Introduction
Pseudohypoparathyroidism (PHP) is a rare inherited disorder characterized by target organ resistance or unresponsiveness to parathyroid hormone (PTH). The syndrome mimics hypoparathyroidism with patients experiencing hypocalcemia and hyperphosphatemia. However, instead of having low PTH levels, elevated levels of PTH are present in serum. PHP is typically classified as either type 1 or type 2, and then type 1 is further subdivided into 1a, 1b, or 1c. Type 1 is distinguishable from type 2 by the abnormal cAMP response to G protein activation seen in type 1, whereas the cAMP response is normal in type 2. PHP 1a and 1c both can exhibit multi-hormone resistance, whereas 1b is localized only to the kidney. Of all the subtypes of PHP, type 1a is thought to be the most common subtype, but PHP type 1b has recently been found to occur with similar frequency.
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
The first consensus on diagnosing and managing pseudohypoparathyroidism was published in 2018 after agreed-upon experts were invited from several countries worldwide.[1] Pseudohypoparathyroidism is subdivided into several categories. It first divides into PHP type 1 and PHP type 2. In general, PHP type 1 forms exhibit blunted cAMP and phosphate excretion in urine in response to PTH (cAMP being a downstream response to activation of G protein-coupled receptor for PTH). In PHP type 2, the cAMP response is normal but blunted phosphate excretion in response to PTH still occurs. Only a few cases of PHP type 2 have been documented, and its pathophysiology remains poorly understood.
PHP type 1 is further divided into several subtypes a,b, and c. PHP type 1b is isolated resistance to PTH in the kidney and normal PTH response in bone without phenotypic expression of Albright hereditary osteodystrophy (AHO). Novel defects are described in a recent study, and more information is being revealed as we have a better analysis of the genome in recent years.[2][3][4][5] Deletion of STX16 in the maternal gene has been shown to cause PTH resistance and progression to be autosomal dominant pseudohypoparathyroidism type 1b.[6] PHP 1a and 1c have PTH resistance, and both express a phenotype of AHO. AHO patients present with short stature, frequently below normal intelligence, obesity, round face, short neck, subcutaneous ossifications, brachydactyly, and shortened metatarsals. The shortened metacarpals often cause an absence of knuckles at the third, fourth and fifth or only some of those digits resulting in the classic "knuckle, knuckle, dimple, knuckle" sign.
PHP type 1a has reduced Gs alpha activity and presents with multi-hormone resistance.
PHP type 1c, much like 1a, presents with multi-hormone resistance, but it is different from type 1a in that normal Gs alpha activity is present.[7]
Epidemiology
Pseudohypoparathyroidism and Albright hereditary osteodystrophy are very rare disorders. The estimated prevalence is between 0.3 and 1.1 cases per 100000 population, depending on geographic location.[1][8]
Pathophysiology
PHP type 1a results when there is a loss of function mutation in GNAS, PRKAR1A, PDE4D, or PDE3A.[9][10] The result is an aberrant response in many G protein-coupled receptors leading to multi-hormone resistance or unresponsiveness. Thus far, over 400 mutations within the GNAS coding region have been identified as causes of PHP 1a.[11] The mutations identified include frameshift, nonsense, missense, insertion, and deletions, as well as others. Of note, research has discovered a temperature-sensitive Gs alpha mutant (p.A366S) that can cause testotoxicosis and precocious puberty.[12]
In addition to PTH resistance, PHP type 1a infrequently exhibits blunted response to thyroid-stimulating hormone (TSH), luteinizing hormone (LH), follicle-stimulating hormone (FSH), and growth hormone-releasing hormone (GHRH) resulting in primary hypothyroidism, primary hypogonadism, and short stature, depending on the affected gene.
The inheritance of GNAS mutation is fairly complex due to paternal genetic imprinting. Individuals who acquire the mutant GNAS allele from their father will only have the phenotypic expression of AHO (termed pseudopseudohypoparathyroidism) and do not have PTH resistance. Individuals who inherit the mutant allele from their mother express both AHO and multi-hormone resistance.
PHP type 1b results from abnormal methylation of GNAS regulatory elements. The abnormal methylation leads to subnormal production of Gs alpha protein expression and a blunted response to PTH.
History and Physical
Pseudohypoparathyroidism typically gets discovered during early childhood. There may or may not be an obvious family history of the disease depending on which parent the child inherited the disease from, as genetic imprinting can hide its expression. The presence of the AHO phenotype should prompt physicians to complete biochemical workup.
Patients experiencing symptoms of hypocalcemia may complain of paresthesias, peri-oral numbness, and muscle cramping or spasms.
A physical exam for PHP type 1b would likely only find symptomatic hypocalcemia. Hypocalcemia can present with a positive Chvostek sign and a positive Trousseau sign.
The Chovostek sign is positive when tapping on the facial nerve leads to the contraction of facial muscles on the same side due to the nerve's hyperexcitable state. A positive Trousseau sign is when inflation of a sphygmomanometer to higher than systolic blood pressure and maintaining that pressure for 3 minutes leads to transient ischemia and carpal pedal spasm due to hypocalcemia.[13]
In rare cases, the patient might present with seizures, and special imaging of the brain will be needed to rule out any cerebral pathology.[14]
In a recent case report, there was a finding of hypokalemia as the presenting sign of pseudohypoparathyroidism.[15]
PHP type 1a and 1c may have signs of hypocalcemia but will also exhibit the AHO phenotype.
Evaluation
Serum calcium, phosphorus, and parathyroid hormone levels should be checked together as well as 25OH Vitamin D levels. The presence of hypocalcemia, hyperphosphatemia, normal 25 hydroxyvitamin D, and elevated parathyroid hormone levels suggest pseudohypoparathyroidism. A synthetic PTH challenge test can be performed (Ellsworth-Howard test) but is not necessary for diagnosis.
An electrocardiogram is likely appropriate if significant hypocalcemia is present to assess for prolonged QT interval and risk for arrhythmia. PHP1 patients should have biochemical testing completed annually, including PTH, calcium, phosphate, TSH, and urine calcium.[1]
Monitoring of appropriate height and growth is important, and testing for growth hormone deficiency should take place even if the height is normal.[16] Screening and treatment for other endocrinopathies, such as hypogonadism, should be individualized.[17]
X-rays of the upper and lower extremities are recommended during the initial evaluation to screen for brachydactyly or other bone malformations.[18][19][20]
In patients with PHP with AHO, genetic testing is essential. Genetic testing and counseling provide information regarding likely disease manifestations and future complications. Additionally, genetic testing can help determine family inheritance patterns and provide insight into the possibility of affected offspring.
Treatment / Management
The overall goals in the treatment of PHP are to maintain normal calcium and normal phosphorus, avoid hypercalciuria and lower PTH levels to normal if possible. It is typically not possible to achieve all electrolyte and hormone level goals; however, getting them as close as possible is recommended.
Calcium and phosphorus goals are usually attainable with the administration of calcitriol (active vitamin D metabolite) with or without calcium supplementation. Dosing is adjusted so that calcium remains at the low end of the normal range. In addition to checking serum calcium, 24-hour urine calcium levels should be maintained in the low to mid-normal range to avoid renal calculi and impaired kidney function.
In patients with persistent hypocalcemia, treatment with calcium supplementation is a requirement.
In the acute setting, the recommendation is for intravenous calcium Patients who are symptomatic (spasms, seizure, etc.), those with prolonged QT interval on electrocardiogram, or if corrected calcium is less than 7.5mg/dL, and the patient is considered to be at high risk should receive IV calcium infusion. Initially, 1 to 2 g of calcium gluconate administration should be over a 10 to 20-minute interval followed by a slow infusion of calcium. The slow infusion can be achieved by diluting 1000 mg elemental calcium into 1 L of 5% dextrose or normal saline and running at a rate of around 1 cc/kg/hr. Oral calcium and vitamin D supplementation should start as soon as possible. Oral calcium carbonate 2-8g elemental calcium/day and oral calcitriol 0.25 to 1.0 mcg twice a day are usually sufficient doses and allow calcium infusion to stop within a few hours.
These patients will require permanent active vitamin D supplementation with or without calcium. Calcitriol 0.25 to 1.0 mcg/day should be used (not other forms of vitamin D due to decreased 1 alpha-hydroxylase activity, which converts vitamin D to an active form). Calcium carbonate or calcium citrate should be given 2 to 8 g elemental calcium daily in divided doses to keep calcium at the low end of normal.[21]
PTH levels require monitoring during treatment with the goal of reduction into the normal range or as close as possible. Lowering PTH attempts to minimize skeletal resorption and later development of osteoporosis.[7]
Treatment with parathyroid hormone or parathyroid hormone analogs is not recommended.
Differential Diagnosis
Pseudohypoparathyroidism presents with hypocalcemia, hyperphosphatemia, elevated PTH levels, normal 25 hydroxyvitamin D, and normal kidney function. Additionally, many patients express the phenotype of Albright hereditary osteodystrophy, but not always. Although no other diseases present identically, many conditions share similar characteristics providing the following differential diagnoses.
- True hypoparathyroidism
- It presents with hypocalcemia and hyperphosphatemia, but the PTH levels are low. True hypoparathyroidism typically classifies as either surgical or non-surgical. The most frequent cause of hypoparathyroidism is surgical, post-parathyroidectomy for treatment of hyperparathyroidism or accidental removal of parathyroid glands during other neck surgeries such as thyroidectomy. Non-surgical hypoparathyroidism can result from a variety of inherited and acquired disorders. Examples include hemochromatosis or other infiltrative diseases and mutations in the PTH gene, CaSR gene, and autoimmune polyendocrine syndrome type 1 (APS1), which presents with the triad of hypoparathyroidism, primary adrenal insufficiency, and mucocutaneous candidiasis.
- Vitamin D deficiency
- This condition presents with hypocalcemia and elevated PTH levels (secondary hyperparathyroidism) but obviously low 25 OH vitamin D levels.
- Worsening chronic kidney disease (CKD)
- Patients with impaired glomerular filtration rate (GFR) present with similar findings. The kidney is the primary site for 1-alpha-hydroxylation of 25 OH vitamin D which produces the active form of the hormone 1,25(OH)2 vitamin D. The impaired conversion to active vitamin D leads to its low levels in patients with CKD. In addition to low active vitamin D levels, higher than normal phosphate frequently accumulates in the blood due to declining GFR. The low active vitamin D levels and high phosphate levels lead to increased parathyroid hormone release.
Prognosis
The prognosis of PHP is variable. In mild forms of the disease, when treated appropriately with calcium and vitamin D, a normal life expectancy is not unreasonable.[8] For others with a more severe expression of the AHO phenotype, the presence of obesity, sleep apnea, and mental retardation can cause significant morbidity and mortality.
Complications
- Chronic hypocalcemia and hyperphosphatemia can lead to parkinsonism which may or may not resolve with appropriate calcium and vitamin D supplementation.[22]
- Children with brachydactyly may suffer from difficulty with fine motor skills.
- There is an increased prevalence of carpal tunnel syndrome seen in AHO patients.[23]
- There are reports of higher than normal, as well as an earlier presentation of spinal stenosis, which can lead to lower extremity paraparesis.[24][25]
- Finally, there is a more than four-fold increased risk for sleep apnea reported in childhood.[26]
Deterrence and Patient Education
Unfortunately, PHP is an inherited disorder, and once an individual has developed the condition, there is no cure. Genetic testing and counseling are recommendations for individuals with PHP, especially if they plan to have children, so they can fully identify their risk of passing on the disease. Patients with PHP should have yearly screening exams, even if they feel well.
Enhancing Healthcare Team Outcomes
Patients with pseudohypoparathyroidism should follow up with an endocrinologist. Regular testing of serum calcium levels is necessary, as well as screening for other hormone resistance. Each patient's care requires individualization to their specific expression of hormone resistance.
Diagnosing, treating, and managing pseudohypoparathyroidism requires an interprofessional healthcare team approach. The patient's family doctor will enlist the services of an endocrinologist, as stated above. A specialist in genetics is also a necessary consult. The involvement of other healthcare team members will vary based on individual patient presentation. If IV interventions are required, the nurse will administer those and monitor the effectiveness of treatment, reporting their findings to the treating clinician. The pharmacist will prepare the IV and, in cases of oral therapy, can counsel the patient on dosing and administration. As treatment progresses, the team must be able to adapt based on patient response and coordinate with the next steps as described above in the Treatment section. With open communication and collaborative effort, the interprofessional team can help drive outcomes to the best possible result for the patient. [Level 5]
Media
(Click Image to Enlarge)
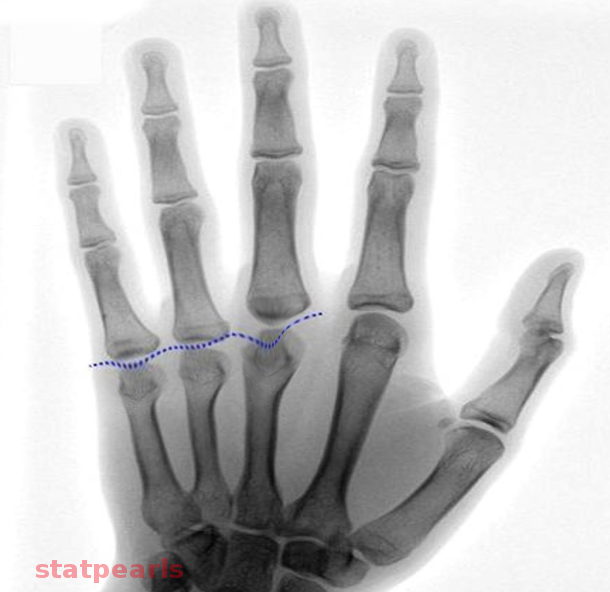
Pseudohypoparathyroidism Image courtesy O.Chaigasame
References
Mantovani G, Bastepe M, Monk D, de Sanctis L, Thiele S, Usardi A, Ahmed SF, Bufo R, Choplin T, De Filippo G, Devernois G, Eggermann T, Elli FM, Freson K, García Ramirez A, Germain-Lee EL, Groussin L, Hamdy N, Hanna P, Hiort O, Jüppner H, Kamenický P, Knight N, Kottler ML, Le Norcy E, Lecumberri B, Levine MA, Mäkitie O, Martin R, Martos-Moreno GÁ, Minagawa M, Murray P, Pereda A, Pignolo R, Rejnmark L, Rodado R, Rothenbuhler A, Saraff V, Shoemaker AH, Shore EM, Silve C, Turan S, Woods P, Zillikens MC, Perez de Nanclares G, Linglart A. Diagnosis and management of pseudohypoparathyroidism and related disorders: first international Consensus Statement. Nature reviews. Endocrinology. 2018 Aug:14(8):476-500. doi: 10.1038/s41574-018-0042-0. Epub [PubMed PMID: 29959430]
Level 3 (low-level) evidenceDanzig J, Li D, Jan de Beur S, Levine MA. High-throughput Molecular Analysis of Pseudohypoparathyroidism 1b Patients Reveals Novel Genetic and Epigenetic Defects. The Journal of clinical endocrinology and metabolism. 2021 Oct 21:106(11):e4603-e4620. doi: 10.1210/clinem/dgab460. Epub [PubMed PMID: 34157100]
Milioto A, Reyes M, Hanna P, Kiuchi Z, Turan S, Zeve D, Agarwal C, Grigelioniene G, Chen A, Mericq V, Frangos M, Ten S, Mantovani G, Salusky IB, Tebben P, Jüppner H. Lack of GNAS Remethylation During Oogenesis May Be a Cause of Sporadic Pseudohypoparathyroidism Type Ib. The Journal of clinical endocrinology and metabolism. 2022 Mar 24:107(4):e1610-e1619. doi: 10.1210/clinem/dgab830. Epub [PubMed PMID: 34791361]
Kiuchi Z, Reyes M, Jüppner H. Preferential Maternal Transmission of STX16-GNAS Mutations Responsible for Autosomal Dominant Pseudohypoparathyroidism Type Ib (PHP1B): Another Example of Transmission Ratio Distortion. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2021 Apr:36(4):696-703. doi: 10.1002/jbmr.4221. Epub 2020 Dec 27 [PubMed PMID: 33247854]
Reyes M, Kagami M, Kawashima S, Pallotta J, Schnabel D, Fukami M, Jüppner H. A Novel GNAS Duplication Associated With Loss-of-Methylation Restricted to Exon A/B Causes Pseudohypoparathyroidism Type Ib (PHP1B). Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2021 Mar:36(3):546-552. doi: 10.1002/jbmr.4209. Epub 2020 Nov 28 [PubMed PMID: 33180333]
Kiuchi Z, Reyes M, Hanna P, Sharma A, DeClue T, Olney RC, Tebben P, Jüppner H. Progression of PTH Resistance in Autosomal Dominant Pseudohypoparathyroidism Type Ib Due to Maternal STX16 Deletions. The Journal of clinical endocrinology and metabolism. 2022 Jan 18:107(2):e681-e687. doi: 10.1210/clinem/dgab660. Epub [PubMed PMID: 34477200]
Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, de Herder WW, Dhatariya K, Dungan K, Hofland J, Kalra S, Kaltsas G, Kapoor N, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrère B, Levy M, McGee EA, McLachlan R, New M, Purnell J, Sahay R, Shah AS, Singer F, Sperling MA, Stratakis CA, Trence DL, Wilson DP, Hendy GN, Cole DEC, Bastepe M. Hypoparathyroidism and Pseudohypoparathyroidism. Endotext. 2000:(): [PubMed PMID: 25905388]
Underbjerg L, Sikjaer T, Mosekilde L, Rejnmark L. Pseudohypoparathyroidism - epidemiology, mortality and risk of complications. Clinical endocrinology. 2016 Jun:84(6):904-11. doi: 10.1111/cen.12948. Epub 2015 Oct 19 [PubMed PMID: 26387561]
Mantovani G, Elli FM. Inactivating PTH/PTHrP Signaling Disorders. Frontiers of hormone research. 2019:51():147-159. doi: 10.1159/000491045. Epub 2018 Nov 19 [PubMed PMID: 30641531]
Jüppner H. Molecular Definition of Pseudohypoparathyroidism Variants. The Journal of clinical endocrinology and metabolism. 2021 May 13:106(6):1541-1552. doi: 10.1210/clinem/dgab060. Epub [PubMed PMID: 33529330]
Long XD, Xiong J, Mo ZH, Dong CS, Jin P. Identification of a novel GNAS mutation in a case of pseudohypoparathyroidism type 1A with normocalcemia. BMC medical genetics. 2018 Jul 30:19(1):132. doi: 10.1186/s12881-018-0648-z. Epub 2018 Jul 30 [PubMed PMID: 30060753]
Level 3 (low-level) evidenceNakamoto JM, Zimmerman D, Jones EA, Loke KY, Siddiq K, Donlan MA, Brickman AS, Van Dop C. Concurrent hormone resistance (pseudohypoparathyroidism type Ia) and hormone independence (testotoxicosis) caused by a unique mutation in the G alpha s gene. Biochemical and molecular medicine. 1996 Jun:58(1):18-24 [PubMed PMID: 8809352]
Level 3 (low-level) evidenceMazoni L, Apicella M, Saponaro F, Mantovani G, Elli FM, Borsari S, Pardi E, Piaggi P, Marcocci C, Cetani F. Pseudohypoparathyroidism: Focus on Cerebral and Renal Calcifications. The Journal of clinical endocrinology and metabolism. 2021 Jul 13:106(8):e3005-e3020. doi: 10.1210/clinem/dgab208. Epub [PubMed PMID: 33780542]
Qi Z, Li Z, Gao Q, Dong L, Lin J, Peng K, Xiang W, Deng B. Characterizing Cerebral Imaging and Electroclinical Features of Five Pseudohypoparathyroidism Cases Presenting with Epileptic Seizures. Behavioural neurology. 2022:2022():8710989. doi: 10.1155/2022/8710989. Epub 2022 Aug 12 [PubMed PMID: 35992960]
Level 2 (mid-level) evidenceHuang S, He Y, Lin X, Sun S, Zheng F. Clinical and genetic analysis of pseudohypoparathyroidism complicated by hypokalemia: a case report and review of the literature. BMC endocrine disorders. 2022 Apr 11:22(1):98. doi: 10.1186/s12902-022-01011-9. Epub 2022 Apr 11 [PubMed PMID: 35410271]
Level 3 (low-level) evidenceGermain-Lee EL. Management of pseudohypoparathyroidism. Current opinion in pediatrics. 2019 Aug:31(4):537-549. doi: 10.1097/MOP.0000000000000783. Epub [PubMed PMID: 31145125]
Level 3 (low-level) evidenceJun JE, Park SY, Jeong IK, Hwang YC, Ahn KJ, Chung HY. Delayed diagnosis of pseudohypoparathyroidism type 1a with rare hypothyroidism since childhood. Oxford medical case reports. 2022 Aug:2022(8):omac080. doi: 10.1093/omcr/omac080. Epub 2022 Aug 18 [PubMed PMID: 35991493]
Level 3 (low-level) evidenceElli FM, Mantovani G. Pseudohypoparathyroidism, acrodysostosis, progressive osseous heteroplasia: different names for the same spectrum of diseases? Endocrine. 2021 Jun:72(3):611-618. doi: 10.1007/s12020-020-02533-9. Epub 2020 Nov 11 [PubMed PMID: 33179219]
Zhang J, Guan M, Zhao S, Wu S, Weng L, Sheng W. A patient with pseudohypoparathyroidism type 1A previously misdiagnosed as hereditary multiple exostosis: A case report. Experimental and therapeutic medicine. 2022 Sep:24(3):597. doi: 10.3892/etm.2022.11534. Epub 2022 Jul 28 [PubMed PMID: 35949342]
Level 3 (low-level) evidenceMatsuura N, Kaname T, Niikawa N, Ooyama Y, Shinohara O, Yokota Y, Ohtsu S, Takubo N, Kitsuda K, Shibayama K, Takada F, Koike A, Sano H, Ito Y, Ishikura K. Acrodysostosis and pseudohypoparathyroidism (PHP): adaptation of Japanese patients with a newly proposed classification and expanding the phenotypic spectrum of variants. Endocrine connections. 2022 Oct 1:11(10):. doi: 10.1530/EC-22-0151. Epub 2022 Sep 22 [PubMed PMID: 36006853]
Antoun J, Williamson D, Hubler M, Shoemaker AH. Calcitriol and Levothyroxine Dosing for Patients With Pseudohypoparathyroidism. Journal of the Endocrine Society. 2021 Dec 1:5(12):bvab161. doi: 10.1210/jendso/bvab161. Epub 2021 Oct 27 [PubMed PMID: 34765856]
Kim YS, Park J, Park Y, Hwang K, Koo DL, Kim D, Seo DW. Intracranial Cortical Calcifications in a Focal Epilepsy Patient with Pseudohypoparathyroidism. Journal of epilepsy research. 2016 Jun:6(1):31-5. doi: 10.14581/jer.16006. Epub 2016 Jun 30 [PubMed PMID: 27390678]
Shoemaker AH, Jüppner H. Nonclassic features of pseudohypoparathyroidism type 1A. Current opinion in endocrinology, diabetes, and obesity. 2017 Feb:24(1):33-38. doi: 10.1097/MED.0000000000000306. Epub [PubMed PMID: 27875418]
Level 3 (low-level) evidencevan Lindert EJ, Bartels RH, Noordam K. Spinal stenosis with paraparesis in albright hereditary osteodystrophy. Case report and review of the literature. Pediatric neurosurgery. 2008:44(4):337-40. doi: 10.1159/000138373. Epub 2008 Jun 13 [PubMed PMID: 18552518]
Level 3 (low-level) evidenceAlam SM, Kelly W. Spinal cord compression associated with pseudohypoparathyroidism. Journal of the Royal Society of Medicine. 1990 Jan:83(1):50-1 [PubMed PMID: 2304055]
Level 3 (low-level) evidenceLandreth H, Malow BA, Shoemaker AH. Increased Prevalence of Sleep Apnea in Children with Pseudohypoparathyroidism Type 1a. Hormone research in paediatrics. 2015:84(1):1-5. doi: 10.1159/000381452. Epub 2015 Apr 23 [PubMed PMID: 25925491]