary hypertension probability into low, intermediate, or high. Tables 3 and 4 provide information about echocardiographic probability and signs suggestive of pulmonary hypertension, respectively.[44][45][46][47]
Table 3: Echocardiographic Pulmonary Hypertension Probability
|
Peak Tricuspid Regurgitant Velocity (m/s) |
Presence of Other Pulmonary Hypertension Signs on Echocardiography |
Echocardiographic Probability of Pulmonary Hypertension |
|
< 2.8 or not measurable |
No |
Low |
|
< 2.8 or not measurable |
Yes |
Intermediate |
|
2.9-3.4 |
No |
Intermediate |
|
2.9-3.4 |
Yes |
High |
|
>3.4 |
Not required |
High |
Table 4: Echocardiographic Signs Suggestive of Pulmonary Hypertension
|
Ventricles |
Right-to-left ventricle basal diameter area ratio >1 |
Interventricular septum flattening (left ventricular eccentricity index > 1.1 in systole, diastole, or both) |
Tricuspid annular plane systolic excursion-systolic pulmonary artery pressure ratio < 0.55 mm / mm Hg |
|
Pulmonary Artery |
RVOT acceleration time <105 ms or mid-systolic notching |
Early diastolic pulmonary regurgitation velocity > 2.2 m/s |
Pulmonary artery > aortic root diameter Pulmonary artery diameter > 25mm |
|
Inferior Vena Cava and Right Atrium |
Inferior vena cava diameter > 21 mm with decreased inspiratory collapse (<50% with a sniff or <20% with quiet respiration) |
Right arterial area (end-systole) > 18 cm2 |
|
RHC should be performed in patients with a high pulmonary hypertension probability on echocardiography, with or without PAH or CTEPH risk factors. In contrast, follow-up imaging should be considered in patients with PAH or CTEPH risk factors and a low pulmonary hypertension probability on echocardiography. However, RHC should be considered in similar patients with a medium pulmonary hypertension probability. An alternative diagnosis should be considered in patients without PAH or CTEPH risk factors and a low pulmonary hypertension probability on echocardiography. Alternative diagnoses or further investigations can be considered in people with medium probability without PAH or CTEPH risk factors.
Besides assessing probability, echocardiography can also detect conditions that may cause pulmonary hypertension, including CHD, valvular heart disease, and left-sided heart dysfunction.
Cardiopulmonary Exercise Testing
Patients with exercise-induced symptoms induced should undergo CPET. In patients with PAH, a low end-tidal partial pressure of carbon dioxide, high ventilatory equivalent for carbon dioxide, low oxygen pulse, and low peak oxygen uptake are typically seen.[48] PAH may be excluded in people with systemic sclerosis with a normal peak oxygen uptake.[49]
Ventilation-Perfusion Scanning
A ventilation-perfusion (V/Q) scan is performed to rule out CTEPH in patients with pulmonary hypertension (see Image. CTEPH Ventilation-Perfusion Scan). This diagnostic test remains the preferred modality since a normal scan can exclude CTEPH with a sensitivity of 90% to 100% and a specificity of 94% to 100%.[50][51] Recent CT, magnetic resonance imaging (MRI), and single photon emission CT advances have narrowed the gap between V/Q scans and these other modalities. The V/Q-single photon emission CT is superior to planar V/Q scan and should be the preferred modality if available.[33] However, some advanced imaging modalities and the expertise to interpret them may not be widely available. Thus, further studies are needed to establish their clinical utility.[52][53][54]
Cardiac Magnetic Resonance Imaging
Cardiac MRI (cMRI) is an incredibly powerful tool that accurately assesses atrial and ventricular function and morphology. This modality can also measure blood flow through the vena cava, pulmonary artery, and aorta, allowing for stroke volume quantification. The test is sensitive for detecting early pulmonary hypertension but does not reliably estimate pulmonary artery pressures. Additionally, cost and availability are considerable barriers to using cMRI.[55]
Abdominal Ultrasound
The main reason for obtaining an abdominal ultrasound is to detect liver abnormalities, portal hypertension, and kidney injury that may arise from chronic pulmonary hypertension. Ultrasonography can help assess the extent of the collateral damage to these organs.[56]
Genetic Testing and Counseling
A trained PAH provider or geneticist should counsel patients with familial PAH, IPAH, HPAH, anorexigenic-associated PAH, pulmonary venoocclusive disease, and pulmonary capillary hemangiomatosis that family members could carry a mutation that increases their PAH risk.[57] Genetic testing can inform the patient and family members to screen for early symptoms and signs to ensure a timely diagnosis. Genetic sequencing technological advancements have led to the development of gene panels that can simultaneously test for several gene mutations.[58] A trained geneticist must provide counseling and help interpret the tests.
Right Heart Catheterization
RHC confirms the diagnosis of pulmonary hypertension. This procedure is ideally performed at a center with technical expertise to obtain high-quality data and avoid morbidity and mortality.[59] RHC can yield enormous information, including right- and left-sided filling pressures, pulmonary artery pressure, PAWP, PVR, and cardiac output.
The following are nuances regarding RHC measurements:
- External pressure transducers must be calibrated to zero at the level of the left atrium.
- PAWP should be measured at the end of normal expiration.[60]
- Cardiac output should be measured using triplicate thermodilution. However, thermodilution may be inaccurate in patients with intracardiac shunts, warranting direct Fick estimation.[61] The information should be interpreted based on the clinical picture and imaging findings, especially echocardiography.
Vasoreactivity testing is an RHC component that should only be done in patients with IPAH, HPAP, and drug-induced IPAH. Inhaled nitric oxide at 10 to 20 parts per million is usually preferred, but intravenous epoprostenol, intravenous adenosine, or inhaled iloprost may also be used. The test is deemed positive if there is an mPAP decrease of at least 10 mm Hg, resulting in a final mPAP of 40 mm Hg or less and either unchanged or increased cardiac output. Patients who test positive are suitable for high-dose calcium channel blocker (CCB) treatments.
Other maneuvers, such as a fluid challenge, may be used to discriminate IPAH from left ventricular diastolic dysfunction. However, this technique needs to be further studied before it becomes the standard of care.[62][63] A left heart catheterization should also be performed in individuals with clinical risk factors for coronary artery disease or echocardiographic signs of left heart systolic or diastolic dysfunction.
RHC contraindications include a recently implanted pacemaker (<1 month), known right atrial or ventricular thrombus, mechanical right heart valve, tricuspid valve clip, and acute infection. The most feared complication of the procedure is pulmonary arterial rupture.
Treatment / Management
The treatment strategy for PAH involves a risk-based approach, considering clinical, functional, exercise, hemodynamic, and right ventricular function parameters. Multiple studies show this treatment approach must be based on baseline methodical risk assessment and follow-up to predict survival or event-free survival.[64][65][66][67]
Patients should be assessed at every visit using a multidimensional approach. Key risk stratification tools include the US Registry to Evaluate Early and Long-term PAH Disease Management (REVEAL) equation and risk score, the 2022 ESC/ERS guidelines risk table categorizing patients as having a low (<5%), intermediate (5 to 20%), or high (>20%) risk based on estimated 1-year mortality, and the French Pulmonary Hypertension Network (FPHN) Registry risk equation. The REVEAL 2.0 risk score calculator refines the original risk assessment tool.[68][69][70][71][72][73][74] However, studies show that the World Health Organization-functional class (WHO-FC), 6-minute walk distance, and BNP (or NT-proBNP) are the strongest survival predictors across all risk scores.[75](A1)
The ESC/ERS risk stratification correlates with the REVEAL 2.0 score: “low risk” has a REVEAL score less than or equal to 6, “intermediate risk” has a REVEAL score of 7 or 8, and “high risk” corresponds to a REVEAL score of at least 9.[66] However, the REVEAL score has many variables and predicts outcomes over a shorter period (1 year), whereas the ESC/ERS system may assign prognostic parameters to multiple risk categories for the same patient. Despite their usefulness, all these stratification systems have limitations. Therefore, a comprehensive risk assessment strategy incorporating clinical judgment is essential for each patient, alongside these scoring tools.[76] The overall treatment goal is to achieve low-risk status. (A1)
General and Supportive Measures
For individuals with PAH under medical therapy, a supervised exercise training program and psychosocial support are recommended. Patients are advised to avoid excessive physical activity causing distressing symptoms. Patients with PAH should obtain vaccination against influenza, SARS-CoV-2, and Streptococcus pneumonia. Optimization of blood iron levels is also recommended. Anticoagulation is generally not recommended in PAH but may be considered individually.
Women of childbearing potential with PAH should be counseled against pregnancy and provided with clear contraceptive advice and psychosocial support. Counseling at an expert pulmonary hypertension center, including genetic counseling, should be given to women who desire to become pregnant or who become pregnant. These individuals may be counseled about adoption and surrogacy with preconception genetic counseling being options they can explore. Pregnancy termination in women with PAH should be performed at expert centers. Endothelin receptor antagonists and riociguat are teratogenic and should not be used during pregnancy. Patients requiring procedures under anesthesia warrant a multidisciplinary consultation at a pulmonary hypertension center to assess the risks and benefits.
Patients with PAH who show signs of fluid retention and right ventricular failure should be treated with diuretic therapy. Angiotensin-converting enzyme inhibitors, angiotensin receptor blockers, β-blockers, angiotensin receptor-neprilysin inhibitors, or ivabradine are not recommended for PAH treatment unless other indications exist, such as hypertension, left heart failure, coronary artery disease, or arrhythmias. Long-term oxygen therapy, including in-flight oxygen, is recommended in patients with PAH with an arterial partial pressure of oxygen less than 60 mm Hg.
Patients with pulmonary hypertension may decompensate, leading to cardiorespiratory arrest. Immediate resuscitative measures must be started for an individual presenting with apnea, unresponsiveness, and pulselessness regardless of cause.
Idiopathic Pulmonary Arterial Hypertension Medical Management
IPAH represents a significant clinical challenge, as it has debilitating symptoms and a poor prognosis if left untreated. Pharmacotherapy plays a central role in IPAH management, aiming to alleviate symptoms, improve exercise capacity, and delay disease progression. IPAH pharmacotherapy requires a personalized approach, with treatment selection guided by disease severity, patient characteristics, and therapeutic response. The drug classes used in treating this condition include CCBs, ERAs, phosphodiesterase type 5 inhibitors (PDE-5 inhibitors or PDE-5is), soluble guanylate cyclase stimulators (sGCSs), prostacyclin analogs (PCAs), and prostacyclin receptor agonists (PRAs).
Calcium channel blockers
High-dose CCBs may be used as first-line therapy in patients with IPAH, HPAH, and drug-induced PAH that test positive during vasoreactivity testing. Amlodipine or nifedipine is preferred in patients who are relatively bradycardic at baseline.[77][78] Diltiazem is preferred in patients who are relatively tachycardic at baseline. Follow-up should be done 3 to 4 months after initiation of therapy. Additional PAH therapy should be started if the patient does not meet treatment goals, ie, low-risk status, classified under WHO-FC I to II, and has marked hemodynamic improvement. CCBs’ common side effects include hypotension and edema. (B2)
Endothelin receptor antagonists
Endothelin-1 causes vasoconstriction and mitogenic effects via the endothelin type A receptors. Endothelin B receptors promote vasodilation by clearing endothelin-1 and accelerating prostacyclin and nitric oxide production. However, nonselective endothelin receptor or selective endothelin A receptor blockade has similar effectiveness in PAH.[79] Ambrisentan is an endothelin receptor type A antagonist, while bosentan and macitentan are dual type A and type B ERAs. Ambrisentan can cause peripheral edema. Both ambrisentan and bosentan can produce LFT abnormalities. Bosentan can reduce warfarin, sildenafil, and tadalafil levels and negate hormonal contraceptives' effects. Macitentan can decrease hemoglobin levels. These agents are potentially teratogenic and must not be used during pregnancy. (B3)
Phosphodiesterase type 5 inhibitors and soluble guanylate cyclase stimulators
These medications primarily act on the nitric oxide pathway. PDE-5is prevent cyclic guanosine monophosphate degradation, resulting in higher nitric oxide levels and vasodilation. These drugs also have antiproliferative effects.[80][81][82] Sildenafil, tadalafil, and vardenafil are PDE-5is studied in PAH treatment. These agents cause vasodilation-related side effects, including flushing, headache, and epistaxis.(A1)
Riociguat is an sGCS that enhances cGMP production and vasodilation. This medication exhibits antiproliferative and antiremodeling properties in animal models.[83] Riociguat is currently approved for patients with CTEPH. The drug’s most common serious adverse effect is syncope. Riociguat should not be combined with PDE-5 inhibitors, as it can precipitate hypotension.
Prostacyclin analogs and prostacyclin receptor agonists
PCAs are potent vasodilators and endogenous platelet aggregation inhibitors with cytoprotective and antiproliferative properties.[83] The prostacyclin pathway is a common pharmacological target in PAH treatment, as it is characteristically dysregulated in this condition.(B3)
Epoprostenol is a synthetic prostacyclin analog with a half-life of 3 to 5 minutes and is administered intravenously. Dosing inconsistencies can arise from pump malfunction and catheter obstruction. Other serious adverse effects of intravenous treatment include local site infection and sepsis.[84] Epoprostenol is usually started at a dose of 2 to 4 ng/kg/min and uptitrated based on tolerance of side effects, including flushing, headache, leg pain, and diarrhea.(A1)
Treprostinil is often administered via intravenous and subcutaneous routes. Oral and inhaled treprostinil formulations are also available. The drug can be administered via an infusion pump and subcutaneous catheter, but the intravenous route is used only for those who cannot tolerate subcutaneous infusion. Infusion site pain is the most common side effect.[85] Subcutaneous treprostinil is started at a dose of 1 to 2 ng/kg/min, and doses are escalated based on tolerance of side effects, including local site pain, flushing, and headache.(A1)
Iloprost is another analog with intravenous formulations available. Selexipag is a PRA and is administered orally.
Idiopathic pulmonary arterial hypertension treatment approach
The medications discussed above target 3 distinct pathways implicated in disease improvement: antagonism of the endothelin system, agonism of the prostacyclin pathway, and agonism of the nitric oxide pathway. Recent updates in hemodynamic definitions for pulmonary hypertension have revised the abnormal mPAP cutoff values from at least 25 to greater than 20 mm Hg and abnormal PVR from greater than 3 to greater than 2 Wood units. While these drugs’ efficacy has been studied in patients with mPAP greater than 25 mm Hg and PVR greater than 3 Wood units, their effectiveness in patients with mPAP between 21 to 24 mm Hg and PVR between 2 to 3 Wood units remains uncertain.
Drawing from global experience and randomized controlled trials, the latest ESC/ERS guidelines recommend specific treatment strategies, which include the following:
- General and supportive measures should be provided to all patients.
- The 3-strata risk stratification system (low, intermediate, and high risk) must be used during the initial evaluation, while the 4-strata system (low, intermediate-low, intermediate-high, and high risk) is recommended for subsequent visits.
- Initial triple therapy with an ERA, PDE-5i, and intravenous or subcutaneous PCA should be started in patients without cardiopulmonary comorbidities classified as high-risk. Lung transplant evaluation should be initiated if the patient remains at high-risk on follow-up.
- Initial therapy should include an ERA and PDE5i in people without cardiopulmonary comorbidities classified as low-to-intermediate risk. A PRA may be added or the PDE5i switched to an sGCS if the patients remain in the intermediate-low risk status. Intravenous or subcutaneous PCA should be added if the patient deteriorates and acquires a high-risk status.
- Initial therapy should include either a PDE5i or ERA in individuals with cardiopulmonary comorbidities. Patients should receive regular follow-up and individualized therapy thereafter.
- Cardiac comorbidities are defined in this context as conditions associated with an increased left ventricular diastolic dysfunction risk, such as obesity, hypertension, diabetes mellitus, and coronary heart disease. Pulmonary comorbidities include parenchymal lung diseases and are often associated with low DLCO (<45% predicted value).
Interventional idiopathic pulmonary arterial hypertension therapy
Balloon atrial septostomy and Potts shunt (a shunt between a left pulmonary artery and descending aorta) can decompress the right heart, improving systemic blood flow and oxygen transportation despite causing desaturation. These complex treatments are rarely performed and have substantial mortality. Thus, these procedures must only be considered in centers with expert providers.[86][87](A1)
Pulmonary artery denervation using intravascular ultrasound catheters and radiofrequency ablation is still in the experimental stages.[88][89] The technique aims to reduce sympathetic overdrive associated with vasoconstriction and vascular remodeling mediated through the baroreceptors at the pulmonary artery bifurcation.[90][91][92][93](A1)
Lung and heart-lung transplantation in idiopathic pulmonary arterial hypertension
Referral to a transplant center should be made in patients with PAH who have an inadequate response to combination therapy, remain at intermediate-high or high risk of death, or have treatment-refractory conditions such as pulmonary venoocclusive disease or pulmonary capillary hemangiomatosis. Most patients receive a bilateral lung transplant, with heart-lung transplant only reserved for patients with noncorrectable cardiac conditions.[94] Patients who survive the early posttransplant period have good long-term outcomes. A study demonstrated a median survival of 10 years in patients who survived the 1st year posttransplant.[95]
Treatment of Group 1 Other Than IPAH and HPAH
Drug-induced PAH should be diagnosed after relevant exposure has been elicited, and other causes have been ruled out. The causative agent should be discontinued immediately if possible, and immediate PAH therapy should be considered in patients classified as intermediate- and high-risk. Individuals classified as low-risk should be reassessed in 3 to 4 months, with PAH therapy reconsidered if the hemodynamics are still abnormal.
The approach to CTD-associated PAH is to treat the underlying condition before proceeding with the IPAH treatment protocol described above. Antiretroviral treatment should be started in HIV-associated PAH. Initial PAH monotherapy may be considered with a sequential combination if necessary.
Patients suspected to have portopulmonary hypertension must have a screening echocardiogram and referred to expert centers. Initial PAH monotherapy may be considered with a sequential combination if necessary. Liver transplantation may be considered individually as long as the PVR becomes normal or near normal with PAH therapy.
PAH from CHD should be treated with closure of the atrial septal defect, ventricular septal defect, or patent ductus arteriosus if the PVR is less than 3 Wood units. Shunt closure may be considered if the PVR is between 3 and 5 Wood units or if the PVR decreases from greater than 5 to less than 5 Wood units after therapy. Shunt closure should not be performed if the PVR exceeds 5 Wood units. Risk assessment is recommended in patients with CHD with defect closure. An initial combination therapy followed by sequential therapy based on risk assessment similar to the approach used for IPAH should be considered.
Risk assessment must be contemplated in patients with Eisenmenger syndrome. Bosentan is recommended in symptomatic patients, with the possibility of adding other medications sequentially if treatment goals are not met.
Management of Pulmonary Hypertension Due to Left Heart Disease
Patients suspected of having PH-LHD should undergo phenotyping to assess the likelihood of LHD causing their pulmonary hypertension. This assessment considers factors such as age, LHD risk factors (eg, obesity, hypertension, dyslipidemia, and diabetes), history of known LHD, prior cardiac interventions, presence of atrial fibrillation, structural LHD, and ECG, echocardiogram, CPET, and cMRI findings. This approach should help in the decision to pursue further invasive assessment.
RHC should only be performed if it will aid treatment decisions. A referral should be made to an expert center for further evaluation if a severe precapillary component is suspected.
People with CpcPH require an individualized treatment approach at an expert center. Current guidelines neither recommend nor discourage PDE5is. However, these medications should not be used in patients with IpcPH.
The cornerstone of therapy in all these conditions is optimizing underlying cardiac disease. This approach improves symptoms and quality of life and helps prevent further worsening of pulmonary hypertension.
Management of Group 3 Pulmonary Hypertension
Pulmonary hypertension in patients with lung disease is classified into severe (PVR > 5 Wood units) or nonsevere (PVR < 5 Wood units).[96][97] However, differentiating between groups 1 and 3 pulmonary hypertension can be challenging. RHC should be performed in patients suspected to have PAH or CTEPH to help with disease phenotyping. Patients being considered for lung surgery, including lung transplant or lung volume reduction, also warrant an RHC. Therapy should focus on treating the underlying disease. Patients with group 3 pulmonary hypertension should ideally be referred to expert centers for individualized treatment and possible transplant. Inhaled treprostinil has been approved in the United States for use in patients with lung disease-associated pulmonary hypertension.
Management of Group 4 Pulmonary Hypertension
Patients with suspected CTEPH should be referred to an expert center where diagnostic confirmation may be performed using RHC and CT with contrast or DSA. Patients should be started on lifelong anticoagulation once the diagnosis is confirmed. An interprofessional team should evaluate patients’ suitability for endarterectomy. If inoperable, balloon pulmonary angioplasty or riociguat treatment may be explored. Pulmonary endarterectomy may leave behind residual disease, warranting balloon pulmonary angioplasty, riociguat treatment, or both.
Management of Group 5 Pulmonary Hypertension
The mainstay of group 5 pulmonary hypertension treatment is managing the underlying condition. Other essential parts of management for patients with this condition include symptomatic treatment, close monitoring, and regular assessment of therapeutic response and disease progression.
Differential Diagnosis
The diagnosis of pulmonary hypertension is commonly delayed because the presenting symptoms overlap with other disease processes. Other conditions to be considered in the differential diagnosis include but are not limited to congestive heart failure, coronary artery disease, pulmonary fibrosis, COPD, valvular heart disease, CHD, and pulmonary embolism.
Prognosis
Progressive right heart failure and death can occur in patients with PAH if left untreated. Recent United States data suggest 1-year mortality as high as 8% for intermediate-risk and 19% for high-risk patients, with 3-year mortality rates being 20% and 55% for those groups, respectively.[98] These data emphasize the importance of early diagnosis and aggressive therapy. Prognosis varies among different PAH subgroups based on underlying etiology. Meanwhile, the presence of pulmonary hypertension and right ventricular dysfunction in patients with LHD is associated with high mortality. In patients with lung disease, even nonsevere pulmonary hypertension negatively impacts survival and is associated with increased hospitalization. People with severe pulmonary hypertension have worse outcomes than those with nonsevere pulmonary hypertension.[99] CTEPH has an excellent long-term prognosis if the disease is operable.
Complications
Complications of pulmonary hypertension include the following:
- Right ventricular dysfunction, leading to right heart failure
- Tachyarrythmias, including atrial fibrillation and atrial flutter; ventricular arrhythmias are less common
- Hemoptysis
- Mechanical complications include pulmonary artery aneurysm, dissection, or rupture, and compression of the left main coronary artery, pulmonary veins, main bronchi, and recurrent laryngeal nerves
- Congestive hepatopathy and cirrhosis
These conditions encompass a wide range of cardiovascular and systemic manifestations that can significantly impact patient outcomes and quality of life. Recognizing and addressing these complications early in the disease course is essential for optimizing patient care and improving long-term outcomes in individuals with pulmonary hypertension.
Deterrence and Patient Education
Primary prevention strategies for pulmonary hypertension aim to avoid developing the condition by addressing risk factors and promoting cardiovascular health. Lifestyle modifications, including regular exercise, maintaining a healthy weight, and adopting a balanced diet, are vital in reducing pulmonary hypertension risk. Smoking cessation is crucial, as tobacco smoking is a significant risk factor for pulmonary hypertension and other cardiovascular diseases. Additionally, managing comorbid conditions such as obstructive sleep apnea, COPD, and CTDs can help prevent the onset of pulmonary hypertension. Furthermore, individuals with genetic predisposition should undergo genetic testing and counseling to evaluate their risk and consider preventive measures.
Pulmonary hypertension typically has a diagnostic delay of more than 2 years from symptom onset, and clinicians should have a high index of suspicion. Promptly initiating a stepwise evaluation and referring patients to expert centers is crucial for the timely diagnosis of pulmonary hypertension, identification of its underlying cause, and initiation of aggressive therapy. Given the high morbidity and mortality associated with the disease, such measures are essential for optimizing patient outcomes.
Secondary prevention focuses on preventing disease progression and reducing complications in individuals already diagnosed with this condition. Regular medical follow-up is essential for monitoring disease progression, treatment response, and early detection of complications. Pharmacological therapy is central to managing pulmonary hypertension to improve symptoms and delay disease progression. Treatments may include oxygen supplementation and diuretic and anticoagulant intake. Patients requiring subcutaneous or intravenous drug therapy need specialized education to manage such medications.
Lifestyle modifications, including regular exercise and a low-salt diet, are essential for improving cardiovascular health and overall well-being in patients with pulmonary hypertension. Avoidance of triggers, such as high altitudes and certain medications, can help prevent exacerbation of symptoms and complications.
Patients should be aware of the typical disease symptoms, such as shortness of breath on exertion, weight gain, and even subtle symptoms, such as fatigue, loss of appetite, chest pain, belly pain, and tiredness. Vigilance is imperative, especially in people with a family history of pulmonary hypertension. Patients must be educated about the potential for a delay in diagnosis, which may require visiting multiple doctors across various institutions before arriving at a definitive diagnosis. The experience can evoke feelings of anxiety and frustration. Additionally, patients may undergo extensive testing, including blood tests, PFTs, sleep studies, echocardiograms, and RHC, to aid in the diagnostic process. Healthcare providers, patients, and caregivers must collaborate to implement effective prevention strategies and optimize outcomes for individuals with this condition.
Pearls and Other Issues
Several key concepts can guide healthcare providers when evaluating and managing pulmonary hypertension. Firstly, early recognition and accurate diagnosis are paramount. Pulmonary hypertension often presents with nonspecific symptoms such as dyspnea and fatigue. Clinicians should maintain a high suspicion index, especially in high-risk patients. Diagnostic evaluation must include a thorough history and physical examination, followed by appropriate testing such as echocardiography, PFTs, and RHC to confirm the diagnosis and assess disease severity.
Secondly, individualized treatment strategies are crucial. Pulmonary hypertension management involves an interprofessional approach, and treatment decisions should be tailored to each patient's underlying condition, functional status, and therapeutic response. Pharmacotherapy with agents targeting the pulmonary vasculature forms the cornerstone of treatment. Lifestyle modifications also play significant roles in symptom management and improving quality of life. Regular follow-up and monitoring are essential to adjust treatment as needed and mitigate potential complications.
Emphasis should be placed on patient education and shared decision-making to enhance treatment adherence and empower patients in managing their condition. Healthcare providers should remain vigilant for potential complications such as right heart failure, arrhythmias, and thromboembolic events, promptly addressing these conditions to prevent disease progression and improve outcomes.
Enhancing Healthcare Team Outcomes
Pulmonary hypertension can occur due to a variety of causes, as described above, but conditions such as PAH and CTEPH require highly specialized interprofessional care. An expert center is designated based on the availability of an interprofessional team, including a cardiologist, pulmonologist, rheumatologist, cardiothoracic surgery, interventional radiology, nurse specialist, pharmacists, social workers, and on-call experts. Such centers have large patient volumes, providing the best standard of care with better clinical outcomes.[100]
Interprofessional care involves coordinated activity and open communication between all care team members, with each contributing from their areas of expertise. Clinicians focus on their particular practice expertise, coordinating their actions with other disciplines. Nurses provide invaluable input, assisting in patient evaluation, helping with surgical procedures when necessary, counseling patients, and coordinating activities between the team members. Pharmacists coordinate medication regimens, perform medication reconciliation, counsel patients on their drugs, and answer clinician questions regarding medications. Other areas, such as mental health professionals, likewise coordinate their activities within the care team. Interprofessional care teams provide the optimal approach for patients with pulmonary hypertension, leading to improved patient outcomes.
Media
(Click Image to Enlarge)
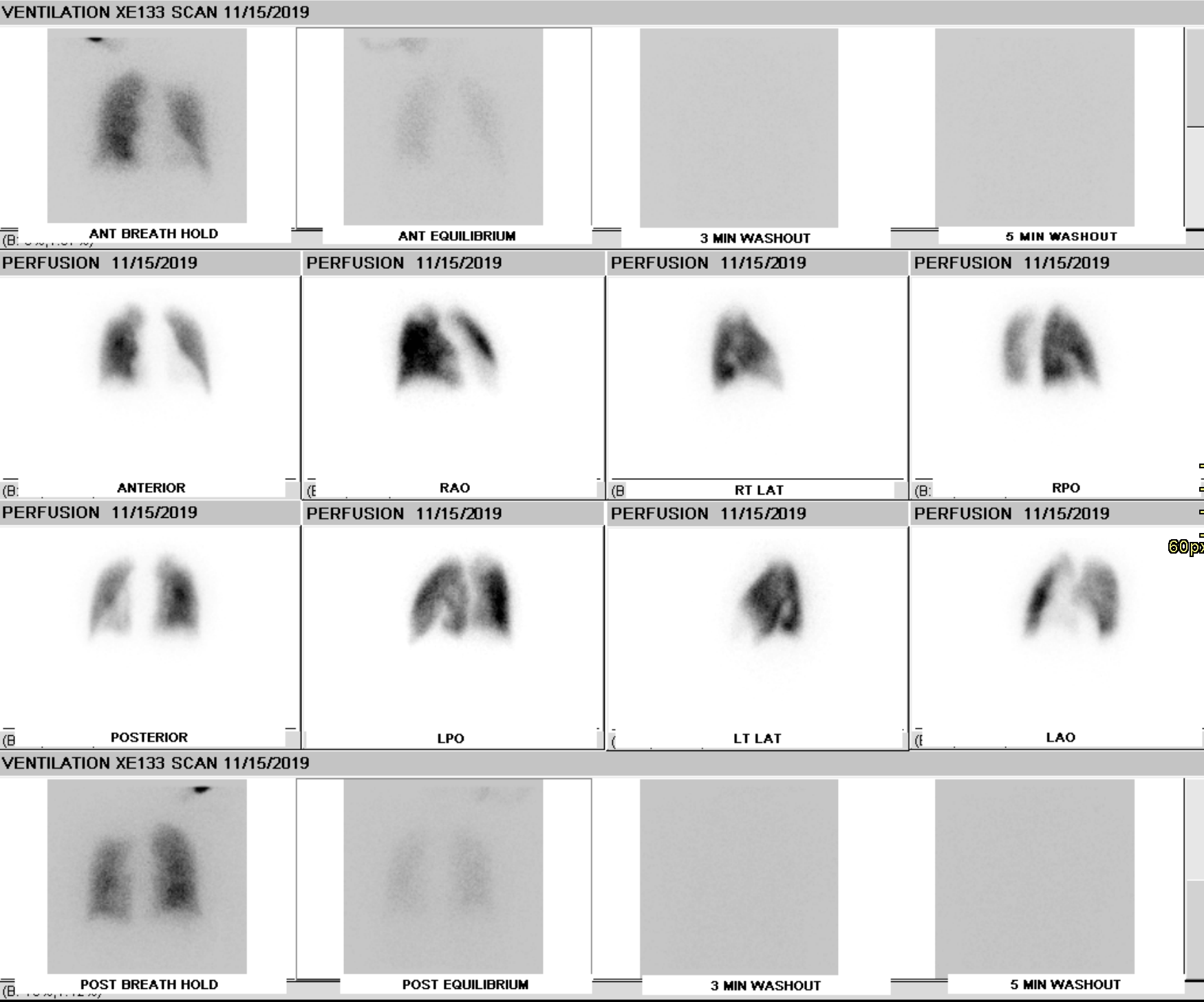
CTEPH Ventilation-Perfusion Scan. A ventilation-perfusion scan showing moderate-sized mismatched perfusion defects seen in chronic thromboembolic pulmonary hypertension.
Contributed by D Tafti, MD
(Click Image to Enlarge)
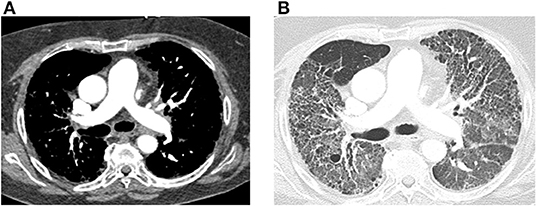
Pulmonary Hypertension From Connective Tissue Disease on CT. Transaxial computed tomography (CT) images of the chest, both with and without contrast, depict findings in a 64-year-old female with connective tissue disease, severe interstitial lung disease, and mixed severe pulmonary hypertension. In Image A, an enlarged main pulmonary arterial size measuring 3.2 cm is noted, compared to the ascending aorta size of 2.9 cm at the same level, indicating pulmonary hypertension. No evidence of pulmonary embolism is observed, with optimal contrast opacification. In Image B, transaxial images in the lung window reveal extensive bilateral diffuse ground-glass opacities and honeycombing, along with intralobular and interstitial thickening and bronchiectasis, consistent with the patient's known history of connective tissue disease-associated non-specific interstitial pneumonitis.
Farrell C, Balasubramaian A, Hays AG, et al. A clinical approach to multimodality imaging in pulmonary hypertension. Front Cardiovasc Med. 2022;8:794706. doi: 10.3389/fcvm.2021.794706.
References
Humbert M, Kovacs G, Hoeper MM, Badagliacca R, Berger RMF, Brida M, Carlsen J, Coats AJS, Escribano-Subias P, Ferrari P, Ferreira DS, Ghofrani HA, Giannakoulas G, Kiely DG, Mayer E, Meszaros G, Nagavci B, Olsson KM, Pepke-Zaba J, Quint JK, Rådegran G, Simonneau G, Sitbon O, Tonia T, Toshner M, Vachiery JL, Vonk Noordegraaf A, Delcroix M, Rosenkranz S, ESC/ERS Scientific Document Group. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. European heart journal. 2022 Oct 11:43(38):3618-3731. doi: 10.1093/eurheartj/ehac237. Epub [PubMed PMID: 36017548]
Condliffe R, Kovacs G. Identifying early pulmonary arterial hypertension in patients with systemic sclerosis. The European respiratory journal. 2018 Apr:51(4):. pii: 1800495. doi: 10.1183/13993003.00495-2018. Epub 2018 Apr 4 [PubMed PMID: 29618608]
Maron BA, Brittain EL, Choudhary G, Gladwin MT. Redefining pulmonary hypertension. The Lancet. Respiratory medicine. 2018 Mar:6(3):168-170. doi: 10.1016/S2213-2600(17)30498-8. Epub 2017 Dec 18 [PubMed PMID: 29269004]
Maron BA, Wertheim BM, Gladwin MT. Under Pressure to Clarify Pulmonary Hypertension Clinical Risk. American journal of respiratory and critical care medicine. 2018 Feb 15:197(4):423-426. doi: 10.1164/rccm.201711-2306ED. Epub [PubMed PMID: 29216444]
Kovacs G, Berghold A, Scheidl S, Olschewski H. Pulmonary arterial pressure during rest and exercise in healthy subjects: a systematic review. The European respiratory journal. 2009 Oct:34(4):888-94. doi: 10.1183/09031936.00145608. Epub 2009 Mar 26 [PubMed PMID: 19324955]
Level 1 (high-level) evidenceSimonneau G, Montani D, Celermajer DS, Denton CP, Gatzoulis MA, Krowka M, Williams PG, Souza R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. The European respiratory journal. 2019 Jan:53(1):. doi: 10.1183/13993003.01913-2018. Epub 2019 Jan 24 [PubMed PMID: 30545968]
Bentley RF, Barker M, Esfandiari S, Wright SP, Valle FH, Granton JT, Mak S. Normal and Abnormal Relationships of Pulmonary Artery to Wedge Pressure During Exercise. Journal of the American Heart Association. 2020 Nov 17:9(22):e016339. doi: 10.1161/JAHA.120.016339. Epub 2020 Nov 6 [PubMed PMID: 33153377]
Eisman AS, Shah RV, Dhakal BP, Pappagianopoulos PP, Wooster L, Bailey C, Cunningham TF, Hardin KM, Baggish AL, Ho JE, Malhotra R, Lewis GD. Pulmonary Capillary Wedge Pressure Patterns During Exercise Predict Exercise Capacity and Incident Heart Failure. Circulation. Heart failure. 2018 May:11(5):e004750. doi: 10.1161/CIRCHEARTFAILURE.117.004750. Epub [PubMed PMID: 29695381]
Garcia-Rivas G, Jerjes-Sánchez C, Rodriguez D, Garcia-Pelaez J, Trevino V. A systematic review of genetic mutations in pulmonary arterial hypertension. BMC medical genetics. 2017 Aug 2:18(1):82. doi: 10.1186/s12881-017-0440-5. Epub 2017 Aug 2 [PubMed PMID: 28768485]
Level 1 (high-level) evidenceSimonneau G, Galiè N, Rubin LJ, Langleben D, Seeger W, Domenighetti G, Gibbs S, Lebrec D, Speich R, Beghetti M, Rich S, Fishman A. Clinical classification of pulmonary hypertension. Journal of the American College of Cardiology. 2004 Jun 16:43(12 Suppl S):5S-12S [PubMed PMID: 15194173]
Hoeper MM, Humbert M, Souza R, Idrees M, Kawut SM, Sliwa-Hahnle K, Jing ZC, Gibbs JS. A global view of pulmonary hypertension. The Lancet. Respiratory medicine. 2016 Apr:4(4):306-22. doi: 10.1016/S2213-2600(15)00543-3. Epub 2016 Mar 12 [PubMed PMID: 26975810]
Leber L, Beaudet A, Muller A. Epidemiology of pulmonary arterial hypertension and chronic thromboembolic pulmonary hypertension: identification of the most accurate estimates from a systematic literature review. Pulmonary circulation. 2021 Jan-Mar:11(1):2045894020977300. doi: 10.1177/2045894020977300. Epub 2021 Jan 7 [PubMed PMID: 33456755]
Level 1 (high-level) evidenceMontani D, Girerd B, Jaïs X, Laveneziana P, Lau EMT, Bouchachi A, Hascoët S, Günther S, Godinas L, Parent F, Guignabert C, Beurnier A, Chemla D, Hervé P, Eyries M, Soubrier F, Simonneau G, Sitbon O, Savale L, Humbert M. Screening for pulmonary arterial hypertension in adults carrying a BMPR2 mutation. The European respiratory journal. 2021 Jul:58(1):. doi: 10.1183/13993003.04229-2020. Epub 2021 Jul 22 [PubMed PMID: 33380512]
Lau EMT, Giannoulatou E, Celermajer DS, Humbert M. Epidemiology and treatment of pulmonary arterial hypertension. Nature reviews. Cardiology. 2017 Oct:14(10):603-614. doi: 10.1038/nrcardio.2017.84. Epub 2017 Jun 8 [PubMed PMID: 28593996]
Rosenkranz S, Gibbs JS, Wachter R, De Marco T, Vonk-Noordegraaf A, Vachiéry JL. Left ventricular heart failure and pulmonary hypertension. European heart journal. 2016 Mar 21:37(12):942-54. doi: 10.1093/eurheartj/ehv512. Epub 2015 Oct 27 [PubMed PMID: 26508169]
Lam CS, Roger VL, Rodeheffer RJ, Borlaug BA, Enders FT, Redfield MM. Pulmonary hypertension in heart failure with preserved ejection fraction: a community-based study. Journal of the American College of Cardiology. 2009 Mar 31:53(13):1119-26. doi: 10.1016/j.jacc.2008.11.051. Epub [PubMed PMID: 19324256]
Tichelbäcker T, Dumitrescu D, Gerhardt F, Stern D, Wissmüller M, Adam M, Schmidt T, Frerker C, Pfister R, Halbach M, Baldus S, Rosenkranz S. Pulmonary hypertension and valvular heart disease. Herz. 2019 Sep:44(6):491-501. doi: 10.1007/s00059-019-4823-6. Epub [PubMed PMID: 31312873]
Weber L, Rickli H, Haager PK, Joerg L, Weilenmann D, Brenner R, Taramasso M, Baier P, Maisano F, Maeder MT. Haemodynamic mechanisms and long-term prognostic impact of pulmonary hypertension in patients with severe aortic stenosis undergoing valve replacement. European journal of heart failure. 2019 Feb:21(2):172-181. doi: 10.1002/ejhf.1322. Epub 2018 Oct 17 [PubMed PMID: 30328215]
Nathan SD, Barbera JA, Gaine SP, Harari S, Martinez FJ, Olschewski H, Olsson KM, Peacock AJ, Pepke-Zaba J, Provencher S, Weissmann N, Seeger W. Pulmonary hypertension in chronic lung disease and hypoxia. The European respiratory journal. 2019 Jan:53(1):. doi: 10.1183/13993003.01914-2018. Epub 2019 Jan 24 [PubMed PMID: 30545980]
Hurdman J, Condliffe R, Elliot CA, Swift A, Rajaram S, Davies C, Hill C, Hamilton N, Armstrong IJ, Billings C, Pollard L, Wild JM, Lawrie A, Lawson R, Sabroe I, Kiely DG. Pulmonary hypertension in COPD: results from the ASPIRE registry. The European respiratory journal. 2013 Jun:41(6):1292-301. doi: 10.1183/09031936.00079512. Epub 2012 Sep 27 [PubMed PMID: 23018917]
Level 2 (mid-level) evidenceDelcroix M, Torbicki A, Gopalan D, Sitbon O, Klok FA, Lang I, Jenkins D, Kim NH, Humbert M, Jais X, Vonk Noordegraaf A, Pepke-Zaba J, Brénot P, Dorfmuller P, Fadel E, Ghofrani HA, Hoeper MM, Jansa P, Madani M, Matsubara H, Ogo T, Grünig E, D'Armini A, Galie N, Meyer B, Corkery P, Meszaros G, Mayer E, Simonneau G. ERS statement on chronic thromboembolic pulmonary hypertension. The European respiratory journal. 2021 Jun:57(6):. pii: 2002828. doi: 10.1183/13993003.02828-2020. Epub 2021 Jun 17 [PubMed PMID: 33334946]
Kramm T, Wilkens H, Fuge J, Schäfers HJ, Guth S, Wiedenroth CB, Weingard B, Huscher D, Pittrow D, Cebotari S, Hoeper MM, Mayer E, Olsson KM. Incidence and characteristics of chronic thromboembolic pulmonary hypertension in Germany. Clinical research in cardiology : official journal of the German Cardiac Society. 2018 Jul:107(7):548-553. doi: 10.1007/s00392-018-1215-5. Epub 2018 Feb 15 [PubMed PMID: 29450722]
Shlobin OA, Kouranos V, Barnett SD, Alhamad EH, Culver DA, Barney J, Cordova FC, Carmona EM, Scholand MB, Wijsenbeek M, Ganesh S, Lower EE, Engel PJ, Wort J, Price L, Wells AU, Nathan SD, Baughman RP. Physiological predictors of survival in patients with sarcoidosis-associated pulmonary hypertension: results from an international registry. The European respiratory journal. 2020 May:55(5):. pii: 1901747. doi: 10.1183/13993003.01747-2019. Epub 2020 May 14 [PubMed PMID: 32139456]
Boucly A, Cottin V, Nunes H, Jaïs X, Tazi A, Prévôt G, Reynaud-Gaubert M, Dromer C, Viacroze C, Horeau-Langlard D, Pison C, Bergot E, Traclet J, Weatherald J, Simonneau G, Valeyre D, Montani D, Humbert M, Sitbon O, Savale L. Management and long-term outcomes of sarcoidosis-associated pulmonary hypertension. The European respiratory journal. 2017 Oct:50(4):. pii: 1700465. doi: 10.1183/13993003.00465-2017. Epub 2017 Oct 19 [PubMed PMID: 29051269]
Galiè N, Humbert M, Vachiery JL, Gibbs S, Lang I, Torbicki A, Simonneau G, Peacock A, Vonk Noordegraaf A, Beghetti M, Ghofrani A, Gomez Sanchez MA, Hansmann G, Klepetko W, Lancellotti P, Matucci M, McDonagh T, Pierard LA, Trindade PT, Zompatori M, Hoeper M. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). The European respiratory journal. 2015 Oct:46(4):903-75. doi: 10.1183/13993003.01032-2015. Epub 2015 Aug 29 [PubMed PMID: 26318161]
Armstrong I, Billings C, Kiely DG, Yorke J, Harries C, Clayton S, Gin-Sing W. The patient experience of pulmonary hypertension: a large cross-sectional study of UK patients. BMC pulmonary medicine. 2019 Mar 21:19(1):67. doi: 10.1186/s12890-019-0827-5. Epub 2019 Mar 21 [PubMed PMID: 30898139]
Level 2 (mid-level) evidenceStrange G, Gabbay E, Kermeen F, Williams T, Carrington M, Stewart S, Keogh A. Time from symptoms to definitive diagnosis of idiopathic pulmonary arterial hypertension: The delay study. Pulmonary circulation. 2013 Jan:3(1):89-94. doi: 10.4103/2045-8932.109919. Epub [PubMed PMID: 23662179]
Rich JD, Thenappan T, Freed B, Patel AR, Thisted RA, Childers R, Archer SL. QTc prolongation is associated with impaired right ventricular function and predicts mortality in pulmonary hypertension. International journal of cardiology. 2013 Aug 10:167(3):669-76. doi: 10.1016/j.ijcard.2012.03.071. Epub 2012 Mar 27 [PubMed PMID: 22459397]
Level 2 (mid-level) evidenceSun PY, Jiang X, Gomberg-Maitland M, Zhao QH, He J, Yuan P, Zhang R, Jing ZC. Prolonged QRS duration: a new predictor of adverse outcome in idiopathic pulmonary arterial hypertension. Chest. 2012 Feb:141(2):374-380. doi: 10.1378/chest.10-3331. Epub 2011 Jul 21 [PubMed PMID: 21778258]
Level 2 (mid-level) evidenceHenkens IR, Gan CT, van Wolferen SA, Hew M, Boonstra A, Twisk JWR, Kamp O, van der Wall EE, Schalij MJ, Vonk Noordegraaf A, Vliegen HW. ECG monitoring of treatment response in pulmonary arterial hypertension patients. Chest. 2008 Dec:134(6):1250-1257. doi: 10.1378/chest.08-0461. Epub 2008 Jul 18 [PubMed PMID: 18641107]
Level 2 (mid-level) evidenceBossone E, Paciocco G, Iarussi D, Agretto A, Iacono A, Gillespie BW, Rubenfire M. The prognostic role of the ECG in primary pulmonary hypertension. Chest. 2002 Feb:121(2):513-8 [PubMed PMID: 11834666]
Wanamaker B, Cascino T, McLaughlin V, Oral H, Latchamsetty R, Siontis KC. Atrial Arrhythmias in Pulmonary Hypertension: Pathogenesis, Prognosis and Management. Arrhythmia & electrophysiology review. 2018 Mar:7(1):43-48. doi: 10.15420/aer.2018.3.2. Epub [PubMed PMID: 29636972]
Ascha M, Renapurkar RD, Tonelli AR. A review of imaging modalities in pulmonary hypertension. Annals of thoracic medicine. 2017 Apr-Jun:12(2):61-73. doi: 10.4103/1817-1737.203742. Epub [PubMed PMID: 28469715]
Hoeper MM, Bogaard HJ, Condliffe R, Frantz R, Khanna D, Kurzyna M, Langleben D, Manes A, Satoh T, Torres F, Wilkins MR, Badesch DB. Definitions and diagnosis of pulmonary hypertension. Journal of the American College of Cardiology. 2013 Dec 24:62(25 Suppl):D42-50. doi: 10.1016/j.jacc.2013.10.032. Epub [PubMed PMID: 24355641]
Level 3 (low-level) evidenceRich S, Dantzker DR, Ayres SM, Bergofsky EH, Brundage BH, Detre KM, Fishman AP, Goldring RM, Groves BM, Koerner SK. Primary pulmonary hypertension. A national prospective study. Annals of internal medicine. 1987 Aug:107(2):216-23 [PubMed PMID: 3605900]
Trip P, Nossent EJ, de Man FS, van den Berk IA, Boonstra A, Groepenhoff H, Leter EM, Westerhof N, Grünberg K, Bogaard HJ, Vonk-Noordegraaf A. Severely reduced diffusion capacity in idiopathic pulmonary arterial hypertension: patient characteristics and treatment responses. The European respiratory journal. 2013 Dec:42(6):1575-85. doi: 10.1183/09031936.00184412. Epub 2013 Aug 15 [PubMed PMID: 23949959]
Level 2 (mid-level) evidenceSun XG, Hansen JE, Oudiz RJ, Wasserman K. Pulmonary function in primary pulmonary hypertension. Journal of the American College of Cardiology. 2003 Mar 19:41(6):1028-35 [PubMed PMID: 12651053]
Mélot C, Naeije R. Pulmonary vascular diseases. Comprehensive Physiology. 2011 Apr:1(2):593-619. doi: 10.1002/cphy.c090014. Epub [PubMed PMID: 23737196]
Level 3 (low-level) evidenceHarbaum L, Fuge J, Kamp JC, Hennigs JK, Simon M, Sinning C, Oqueka T, Grimminger J, Olsson KM, Hoeper MM, Klose H. Blood carbon dioxide tension and risk in pulmonary arterial hypertension. International journal of cardiology. 2020 Nov 1:318():131-137. doi: 10.1016/j.ijcard.2020.06.069. Epub 2020 Jul 4 [PubMed PMID: 32634498]
Jilwan FN, Escourrou P, Garcia G, Jaïs X, Humbert M, Roisman G. High occurrence of hypoxemic sleep respiratory disorders in precapillary pulmonary hypertension and mechanisms. Chest. 2013 Jan:143(1):47-55. doi: 10.1378/chest.11-3124. Epub [PubMed PMID: 22878784]
Swift AJ, Dwivedi K, Johns C, Garg P, Chin M, Currie BJ, Rothman AM, Capener D, Shahin Y, Elliot CA, Charalampopolous T, Sabroe I, Rajaram S, Hill C, Wild JM, Condliffe R, Kiely DG. Diagnostic accuracy of CT pulmonary angiography in suspected pulmonary hypertension. European radiology. 2020 Sep:30(9):4918-4929. doi: 10.1007/s00330-020-06846-1. Epub 2020 Apr 27 [PubMed PMID: 32342182]
Dong C, Zhou M, Liu D, Long X, Guo T, Kong X. Diagnostic accuracy of computed tomography for chronic thromboembolic pulmonary hypertension: a systematic review and meta-analysis. PloS one. 2015:10(4):e0126985. doi: 10.1371/journal.pone.0126985. Epub 2015 Apr 29 [PubMed PMID: 25923810]
Level 1 (high-level) evidenceRajaram S, Swift AJ, Capener D, Telfer A, Davies C, Hill C, Condliffe R, Elliot C, Hurdman J, Kiely DG, Wild JM. Diagnostic accuracy of contrast-enhanced MR angiography and unenhanced proton MR imaging compared with CT pulmonary angiography in chronic thromboembolic pulmonary hypertension. European radiology. 2012 Feb:22(2):310-7. doi: 10.1007/s00330-011-2252-x. Epub 2011 Sep 2 [PubMed PMID: 21887483]
Level 2 (mid-level) evidenceGaliè N, Humbert M, Vachiery JL, Gibbs S, Lang I, Torbicki A, Simonneau G, Peacock A, Vonk Noordegraaf A, Beghetti M, Ghofrani A, Gomez Sanchez MA, Hansmann G, Klepetko W, Lancellotti P, Matucci M, McDonagh T, Pierard LA, Trindade PT, Zompatori M, Hoeper M, ESC Scientific Document Group. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). European heart journal. 2016 Jan 1:37(1):67-119. doi: 10.1093/eurheartj/ehv317. Epub 2015 Aug 29 [PubMed PMID: 26320113]
Lang RM, Badano LP, Mor-Avi V, Afilalo J, Armstrong A, Ernande L, Flachskampf FA, Foster E, Goldstein SA, Kuznetsova T, Lancellotti P, Muraru D, Picard MH, Rietzschel ER, Rudski L, Spencer KT, Tsang W, Voigt JU. Recommendations for cardiac chamber quantification by echocardiography in adults: an update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. Journal of the American Society of Echocardiography : official publication of the American Society of Echocardiography. 2015 Jan:28(1):1-39.e14. doi: 10.1016/j.echo.2014.10.003. Epub [PubMed PMID: 25559473]
Rudski LG, Lai WW, Afilalo J, Hua L, Handschumacher MD, Chandrasekaran K, Solomon SD, Louie EK, Schiller NB. Guidelines for the echocardiographic assessment of the right heart in adults: a report from the American Society of Echocardiography endorsed by the European Association of Echocardiography, a registered branch of the European Society of Cardiology, and the Canadian Society of Echocardiography. Journal of the American Society of Echocardiography : official publication of the American Society of Echocardiography. 2010 Jul:23(7):685-713; quiz 786-8. doi: 10.1016/j.echo.2010.05.010. Epub [PubMed PMID: 20620859]
Foale R, Nihoyannopoulos P, McKenna W, Kleinebenne A, Nadazdin A, Rowland E, Smith G. Echocardiographic measurement of the normal adult right ventricle. British heart journal. 1986 Jul:56(1):33-44 [PubMed PMID: 3730205]
Sun XG, Hansen JE, Oudiz RJ, Wasserman K. Exercise pathophysiology in patients with primary pulmonary hypertension. Circulation. 2001 Jul 24:104(4):429-35 [PubMed PMID: 11468205]
Dumitrescu D, Nagel C, Kovacs G, Bollmann T, Halank M, Winkler J, Hellmich M, Grünig E, Olschewski H, Ewert R, Rosenkranz S. Cardiopulmonary exercise testing for detecting pulmonary arterial hypertension in systemic sclerosis. Heart (British Cardiac Society). 2017 May:103(10):774-782. doi: 10.1136/heartjnl-2016-309981. Epub 2017 Jan 6 [PubMed PMID: 28062514]
He J, Fang W, Lv B, He JG, Xiong CM, Liu ZH, He ZX. Diagnosis of chronic thromboembolic pulmonary hypertension: comparison of ventilation/perfusion scanning and multidetector computed tomography pulmonary angiography with pulmonary angiography. Nuclear medicine communications. 2012 May:33(5):459-63. doi: 10.1097/MNM.0b013e32835085d9. Epub [PubMed PMID: 22262242]
Tunariu N, Gibbs SJ, Win Z, Gin-Sing W, Graham A, Gishen P, Al-Nahhas A. Ventilation-perfusion scintigraphy is more sensitive than multidetector CTPA in detecting chronic thromboembolic pulmonary disease as a treatable cause of pulmonary hypertension. Journal of nuclear medicine : official publication, Society of Nuclear Medicine. 2007 May:48(5):680-4 [PubMed PMID: 17475953]
Level 2 (mid-level) evidenceJohns CS, Swift AJ, Rajaram S, Hughes PJC, Capener DJ, Kiely DG, Wild JM. Lung perfusion: MRI vs. SPECT for screening in suspected chronic thromboembolic pulmonary hypertension. Journal of magnetic resonance imaging : JMRI. 2017 Dec:46(6):1693-1697. doi: 10.1002/jmri.25714. Epub 2017 Apr 4 [PubMed PMID: 28376242]
Meng JJ, Zhang LJ, Wang Q, Fang W, Dai HJ, Yan J, Wang T, Yao ZM, He J, Li M, Mi HZ, Jiao J, Zheng YM. [A comparison of ventilation/perfusion single photon emission CT and CT pulmonary angiography for diagnosis of pulmonary embolism]. Zhonghua jie he he hu xi za zhi = Zhonghua jiehe he huxi zazhi = Chinese journal of tuberculosis and respiratory diseases. 2013 Mar:36(3):177-81 [PubMed PMID: 23856139]
Rajaram S, Swift AJ, Telfer A, Hurdman J, Marshall H, Lorenz E, Capener D, Davies C, Hill C, Elliot C, Condliffe R, Wild JM, Kiely DG. 3D contrast-enhanced lung perfusion MRI is an effective screening tool for chronic thromboembolic pulmonary hypertension: results from the ASPIRE Registry. Thorax. 2013 Jul:68(7):677-8. doi: 10.1136/thoraxjnl-2012-203020. Epub 2013 Jan 24 [PubMed PMID: 23349220]
Level 3 (low-level) evidenceSwift AJ, Lu H, Uthoff J, Garg P, Cogliano M, Taylor J, Metherall P, Zhou S, Johns CS, Alabed S, Condliffe RA, Lawrie A, Wild JM, Kiely DG. A machine learning cardiac magnetic resonance approach to extract disease features and automate pulmonary arterial hypertension diagnosis. European heart journal. Cardiovascular Imaging. 2021 Jan 22:22(2):236-245. doi: 10.1093/ehjci/jeaa001. Epub [PubMed PMID: 31998956]
Rosenkranz S, Howard LS, Gomberg-Maitland M, Hoeper MM. Systemic Consequences of Pulmonary Hypertension and Right-Sided Heart Failure. Circulation. 2020 Feb 25:141(8):678-693. doi: 10.1161/CIRCULATIONAHA.116.022362. Epub 2020 Feb 24 [PubMed PMID: 32091921]
Morrell NW, Aldred MA, Chung WK, Elliott CG, Nichols WC, Soubrier F, Trembath RC, Loyd JE. Genetics and genomics of pulmonary arterial hypertension. The European respiratory journal. 2019 Jan:53(1):. doi: 10.1183/13993003.01899-2018. Epub 2019 Jan 24 [PubMed PMID: 30545973]
Song J, Eichstaedt CA, Viales RR, Benjamin N, Harutyunova S, Fischer C, Grünig E, Hinderhofer K. Identification of genetic defects in pulmonary arterial hypertension by a new gene panel diagnostic tool. Clinical science (London, England : 1979). 2016 Nov 1:130(22):2043-2052. doi: 10.1042/CS20160531. Epub 2016 Sep 9 [PubMed PMID: 27613157]
Hoeper MM, Lee SH, Voswinckel R, Palazzini M, Jais X, Marinelli A, Barst RJ, Ghofrani HA, Jing ZC, Opitz C, Seyfarth HJ, Halank M, McLaughlin V, Oudiz RJ, Ewert R, Wilkens H, Kluge S, Bremer HC, Baroke E, Rubin LJ. Complications of right heart catheterization procedures in patients with pulmonary hypertension in experienced centers. Journal of the American College of Cardiology. 2006 Dec 19:48(12):2546-52 [PubMed PMID: 17174196]
Level 2 (mid-level) evidenceKovacs G, Avian A, Pienn M, Naeije R, Olschewski H. Reading pulmonary vascular pressure tracings. How to handle the problems of zero leveling and respiratory swings. American journal of respiratory and critical care medicine. 2014 Aug 1:190(3):252-7. doi: 10.1164/rccm.201402-0269PP. Epub [PubMed PMID: 24869464]
Hoeper MM, Maier R, Tongers J, Niedermeyer J, Hohlfeld JM, Hamm M, Fabel H. Determination of cardiac output by the Fick method, thermodilution, and acetylene rebreathing in pulmonary hypertension. American journal of respiratory and critical care medicine. 1999 Aug:160(2):535-41 [PubMed PMID: 10430725]
Robbins IM, Hemnes AR, Pugh ME, Brittain EL, Zhao DX, Piana RN, Fong PP, Newman JH. High prevalence of occult pulmonary venous hypertension revealed by fluid challenge in pulmonary hypertension. Circulation. Heart failure. 2014 Jan:7(1):116-22. doi: 10.1161/CIRCHEARTFAILURE.113.000468. Epub 2013 Dec 2 [PubMed PMID: 24297689]
Level 2 (mid-level) evidenceFox BD, Shimony A, Langleben D, Hirsch A, Rudski L, Schlesinger R, Eisenberg MJ, Joyal D, Hudson M, Boutet K, Serban A, Masetto A, Baron M. High prevalence of occult left heart disease in scleroderma-pulmonary hypertension. The European respiratory journal. 2013 Oct:42(4):1083-91. doi: 10.1183/09031936.00091212. Epub 2012 Dec 20 [PubMed PMID: 23258775]
Level 2 (mid-level) evidenceHoeper MM, Pittrow D, Opitz C, Gibbs JSR, Rosenkranz S, Grünig E, Olsson KM, Huscher D. Risk assessment in pulmonary arterial hypertension. The European respiratory journal. 2018 Mar:51(3):. pii: 1702606. doi: 10.1183/13993003.02606-2017. Epub 2018 Mar 29 [PubMed PMID: 29599117]
Boucly A, Weatherald J, Savale L, Jaïs X, Cottin V, Prevot G, Picard F, de Groote P, Jevnikar M, Bergot E, Chaouat A, Chabanne C, Bourdin A, Parent F, Montani D, Simonneau G, Humbert M, Sitbon O. Risk assessment, prognosis and guideline implementation in pulmonary arterial hypertension. The European respiratory journal. 2017 Aug:50(2):. pii: 1700889. doi: 10.1183/13993003.00889-2017. Epub 2017 Aug 3 [PubMed PMID: 28775050]
Hoeper MM, Kramer T, Pan Z, Eichstaedt CA, Spiesshoefer J, Benjamin N, Olsson KM, Meyer K, Vizza CD, Vonk-Noordegraaf A, Distler O, Opitz C, Gibbs JSR, Delcroix M, Ghofrani HA, Huscher D, Pittrow D, Rosenkranz S, Grünig E. Mortality in pulmonary arterial hypertension: prediction by the 2015 European pulmonary hypertension guidelines risk stratification model. The European respiratory journal. 2017 Aug:50(2):. pii: 1700740. doi: 10.1183/13993003.00740-2017. Epub 2017 Aug 3 [PubMed PMID: 28775047]
Kylhammar D, Kjellström B, Hjalmarsson C, Jansson K, Nisell M, Söderberg S, Wikström G, Rådegran G. A comprehensive risk stratification at early follow-up determines prognosis in pulmonary arterial hypertension. European heart journal. 2018 Dec 14:39(47):4175-4181. doi: 10.1093/eurheartj/ehx257. Epub [PubMed PMID: 28575277]
Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habib G, Gressin V, Yaïci A, Weitzenblum E, Cordier JF, Chabot F, Dromer C, Pison C, Reynaud-Gaubert M, Haloun A, Laurent M, Hachulla E, Cottin V, Degano B, Jaïs X, Montani D, Souza R, Simonneau G. Survival in patients with idiopathic, familial, and anorexigen-associated pulmonary arterial hypertension in the modern management era. Circulation. 2010 Jul 13:122(2):156-63. doi: 10.1161/CIRCULATIONAHA.109.911818. Epub 2010 Jun 28 [PubMed PMID: 20585011]
Humbert M, Sitbon O, Yaïci A, Montani D, O'Callaghan DS, Jaïs X, Parent F, Savale L, Natali D, Günther S, Chaouat A, Chabot F, Cordier JF, Habib G, Gressin V, Jing ZC, Souza R, Simonneau G, French Pulmonary Arterial Hypertension Network. Survival in incident and prevalent cohorts of patients with pulmonary arterial hypertension. The European respiratory journal. 2010 Sep:36(3):549-55. doi: 10.1183/09031936.00057010. Epub 2010 Jun 18 [PubMed PMID: 20562126]
Level 2 (mid-level) evidenceBenza RL, Miller DP, Gomberg-Maitland M, Frantz RP, Foreman AJ, Coffey CS, Frost A, Barst RJ, Badesch DB, Elliott CG, Liou TG, McGoon MD. Predicting survival in pulmonary arterial hypertension: insights from the Registry to Evaluate Early and Long-Term Pulmonary Arterial Hypertension Disease Management (REVEAL). Circulation. 2010 Jul 13:122(2):164-72. doi: 10.1161/CIRCULATIONAHA.109.898122. Epub 2010 Jun 28 [PubMed PMID: 20585012]
Benza RL, Gomberg-Maitland M, Miller DP, Frost A, Frantz RP, Foreman AJ, Badesch DB, McGoon MD. The REVEAL Registry risk score calculator in patients newly diagnosed with pulmonary arterial hypertension. Chest. 2012 Feb:141(2):354-362. doi: 10.1378/chest.11-0676. Epub 2011 Jun 16 [PubMed PMID: 21680644]
Benza RL, Miller DP, Foreman AJ, Frost AE, Badesch DB, Benton WW, McGoon MD. Prognostic implications of serial risk score assessments in patients with pulmonary arterial hypertension: a Registry to Evaluate Early and Long-Term Pulmonary Arterial Hypertension Disease Management (REVEAL) analysis. The Journal of heart and lung transplantation : the official publication of the International Society for Heart Transplantation. 2015 Mar:34(3):356-61. doi: 10.1016/j.healun.2014.09.016. Epub 2014 Sep 28 [PubMed PMID: 25447572]
Level 2 (mid-level) evidenceChakinala MM, Coyne DW, Benza RL, Frost AE, McGoon MD, Hartline BK, Frantz RP, Selej M, Zhao C, Mink DR, Farber HW. Impact of declining renal function on outcomes in pulmonary arterial hypertension: A REVEAL registry analysis. The Journal of heart and lung transplantation : the official publication of the International Society for Heart Transplantation. 2018 Jun:37(6):696-705. doi: 10.1016/j.healun.2017.10.028. Epub 2017 Nov 6 [PubMed PMID: 29174533]
Frost AE, Badesch DB, Miller DP, Benza RL, Meltzer LA, McGoon MD. Evaluation of the predictive value of a clinical worsening definition using 2-year outcomes in patients with pulmonary arterial hypertension: a REVEAL Registry analysis. Chest. 2013 Nov:144(5):1521-1529. doi: 10.1378/chest.12-3023. Epub [PubMed PMID: 23907471]
Level 1 (high-level) evidenceBenza RL, Kanwar MK, Raina A, Scott JV, Zhao CL, Selej M, Elliott CG, Farber HW. Development and Validation of an Abridged Version of the REVEAL 2.0 Risk Score Calculator, REVEAL Lite 2, for Use in Patients With Pulmonary Arterial Hypertension. Chest. 2021 Jan:159(1):337-346. doi: 10.1016/j.chest.2020.08.2069. Epub 2020 Sep 1 [PubMed PMID: 32882243]
Level 1 (high-level) evidenceWeatherald J, Sitbon O, Humbert M. Validation of a risk assessment instrument for pulmonary arterial hypertension. European heart journal. 2018 Dec 14:39(47):4182-4185. doi: 10.1093/eurheartj/ehx301. Epub [PubMed PMID: 28637288]
Level 1 (high-level) evidenceSitbon O, Humbert M, Jaïs X, Ioos V, Hamid AM, Provencher S, Garcia G, Parent F, Hervé P, Simonneau G. Long-term response to calcium channel blockers in idiopathic pulmonary arterial hypertension. Circulation. 2005 Jun 14:111(23):3105-11 [PubMed PMID: 15939821]
Level 2 (mid-level) evidenceRich S, Kaufmann E, Levy PS. The effect of high doses of calcium-channel blockers on survival in primary pulmonary hypertension. The New England journal of medicine. 1992 Jul 9:327(2):76-81 [PubMed PMID: 1603139]
Clozel M, Maresta A, Humbert M. Endothelin receptor antagonists. Handbook of experimental pharmacology. 2013:218():199-227. doi: 10.1007/978-3-642-38664-0_9. Epub [PubMed PMID: 24092342]
Level 3 (low-level) evidenceWharton J, Strange JW, Møller GM, Growcott EJ, Ren X, Franklyn AP, Phillips SC, Wilkins MR. Antiproliferative effects of phosphodiesterase type 5 inhibition in human pulmonary artery cells. American journal of respiratory and critical care medicine. 2005 Jul 1:172(1):105-13 [PubMed PMID: 15817798]
Tantini B, Manes A, Fiumana E, Pignatti C, Guarnieri C, Zannoli R, Branzi A, Galié N. Antiproliferative effect of sildenafil on human pulmonary artery smooth muscle cells. Basic research in cardiology. 2005 Mar:100(2):131-8 [PubMed PMID: 15739122]
Ghofrani HA, Voswinckel R, Reichenberger F, Olschewski H, Haredza P, Karadaş B, Schermuly RT, Weissmann N, Seeger W, Grimminger F. Differences in hemodynamic and oxygenation responses to three different phosphodiesterase-5 inhibitors in patients with pulmonary arterial hypertension: a randomized prospective study. Journal of the American College of Cardiology. 2004 Oct 6:44(7):1488-96 [PubMed PMID: 15464333]
Level 1 (high-level) evidenceJones DA, Benjamin CW, Linseman DA. Activation of thromboxane and prostacyclin receptors elicits opposing effects on vascular smooth muscle cell growth and mitogen-activated protein kinase signaling cascades. Molecular pharmacology. 1995 Nov:48(5):890-6 [PubMed PMID: 7476920]
Level 3 (low-level) evidenceDoran AK, Ivy DD, Barst RJ, Hill N, Murali S, Benza RL, Scientific Leadership Council of the Pulmonary Hypertension Association. Guidelines for the prevention of central venous catheter-related blood stream infections with prostanoid therapy for pulmonary arterial hypertension. International journal of clinical practice. Supplement. 2008 Jul:(160):5-9. doi: 10.1111/j.1742-1241.2008.01811.x. Epub [PubMed PMID: 18638170]
Level 1 (high-level) evidenceSimonneau G, Barst RJ, Galie N, Naeije R, Rich S, Bourge RC, Keogh A, Oudiz R, Frost A, Blackburn SD, Crow JW, Rubin LJ, Treprostinil Study Group. Continuous subcutaneous infusion of treprostinil, a prostacyclin analogue, in patients with pulmonary arterial hypertension: a double-blind, randomized, placebo-controlled trial. American journal of respiratory and critical care medicine. 2002 Mar 15:165(6):800-4 [PubMed PMID: 11897647]
Level 1 (high-level) evidenceKhan MS, Memon MM, Amin E, Yamani N, Khan SU, Figueredo VM, Deo S, Rich JD, Benza RL, Krasuski RA. Use of Balloon Atrial Septostomy in Patients With Advanced Pulmonary Arterial Hypertension: A Systematic Review and Meta-Analysis. Chest. 2019 Jul:156(1):53-63. doi: 10.1016/j.chest.2019.03.003. Epub 2019 Mar 23 [PubMed PMID: 30910639]
Level 1 (high-level) evidenceSandoval J, Gaspar J, Pulido T, Bautista E, Martínez-Guerra ML, Zeballos M, Palomar A, Gómez A. Graded balloon dilation atrial septostomy in severe primary pulmonary hypertension. A therapeutic alternative for patients nonresponsive to vasodilator treatment. Journal of the American College of Cardiology. 1998 Aug:32(2):297-304 [PubMed PMID: 9708453]
Rothman AMK, Vachiery JL, Howard LS, Mikhail GW, Lang IM, Jonas M, Kiely DG, Shav D, Shabtay O, Avriel A, Lewis GD, Rosenzweig EB, Kirtane AJ, Kim NH, Mahmud E, McLaughlain VV, Chetcuti S, Leon MB, Ben-Yehuda O, Rubin LJ. Intravascular Ultrasound Pulmonary Artery Denervation to Treat Pulmonary Arterial Hypertension (TROPHY1): Multicenter, Early Feasibility Study. JACC. Cardiovascular interventions. 2020 Apr 27:13(8):989-999. doi: 10.1016/j.jcin.2019.12.027. Epub [PubMed PMID: 32327095]
Level 2 (mid-level) evidenceChen SL, Zhang FF, Xu J, Xie DJ, Zhou L, Nguyen T, Stone GW. Pulmonary artery denervation to treat pulmonary arterial hypertension: the single-center, prospective, first-in-man PADN-1 study (first-in-man pulmonary artery denervation for treatment of pulmonary artery hypertension). Journal of the American College of Cardiology. 2013 Sep 17:62(12):1092-1100. doi: 10.1016/j.jacc.2013.05.075. Epub 2013 Jul 10 [PubMed PMID: 23850902]
Rothman A, Jonas M, Castel D, Tzafriri AR, Traxler H, Shav D, Leon MB, Ben-Yehuda O, Rubin L. Pulmonary artery denervation using catheter-based ultrasonic energy. EuroIntervention : journal of EuroPCR in collaboration with the Working Group on Interventional Cardiology of the European Society of Cardiology. 2019 Oct 20:15(8):722-730. doi: 10.4244/EIJ-D-18-01082. Epub [PubMed PMID: 31062694]
Ciarka A, Doan V, Velez-Roa S, Naeije R, van de Borne P. Prognostic significance of sympathetic nervous system activation in pulmonary arterial hypertension. American journal of respiratory and critical care medicine. 2010 Jun 1:181(11):1269-75. doi: 10.1164/rccm.200912-1856OC. Epub 2010 Mar 1 [PubMed PMID: 20194810]
Velez-Roa S, Ciarka A, Najem B, Vachiery JL, Naeije R, van de Borne P. Increased sympathetic nerve activity in pulmonary artery hypertension. Circulation. 2004 Sep 7:110(10):1308-12 [PubMed PMID: 15337703]
Level 1 (high-level) evidenceJuratsch CE, Jengo JA, Castagna J, Laks MM. Experimental pulmonary hypertension produced by surgical and chemical denervation of the pulmonary vasculature. Chest. 1980 Apr:77(4):525-30 [PubMed PMID: 7357977]
Level 3 (low-level) evidenceChristie JD, Edwards LB, Kucheryavaya AY, Benden C, Dipchand AI, Dobbels F, Kirk R, Rahmel AO, Stehlik J, Hertz MI, International Society of Heart and Lung Transplantation. The Registry of the International Society for Heart and Lung Transplantation: 29th adult lung and heart-lung transplant report-2012. The Journal of heart and lung transplantation : the official publication of the International Society for Heart Transplantation. 2012 Oct:31(10):1073-86. doi: 10.1016/j.healun.2012.08.004. Epub [PubMed PMID: 22975097]
Yusen RD, Edwards LB, Kucheryavaya AY, Benden C, Dipchand AI, Goldfarb SB, Levvey BJ, Lund LH, Meiser B, Rossano JW, Stehlik J. The Registry of the International Society for Heart and Lung Transplantation: Thirty-second Official Adult Lung and Heart-Lung Transplantation Report--2015; Focus Theme: Early Graft Failure. The Journal of heart and lung transplantation : the official publication of the International Society for Heart Transplantation. 2015 Oct:34(10):1264-77. doi: 10.1016/j.healun.2015.08.014. Epub 2015 Sep 3 [PubMed PMID: 26454740]
Olsson KM, Hoeper MM, Pausch C, Grünig E, Huscher D, Pittrow D, Rosenkranz S, Gall H. Pulmonary vascular resistance predicts mortality in patients with pulmonary hypertension associated with interstitial lung disease: results from the COMPERA registry. The European respiratory journal. 2021 Aug:58(2):. pii: 2101483. doi: 10.1183/13993003.01483-2021. Epub 2021 Aug 26 [PubMed PMID: 34385266]
Zeder K, Avian A, Bachmaier G, Douschan P, Foris V, Sassmann T, Troester N, Brcic L, Fuchsjaeger M, Marsh LM, Maron BA, Olschewski H, Kovacs G. Elevated pulmonary vascular resistance predicts mortality in COPD patients. The European respiratory journal. 2021 Aug:58(2):. pii: 2100944. doi: 10.1183/13993003.00944-2021. Epub 2021 Aug 26 [PubMed PMID: 33986032]
Chang KY, Duval S, Badesch DB, Bull TM, Chakinala MM, De Marco T, Frantz RP, Hemnes A, Mathai SC, Rosenzweig EB, Ryan JJ, Thenappan T, PHAR Investigators *. Mortality in Pulmonary Arterial Hypertension in the Modern Era: Early Insights From the Pulmonary Hypertension Association Registry. Journal of the American Heart Association. 2022 May 3:11(9):e024969. doi: 10.1161/JAHA.121.024969. Epub 2022 Apr 27 [PubMed PMID: 35475351]
Vizza CD, Hoeper MM, Huscher D, Pittrow D, Benjamin N, Olsson KM, Ghofrani HA, Held M, Klose H, Lange T, Rosenkranz S, Dumitrescu D, Badagliacca R, Claussen M, Halank M, Vonk-Noordegraaf A, Skowasch D, Ewert R, Gibbs JSR, Delcroix M, Skride A, Coghlan G, Ulrich S, Opitz C, Kaemmerer H, Distler O, Grünig E. Pulmonary Hypertension in Patients With COPD: Results From the Comparative, Prospective Registry of Newly Initiated Therapies for Pulmonary Hypertension (COMPERA). Chest. 2021 Aug:160(2):678-689. doi: 10.1016/j.chest.2021.02.012. Epub 2021 Feb 11 [PubMed PMID: 33581097]
Level 2 (mid-level) evidenceSaunders H, Helgeson SA, Abdelrahim A, Rottman-Pietrzak K, Reams V, Zeiger TK, Moss JE, Burger CD. Comparing Diagnosis and Treatment of Pulmonary Hypertension Patients at a Pulmonary Hypertension Center versus Community Centers. Diseases (Basel, Switzerland). 2022 Jan 7:10(1):. doi: 10.3390/diseases10010005. Epub 2022 Jan 7 [PubMed PMID: 35076491]