Introduction
Progressive supranuclear palsy (PSP) is an uncommon neurological disorder that affects movement, gait, balance, speech, swallowing, vision, eye movements, mood, behavior, and cognition. Steele, Richardson, and Olszewski described the syndrome in 1964 as an unusual constellation of supranuclear gaze palsy, progressive axial rigidity, pseudobulbar palsy, and mild dementia.[1] This disease is now a well-recognized atypical parkinsonian syndrome (or Parkinson-plus disorder).
In 1972, Steele predicted clinical variants of the syndrome were likely to occur as the disease affected different brainstem nuclei at different times and to different degrees.[2] Since then, different phenotypes have been characterized and linked to the severity of abnormal tau accumulation and neuronal loss in various brain regions.[3] Different progressive supranuclear palsies, regardless of clinical characteristics, share similar neuropathologic features.[4]
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
The cause of progressive supranuclear palsy is unknown. Advanced age [5] and environmental factors such as exposure to toxins are theorized causes. [6][7] The tau protein aggregates may be due to an unconventional infectious agent, random genetic mutations, or some unknown chemical in the food, air, or water which slowly damages certain vulnerable areas of the brain.
Epidemiology
Recent studies have reported the prevalence of progressive supranuclear palsy to be 5.8 to 6.5 per 100,000.[5][8][9] The annual incidence rate of progressive supranuclear palsy ranged from 0.3 to 0.4 per 100,000 in earlier studies.[10][11][12]. However, a study published in 1999 reported an annual incidence rate of 1.1 per 100,000.[13][14] The greater incidence found in the latter study is likely due to better case ascertainment with increased recognition of the disorder and its clinical variants.[13] The annual incidence increases with age from 1.7 cases per 100,000 at ages 50 to 59 years to 14.7 per 100,000 at 80 to 89 years.[14]. The mean age of onset for progressive supranuclear palsy is approximately 65 years [15][16][17], which is older than in idiopathic Parkinson disease. Virtually no cases of autopsy-confirmed progressive supranuclear palsy have been reported in patients younger than age 40 years.[18]. The original report noted a strong male predominance of approximately 8 to 1.[1] However, later reports have found no clear gender predominance in progressive supranuclear palsy.[5][19][20][21]
Pathophysiology
The defining histopathologic feature of progressive supranuclear palsy is an intracerebral aggregation of the microtubule-associated protein tau with preferential involvement of the subthalamic nucleus, pallidum, striatum, red nucleus, substantia nigra, pontine tegmentum, oculomotor nucleus, medulla, and dentate nucleus.[3] The aggregates predominantly contain tau isoforms with four microtubule-binding repeats (4R-tau) in neurofibrillary tangles, oligodendrocytic coils, and astrocytic tufts.[4][22][23] Normally, tau is phosphorylated on a series of serine and threonine residues, regulated by numerous kinases and phosphatases. In progressive supranuclear palsy and other tauopathies, the tau protein is hyperphosphorylated, which causes it to lose its affinity for microtubules and become resistant to proteolysis. This results in the accumulation of tau and the formation of neurofibrillary tangles.[24] Definite diagnosis of progressive supranuclear palsy currently requires neuropathological examination.[4][25]
Macroscopic Pathology
The gross examination of the brain in progressive supranuclear palsy often shows atrophy of the frontal lobe (especially the precentral gyrus), midbrain (especially the tectum), and to a lesser degree, the pons. The subthalamic nucleus is smaller than expected and maybe discolored gray.[25] The superior cerebellar peduncle and the hilum of the cerebellar dentate nucleus are usually atrophic and have a gray discoloration due to myelinated fiber loss.
History and Physical
Progressive supranuclear palsy is an akinetic-rigid form of parkinsonism. The onset of the disease is insidious and symmetric in most patients. Early in the course of the disease, motor abnormalities are typically axial and not appendicular. Gait difficulty and falls are the most common initial manifestation. Other modes of onset include non-specific dizziness, generalized motor slowing, and personality change. Only occasionally, the onset may be resting tremor.[26] As the disease progresses, other neurologic manifestations occur, including worsening parkinsonism, dysarthria, dysphagia, frontal cognitive difficulties, and eye movement abnormalities. Although supranuclear ophthalmoplegia is the hallmark of progressive supranuclear palsy, some patients only manifest this late in the progression of the disease. Slowed vertical saccades may be the only eye movement manifestation early on. There are a number of clinical subtypes of progressive supranuclear palsy, including classic progressive supranuclear palsy-Richardson syndrome (PSP-RS), progressive supranuclear palsy-parkinsonism (PSP-P), progressive supranuclear palsy-pure akinesia with gait freezing (PSP-PAGF), progressive supranuclear palsy-corticobasal syndrome (PSP-CBS), progressive supranuclear palsy-behavioral variant of frontotemporal dementia (PSP-bvFTD) and progressive supranuclear palsy-progressive non-fluent aphasia (PSP-PNFA).[3]
Postural Instability and Falls
Patients with classic progressive supranuclear palsy, PSP-RS, have a stiff and broad-based gait, with an inclination to have their knees and trunk extended (as opposed to the flexed posture of idiopathic Parkinson disease), and arms slightly abducted. Instead of turning en bloc, as seen in Parkinson disease, they tend to pivot quickly, which further increases their risk of falls. This is sometimes referred to as the "drunken sailor gait." When they fall, it is usually backward due to a lack of postural reflexes.
Oculomotor Findings
Although extraocular movement abnormalities, specifically supranuclear ophthalmoplegia, are the hallmark of progressive supranuclear palsy, only a minority of patients present with gaze palsy. Often the first ocular motility abnormality noted on physical examination is impairment of vertical saccades. Eventually, most patients with progressive supranuclear palsy show slow or absent vertical saccades.[27][28] Typically, downgaze is affected before the upgaze. Horizontal, visually guided saccades .are also often impaired.[29] Symptomatic eye movement abnormalities begin at a median of 4 years after disease onset.[27][28][27] Other oculomotor findings in progressive supranuclear palsy include saccadic intrusions into fixation ("square wave jerks"), loss of optokinetic nystagmus (particularly in the vertical direction), loss of convergence, blepharospasm, and eyelid-opening apraxia. The patient is often unable to suppress the horizontal vestibulo-ocular reflex.[28] The vestibulo-ocular reflex is present until late in the disease, but with disease progression and brainstem involvement, vestibulo-ocular reflexes may be lost. Eventually, supranuclear gaze palsy may be present in all directions, and toward the end of the disease, the eyes may be virtually immobile.
The combination of rare blinking, facial dystonia, and gaze abnormalities leads to the development of a classic facial expression of perpetual surprise or astonishment.[30] Vertical gaze impairment commonly leads to reading problems, spilling food while eating, and contributing to falls while walking.[31]
Motor Involvement
Bradykinesia, with marked micrographia, is the primary feature of parkinsonism present in all types of progressive supranuclear palsy. Rigidity in patients with progressive supranuclear palsy is usually more apparent in axial than limb muscles, especially the neck and upper trunk. On examination, it can be demonstrated by resistance to passive movement of the neck.
There can be dystonia of the neck, typically retrocollis. The face of patients with progressive supranuclear palsy is often stiff, immobile, and deeply furrowed (the look of surprise) also due to dystonia.[21] About one-third of patients with progressive supranuclear palsy develop pyramidal signs, including hyperreflexia and Babinski signs. A jaw jerk and other frontal release signs can be present. Spastic dysarthria, dysphonia, and dysphagia can be profound in the middle to later stages of the disease.
Cognitive and Behavioral Abnormalities
The neuropsychological profile of progressive supranuclear palsy primarily involves frontal lobe dysfunction. This includes impaired executive function making it difficult to shift between mental tasks,[32], and spontaneous motor behaviors, such as palilalia, motor perseveration, compulsive spitting, among others.
The presence of severe frontal cognitive deficits is a common finding in these patients.[33][34][35] Executive dysfunction may be the presenting symptom of progressive supranuclear palsy in some patients but is more characteristic of the later stages of the disease. Ideomotor apraxia may be a rare manifestation of progressive supranuclear palsy, although it is more common and severe in patients with corticobasal degeneration, a syndrome that can overlap clinically with progressive supranuclear palsy [36]
Behavioral abnormalities are also common in patients with progressive supranuclear palsy. In a case series of 22 patients with progressive supranuclear palsy, the most common behavioral symptoms were apathy (91%), disinhibition (36%), dysphoria (18%), and anxiety (18%).[37] One study of 188 patients with progressive supranuclear palsy demonstrated depression in 50% and anxiety in 37% [38], while another study of 74 patients with progressive supranuclear palsy reported obsessive-compulsive symptoms in 24%.[39] Impulsivity often induces patients to try to perform motor tasks that they are not able to do, such as getting up and walking on their own. This can lead to falls.
Pseudobulbar palsy is another characteristic feature of progressive supranuclear palsy. Emotional incontinence is less common than in other forms of pseudobulbar palsy [21], but patients with progressive supranuclear palsy commonly manifest the characteristic hoarse groaning voice along with moaning. Speech perseveration and anomia can be observed.[30]
Sleep Disturbances
Early or late insomnia and difficulties in maintaining sleep have all been reported in patients with progressive supranuclear palsy. Marked rigidity may result in the inability to remain comfortable in bed, further contributing to sleep complaints. Rapid eye movement sleep behavior disorder (RBD) is infrequently associated with progressive supranuclear palsy.[40] This negative finding, similar to preserved olfaction, can help differentiate progressive supranuclear palsy, a tauopathy, from Parkinson disease and multiple system atrophy, both of which are synucleinopathies and commonly demonstrate symptoms of RBD.
Patients with the classic PSP-RS usually develop initial symptoms in their mid-60s, and the disease advances from symptom onset to death in 7 years, on average.[41] Clinical subtypes of PSP-P and PSP-PAGF have a more benign course with a survival period of a decade or more.[41] Both subtypes have an overall tau burden less than that of PSP-RS, and the distribution of abnormal tau is relatively limited to the brain stem.[42][43] The phenotypes of PSP-P and PSP-PAGF can be referred to as the "brain stem" variants of progressive supranuclear palsy, as opposed to the "cortical" variants, which present with predominant cortical features, including PSP-CBS, PSP-bvFTD, and PSP-PNFA.[3]
Evaluation
The International Parkinson and Movement Disorder Society (MDS) progressive supranuclear palsy study group published the MDS-PSP criteria in 2017[18] in appreciation of the spectrum of clinical phenotypes associated with progressive supranuclear palsy pathology. Until the publication of these new criteria, the clinical criteria from the National Institute of Neurological Disorders and Stroke and the Society for PSP (NINDS-SPSP) were the most widely used criteria for in-life progressive supranuclear palsy diagnosis.[25] The NINDS-SPSP criteria require vertical supranuclear gaze palsy and prominent postural instability with falls in the first year of disease onset for diagnosis of probable progressive supranuclear palsy. For possible progressive supranuclear palsy, vertical supranuclear gaze palsy or slowed vertical saccades plus postural instability with first-year falls are required. Supportive criteria include proximal more than distal symmetric akinesia or rigidity, abnormal neck posturing (particularly retrocollis), poor levodopa responsiveness, early dysphagia, and/or dysarthria and early onset of specific cognitive-behavioral features.[25]
Radiographic Features
Neuroimaging in patients with progressive supranuclear palsy by MRI shows atrophy and signal increase in the midbrain, degeneration of the red nucleus, atrophy of the quadrigeminal plate, enlargement of the aqueduct and third ventricle, atrophy of the pons and cerebellum, and signal increase in the inferior olives. With the progression of the disease, frontal and temporal lobe atrophy may develop. While no feature on MRI or functional imaging studies is entirely specific for progressive supranuclear palsy, the “hummingbird sign” is often present, in which the shape of the rostral midbrain atrophy on mid-sagittal images looks like a hummingbird (see Image. Rostral Midbrain Atrophy in Progressive Supranuclear Palsy).[44] Midbrain to pons ratio of less than 0.52 using AP diameter measurements was useful in differentiating PSP-RS from MSA. Other progressive supranuclear palsy subtypes were not included in the study.[45] Fronto-subcortical hypometabolism on fluorodeoxyglucose positron emission tomography (FDG-PET) imaging is also a typical finding in progressive supranuclear palsy. Dopamine transporter single-photon emission computed tomography (SPECT) imaging, also known as DaTscan imaging, shows reduced tracer uptake in the striatum. This is a useful finding to distinguish progressive supranuclear palsy from other mimics such as cerebrovascular disease and normal pressure hydrocephalus.[46] DaTscan cannot, however, distinguish between Parkinson's disease and progressive supranuclear palsy, both of which would show reduced uptake. PET imaging with tau ligand is an encouraging radiologic tool for diagnostic and longitudinal follow-up.[47]
Treatment / Management
The management of the cognitive, motor and gait aspects of progressive supranuclear palsy is challenging, and the treatment for individuals suspected to have progressive supranuclear palsy remains symptomatic and supportive, with ongoing clinical trials striving to identify disease-modifying therapies often targeting the underlying tau pathology. For motor (parkinsonian) symptoms, levodopa combined with a dopa decarboxylase inhibitor (e.g., carbidopa) is generally tried, with typically modest to no success in most progressive supranuclear palsy phenotypes but potential benefit in the PSP-P predominance type. Overall, evidence for mild to moderate benefits with levodopa is low [48], but given limited therapeutic options, levodopa is generally tried at doses of up to 1000 mg daily. Other dopaminergic agents are rarely of benefit; amantadine is sometimes tried with limited supportive evidence.[48] Botulinum toxin injections can be used for focal dystonias, including apraxia of eyelid opening.[48]
The potential value of physical therapy is increasing interest, particularly given evidence of benefit for individuals with Parkinson's disease. A recent trial showed improvement in the Progressive Supranuclear Palsy Rating Scale (PSPRS).[49] Another study also suggested a potential benefit of the Lee Silverman Voice Treatment (LSVT) in individuals with progressive supranuclear palsy, though benefits in progressive supranuclear palsy were less frequently significant than those observed in Parkinson's disease patients.[50] Physical and occupational therapy with particular attention to balance training and “learning to fall” can minimize the potential for injury and provide patients with tools for maneuvering after a fall has occurred. Counseling on food preparation and swallowing to avoid aspiration is indicated in many. The use of a walker is often necessary for safe ambulation and transfers. At late stages, even using a walker is not enough to maintain balance, and patients may need to use a wheelchair exclusively.(B2)
While case reports and series suggest promising experiences with unilateral or bilateral pedunculopontine nucleus (PPN) deep brain stimulation (DBS) in patients with suspected progressive supranuclear palsy, a recently published randomized controlled trial of unilateral PPN DBS in 8 individuals with PSP-RS showed no benefit in gait, postural stability, and fall PSPRS subitems when comparing ON and OFF stimulation conditions at 6- and 12-month follow-up.[51] DBS is currently not recommended for progressive supranuclear palsy outside of research settings.[48](A1)
There are no accepted treatments for the cognitive symptoms in individuals with suspected progressive supranuclear palsy, with small trials and case series of cholinesterase inhibitors suggesting that these drugs may help cognition but worsen motor function.[48] It is critical to address potentially treatable symptoms in progressive supranuclear palsy such as depression, but no progressive supranuclear palsy-specific recommendations for such symptomatic management exist.
To date, studies of potentially disease-modifying therapies have failed to demonstrate efficacy in individuals suspected to have progressive supranuclear palsy. Randomized, placebo-controlled trials of riluzole,[52] davunetide,[53] tideglusib [54], sodium valproate,[55] and rasagiline[56] showed no impact on primary endpoints tracking disease progression, though study limitations include sample size (for some studies) and lack of evidence that the agents had the intended effect through theorized mechanisms. Though there have been negative double-blind studies of CoQ10 [57], there has been a positive double-blind study using the liposomal form [58](A1)
Regardless of investigational and symptomatic treatment approaches used throughout the disease course, palliative care is an important component of progressive supranuclear palsy treatment with hospice as a valuable resource in the late stages.
Current investigations of tau-focused progressive supranuclear palsy therapies include TPI-287, a microtubule stabilizer, C2N-8E12/ABBV-8E12, and BMS-986168/BIIB092, both anti-tau monoclonal antibodies, and salsalate, a tau acetylation inhibitor. Microtubule stabilizers are hoped to compensate for microtubule dysfunction associated with loss of tau function; anti-tau monoclonal antibodies are hoped to impede the spread of pathogenic tau, and tau acetylation inhibitors are hoped to inhibit acetylation of soluble tau and thus limit hyperphosphorylation.
Differential Diagnosis
The differential diagnosis of progressive supranuclear palsy includes corticobasal ganglionic degeneration (CBGD), Parkinson disease, multiple system atrophy (MSA) of the parkinsonian type, and Pick disease. Microvascular disease of the brain stem and tumors of the midbrain area can mimic progressive supranuclear palsy.
Other conditions that may produce similar symptoms include Whipple disease, mitochondrial myopathies, Wilson disease, and adult-onset Niemann-Pick disease. With the bulbar onset of symptoms, myasthenia gravis and amyotrophic lateral sclerosis should be included in the differential diagnosis.
Prognosis
Disease progression in progressive supranuclear palsy usually occurs fairly rapidly and relentlessly.[57] Most patients become dependent on care within 3 or 4 years from presentation. The disorder culminates in death at a median of 6 to 9 years after the diagnosis.[59][60][61] In addition, quality of life is significantly reduced.[62]
In a 2017 systematic review and meta-analysis, predictors of shorter survival included the progressive supranuclear palsy-Richardson (PSP-RS) phenotype compared with the progressive supranuclear palsy-parkinsonism (PSP-P) phenotype, early falls, and early cognitive symptoms (both more common in the PSP-RS phenotype).[63] Early-onset of dysphagia, which is seen in both the PSP-RS and PSP-P phenotypes, was also predictive of shorter survival. The prognostic effect of the presence of supranuclear gaze palsy was inconsistent across studies. As often seen in the early stages of the PSP-P phenotype, Levodopa response did not predict longer survival.
Pearls and Other Issues
- Unlike typical Parkinson disease, falls begin within the first year of progressive supranuclear palsy, and by year 3, they are common unless precautions are taken to prevent them.
- Downgaze is affected before upgaze, whereas; lateral eye movements are usually preserved in progressive supranuclear palsy.
- Apathy seems to predominate over other neurobehavioral abnormalities in progressive supranuclear palsy.
- Patients with progressive supranuclear palsy develop frontal cognitive impairment.
- The pathology of progressive supranuclear palsy is characterized by widespread neurodegeneration associated with tau protein deposition in subcortical regions.
- The first therapeutic step in progressive supranuclear palsy is identifying and prioritizing the symptoms that can be treated.
Enhancing Healthcare Team Outcomes
Evidence-Based Outcomes
Disease progression in progressive supranuclear palsy usually occurs fairly rapidly and relentlessly.[57] Most patients become dependent on care within 3 or 4 years from the presentation. The disorder culminates in death at a median of 6 to 9 years after the diagnosis.[59][60][61] Quality of life is significantly reduced.[62]
An Interprofessional Approach
Given the rapid progression of the disease and rapidly deteriorating quality of life, the education of the patient and family is critical. The home care nurse should closely monitor the patient with particular attention to daily living activities. The pharmacist should pay particular attention to the potential side effects of any medications. Only an integrated systemic care plan can help improve the quality of life and reduce the morbidity of the disease.
At each clinic visit, the nurse should focus on educating the family and ensuring that all concerns are answered. Today there are support groups for patients with progressive supranuclear palsy that help provide motivation and the latest information.
It is highly recommended that physical therapy and an occupational therapist be involved early in the disease process in helping maximize function, ambulation and facilitate the use of an ambulatory device.
Without a cure, the majority of patients are dead within seven years, but with an interprofessional approach, health care workers can make a significant difference in the quality of life.[64] [Level 5]
Media
(Click Image to Enlarge)
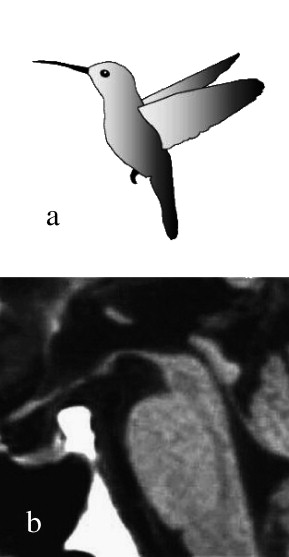
Rostral Midbrain Atrophy in Progressive Supranuclear Palsy. The image shows a hummingbird (a) and the midsagittal plain MRI in PSP (b). The region, including the most rostral midbrain, the midbrain tegmentum, the pontine base, and the cerebellum appears to correspond to the bill, crown, body, and wing, respectively, of a hummingbird (Hummingbird sign).
Adapted from Kato N, Arai K, Hattori T. Study of the rostral midbrain atrophy in progressive supranuclear palsy. J Neurol Sci. 2003;210(1–2):57–60.
References
STEELE JC, RICHARDSON JC, OLSZEWSKI J. PROGRESSIVE SUPRANUCLEAR PALSY. A HETEROGENEOUS DEGENERATION INVOLVING THE BRAIN STEM, BASAL GANGLIA AND CEREBELLUM WITH VERTICAL GAZE AND PSEUDOBULBAR PALSY, NUCHAL DYSTONIA AND DEMENTIA. Archives of neurology. 1964 Apr:10():333-59 [PubMed PMID: 14107684]
Steele JC. Progressive supranuclear palsy. Brain : a journal of neurology. 1972:95(4):693-704 [PubMed PMID: 4647151]
Dickson DW, Ahmed Z, Algom AA, Tsuboi Y, Josephs KA. Neuropathology of variants of progressive supranuclear palsy. Current opinion in neurology. 2010 Aug:23(4):394-400. doi: 10.1097/WCO.0b013e32833be924. Epub [PubMed PMID: 20610990]
Level 3 (low-level) evidenceLitvan I, Hauw JJ, Bartko JJ, Lantos PL, Daniel SE, Horoupian DS, McKee A, Dickson D, Bancher C, Tabaton M, Jellinger K, Anderson DW. Validity and reliability of the preliminary NINDS neuropathologic criteria for progressive supranuclear palsy and related disorders. Journal of neuropathology and experimental neurology. 1996 Jan:55(1):97-105 [PubMed PMID: 8558176]
Golbe LI, Davis PH, Schoenberg BS, Duvoisin RC. Prevalence and natural history of progressive supranuclear palsy. Neurology. 1988 Jul:38(7):1031-4 [PubMed PMID: 3386818]
Golbe LI, Rubin RS, Cody RP, Belsh JM, Duvoisin RC, Grosmann C, Lepore FE, Mark MH, Sachdeo RC, Sage JI, Zimmerman TR Jr. Follow-up study of risk factors in progressive supranuclear palsy. Neurology. 1996 Jul:47(1):148-54 [PubMed PMID: 8710069]
Level 2 (mid-level) evidenceLitvan I, Lees PS, Cunningham CR, Rai SN, Cambon AC, Standaert DG, Marras C, Juncos J, Riley D, Reich S, Hall D, Kluger B, Bordelon Y, Shprecher DR, for ENGENE-PSP. Environmental and occupational risk factors for progressive supranuclear palsy: Case-control study. Movement disorders : official journal of the Movement Disorder Society. 2016 May:31(5):644-52. doi: 10.1002/mds.26512. Epub 2016 Feb 8 [PubMed PMID: 26854325]
Level 2 (mid-level) evidenceKawashima M, Miyake M, Kusumi M, Adachi Y, Nakashima K. Prevalence of progressive supranuclear palsy in Yonago, Japan. Movement disorders : official journal of the Movement Disorder Society. 2004 Oct:19(10):1239-40 [PubMed PMID: 15390010]
Schrag A, Ben-Shlomo Y, Quinn NP. Prevalence of progressive supranuclear palsy and multiple system atrophy: a cross-sectional study. Lancet (London, England). 1999 Nov 20:354(9192):1771-5 [PubMed PMID: 10577638]
Level 2 (mid-level) evidenceRajput AH, Offord KP, Beard CM, Kurland LT. Epidemiology of parkinsonism: incidence, classification, and mortality. Annals of neurology. 1984 Sep:16(3):278-82 [PubMed PMID: 6333204]
Level 2 (mid-level) evidenceRadhakrishnan K, Thacker AK, Maloo JC, Gerryo SE, Mousa ME. Descriptive epidemiology of some rare neurological diseases in Benghazi, Libya. Neuroepidemiology. 1988:7(3):159-64 [PubMed PMID: 3405368]
Level 2 (mid-level) evidenceMastaglia FL, Grainger K, Kee F, Sadka M, Lefroy R. Progressive supranuclear palsy (the Steele-Richardson-Olszewski syndrome) clinical and electrophysiological observations in eleven cases. Proceedings of the Australian Association of Neurologists. 1973:10(0):35-44 [PubMed PMID: 4792160]
Level 3 (low-level) evidenceBower JH, Maraganore DM, McDonnell SK, Rocca WA. Incidence of progressive supranuclear palsy and multiple system atrophy in Olmsted County, Minnesota, 1976 to 1990. Neurology. 1997 Nov:49(5):1284-8 [PubMed PMID: 9371909]
Wenning GK, Litvan I, Tolosa E. Milestones in atypical and secondary Parkinsonisms. Movement disorders : official journal of the Movement Disorder Society. 2011 May:26(6):1083-95. doi: 10.1002/mds.23713. Epub [PubMed PMID: 21626553]
Respondek G, Stamelou M, Kurz C, Ferguson LW, Rajput A, Chiu WZ, van Swieten JC, Troakes C, Al Sarraj S, Gelpi E, Gaig C, Tolosa E, Oertel WH, Giese A, Roeber S, Arzberger T, Wagenpfeil S, Höglinger GU, Movement Disorder Society-endorsed PSP Study Group. The phenotypic spectrum of progressive supranuclear palsy: a retrospective multicenter study of 100 definite cases. Movement disorders : official journal of the Movement Disorder Society. 2014 Dec:29(14):1758-66. doi: 10.1002/mds.26054. Epub 2014 Nov 5 [PubMed PMID: 25370486]
Level 2 (mid-level) evidenceCoyle-Gilchrist IT, Dick KM, Patterson K, Vázquez Rodríquez P, Wehmann E, Wilcox A, Lansdall CJ, Dawson KE, Wiggins J, Mead S, Brayne C, Rowe JB. Prevalence, characteristics, and survival of frontotemporal lobar degeneration syndromes. Neurology. 2016 May 3:86(18):1736-43. doi: 10.1212/WNL.0000000000002638. Epub 2016 Apr 1 [PubMed PMID: 27037234]
Respondek G, Kurz C, Arzberger T, Compta Y, Englund E, Ferguson LW, Gelpi E, Giese A, Irwin DJ, Meissner WG, Nilsson C, Pantelyat A, Rajput A, van Swieten JC, Troakes C, Josephs KA, Lang AE, Mollenhauer B, Müller U, Whitwell JL, Antonini A, Bhatia KP, Bordelon Y, Corvol JC, Colosimo C, Dodel R, Grossman M, Kassubek J, Krismer F, Levin J, Lorenzl S, Morris H, Nestor P, Oertel WH, Rabinovici GD, Rowe JB, van Eimeren T, Wenning GK, Boxer A, Golbe LI, Litvan I, Stamelou M, Höglinger GU, Movement Disorder Society-Endorsed PSP Study Group. Which ante mortem clinical features predict progressive supranuclear palsy pathology? Movement disorders : official journal of the Movement Disorder Society. 2017 Jul:32(7):995-1005. doi: 10.1002/mds.27034. Epub 2017 May 13 [PubMed PMID: 28500752]
Höglinger GU, Respondek G, Stamelou M, Kurz C, Josephs KA, Lang AE, Mollenhauer B, Müller U, Nilsson C, Whitwell JL, Arzberger T, Englund E, Gelpi E, Giese A, Irwin DJ, Meissner WG, Pantelyat A, Rajput A, van Swieten JC, Troakes C, Antonini A, Bhatia KP, Bordelon Y, Compta Y, Corvol JC, Colosimo C, Dickson DW, Dodel R, Ferguson L, Grossman M, Kassubek J, Krismer F, Levin J, Lorenzl S, Morris HR, Nestor P, Oertel WH, Poewe W, Rabinovici G, Rowe JB, Schellenberg GD, Seppi K, van Eimeren T, Wenning GK, Boxer AL, Golbe LI, Litvan I, Movement Disorder Society-endorsed PSP Study Group. Clinical diagnosis of progressive supranuclear palsy: The movement disorder society criteria. Movement disorders : official journal of the Movement Disorder Society. 2017 Jun:32(6):853-864. doi: 10.1002/mds.26987. Epub 2017 May 3 [PubMed PMID: 28467028]
Santacruz P, Uttl B, Litvan I, Grafman J. Progressive supranuclear palsy: a survey of the disease course. Neurology. 1998 Jun:50(6):1637-47 [PubMed PMID: 9633705]
Level 3 (low-level) evidenceDe Bruin VM, Lees AJ. Subcortical neurofibrillary degeneration presenting as Steele-Richardson-Olszewski and other related syndromes: a review of 90 pathologically verified cases. Movement disorders : official journal of the Movement Disorder Society. 1994 Jul:9(4):381-9 [PubMed PMID: 7969203]
Level 3 (low-level) evidenceKristensen MO. Progressive supranuclear palsy--20 years later. Acta neurologica Scandinavica. 1985 Mar:71(3):177-89 [PubMed PMID: 3993325]
Kovacs GG. Invited review: Neuropathology of tauopathies: principles and practice. Neuropathology and applied neurobiology. 2015 Feb:41(1):3-23. doi: 10.1111/nan.12208. Epub [PubMed PMID: 25495175]
Dickson DW. Neuropathologic differentiation of progressive supranuclear palsy and corticobasal degeneration. Journal of neurology. 1999 Sep:246 Suppl 2():II6-15 [PubMed PMID: 10525997]
Hauw JJ, Daniel SE, Dickson D, Horoupian DS, Jellinger K, Lantos PL, McKee A, Tabaton M, Litvan I. Preliminary NINDS neuropathologic criteria for Steele-Richardson-Olszewski syndrome (progressive supranuclear palsy). Neurology. 1994 Nov:44(11):2015-9 [PubMed PMID: 7969952]
Litvan I, Agid Y, Calne D, Campbell G, Dubois B, Duvoisin RC, Goetz CG, Golbe LI, Grafman J, Growdon JH, Hallett M, Jankovic J, Quinn NP, Tolosa E, Zee DS. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): report of the NINDS-SPSP international workshop. Neurology. 1996 Jul:47(1):1-9 [PubMed PMID: 8710059]
Birdi S, Rajput AH, Fenton M, Donat JR, Rozdilsky B, Robinson C, Macaulay R, George D. Progressive supranuclear palsy diagnosis and confounding features: report on 16 autopsied cases. Movement disorders : official journal of the Movement Disorder Society. 2002 Nov:17(6):1255-64 [PubMed PMID: 12465065]
Level 3 (low-level) evidenceChu FC, Reingold DB, Cogan DG, Williams AC. The eye movement disorders of progressive supranuclear palsy. Ophthalmology. 1979 Mar:86(3):422-8 [PubMed PMID: 530592]
Chen AL, Riley DE, King SA, Joshi AC, Serra A, Liao K, Cohen ML, Otero-Millan J, Martinez-Conde S, Strupp M, Leigh RJ. The disturbance of gaze in progressive supranuclear palsy: implications for pathogenesis. Frontiers in neurology. 2010:1():147. doi: 10.3389/fneur.2010.00147. Epub 2010 Dec 3 [PubMed PMID: 21188269]
Pierrot-Deseilligny C, Rivaud S, Pillon B, Fournier E, Agid Y. Lateral visually-guided saccades in progressive supranuclear palsy. Brain : a journal of neurology. 1989 Apr:112 ( Pt 2)():471-87 [PubMed PMID: 2706440]
Litvan I. Update on progressive supranuclear palsy. Current neurology and neuroscience reports. 2004 Jul:4(4):296-302 [PubMed PMID: 15217544]
Level 3 (low-level) evidenceBoeve BF. Progressive supranuclear palsy. Parkinsonism & related disorders. 2012 Jan:18 Suppl 1():S192-4. doi: 10.1016/S1353-8020(11)70060-8. Epub [PubMed PMID: 22166432]
Level 3 (low-level) evidenceRobbins TW, James M, Owen AM, Lange KW, Lees AJ, Leigh PN, Marsden CD, Quinn NP, Summers BA. Cognitive deficits in progressive supranuclear palsy, Parkinson's disease, and multiple system atrophy in tests sensitive to frontal lobe dysfunction. Journal of neurology, neurosurgery, and psychiatry. 1994 Jan:57(1):79-88 [PubMed PMID: 8301310]
Level 3 (low-level) evidenceGolbe LI, Boeve BF, Keegan BM, Parisi JE. An 81-year-old man with imbalance and memory impairment. Neurology. 2007 Apr 3:68(14):1147-52 [PubMed PMID: 17404198]
Level 3 (low-level) evidencePillon B, Dubois B, Lhermitte F, Agid Y. Heterogeneity of cognitive impairment in progressive supranuclear palsy, Parkinson's disease, and Alzheimer's disease. Neurology. 1986 Sep:36(9):1179-85 [PubMed PMID: 3748383]
Dubois B, Pillon B, Legault F, Agid Y, Lhermitte F. Slowing of cognitive processing in progressive supranuclear palsy. A comparison with Parkinson's disease. Archives of neurology. 1988 Nov:45(11):1194-9 [PubMed PMID: 3190499]
Pharr V, Uttl B, Stark M, Litvan I, Fantie B, Grafman J. Comparison of apraxia in corticobasal degeneration and progressive supranuclear palsy. Neurology. 2001 Apr 10:56(7):957-63 [PubMed PMID: 11294936]
Litvan I, Mega MS, Cummings JL, Fairbanks L. Neuropsychiatric aspects of progressive supranuclear palsy. Neurology. 1996 Nov:47(5):1184-9 [PubMed PMID: 8909427]
Schrag A, Sheikh S, Quinn NP, Lees AJ, Selai C, Mathias C, Litvan I, Lang AE, Bower JH, Burn DJ, Low P, Jahanshahi M. A comparison of depression, anxiety, and health status in patients with progressive supranuclear palsy and multiple system atrophy. Movement disorders : official journal of the Movement Disorder Society. 2010 Jun 15:25(8):1077-81. doi: 10.1002/mds.22794. Epub [PubMed PMID: 20535826]
Fukui T, Lee E, Hosoda H, Okita K. Obsessive-compulsive behavior as a symptom of dementia in progressive supranuclear palsy. Dementia and geriatric cognitive disorders. 2010:30(2):179-88. doi: 10.1159/000310351. Epub 2010 Aug 26 [PubMed PMID: 20798538]
Boeve BF, Silber MH, Parisi JE, Dickson DW, Ferman TJ, Benarroch EE, Schmeichel AM, Smith GE, Petersen RC, Ahlskog JE, Matsumoto JY, Knopman DS, Schenck CH, Mahowald MW. Synucleinopathy pathology and REM sleep behavior disorder plus dementia or parkinsonism. Neurology. 2003 Jul 8:61(1):40-5 [PubMed PMID: 12847154]
Level 2 (mid-level) evidenceWilliams DR, de Silva R, Paviour DC, Pittman A, Watt HC, Kilford L, Holton JL, Revesz T, Lees AJ. Characteristics of two distinct clinical phenotypes in pathologically proven progressive supranuclear palsy: Richardson's syndrome and PSP-parkinsonism. Brain : a journal of neurology. 2005 Jun:128(Pt 6):1247-58 [PubMed PMID: 15788542]
Williams DR, Holton JL, Strand C, Pittman A, de Silva R, Lees AJ, Revesz T. Pathological tau burden and distribution distinguishes progressive supranuclear palsy-parkinsonism from Richardson's syndrome. Brain : a journal of neurology. 2007 Jun:130(Pt 6):1566-76 [PubMed PMID: 17525140]
Williams DR, Holton JL, Strand K, Revesz T, Lees AJ. Pure akinesia with gait freezing: a third clinical phenotype of progressive supranuclear palsy. Movement disorders : official journal of the Movement Disorder Society. 2007 Nov 15:22(15):2235-41 [PubMed PMID: 17712855]
Level 3 (low-level) evidenceKato N, Arai K, Hattori T. Study of the rostral midbrain atrophy in progressive supranuclear palsy. Journal of the neurological sciences. 2003 Jun 15:210(1-2):57-60 [PubMed PMID: 12736089]
Massey LA, Jäger HR, Paviour DC, O'Sullivan SS, Ling H, Williams DR, Kallis C, Holton J, Revesz T, Burn DJ, Yousry T, Lees AJ, Fox NC, Micallef C. The midbrain to pons ratio: a simple and specific MRI sign of progressive supranuclear palsy. Neurology. 2013 May 14:80(20):1856-61. doi: 10.1212/WNL.0b013e318292a2d2. Epub 2013 Apr 24 [PubMed PMID: 23616165]
Level 2 (mid-level) evidenceKägi G, Bhatia KP, Tolosa E. The role of DAT-SPECT in movement disorders. Journal of neurology, neurosurgery, and psychiatry. 2010 Jan:81(1):5-12. doi: 10.1136/jnnp.2008.157370. Epub [PubMed PMID: 20019219]
Kepe V, Bordelon Y, Boxer A, Huang SC, Liu J, Thiede FC, Mazziotta JC, Mendez MF, Donoghue N, Small GW, Barrio JR. PET imaging of neuropathology in tauopathies: progressive supranuclear palsy. Journal of Alzheimer's disease : JAD. 2013:36(1):145-53. doi: 10.3233/JAD-130032. Epub [PubMed PMID: 23579330]
Stamelou M, Höglinger G. A Review of Treatment Options for Progressive Supranuclear Palsy. CNS drugs. 2016 Jul:30(7):629-36. doi: 10.1007/s40263-016-0347-2. Epub [PubMed PMID: 27222018]
Golbe LI, Ohman-Strickland PA. A clinical rating scale for progressive supranuclear palsy. Brain : a journal of neurology. 2007 Jun:130(Pt 6):1552-65 [PubMed PMID: 17405767]
Level 2 (mid-level) evidenceSale P, Castiglioni D, De Pandis MF, Torti M, Dall'armi V, Radicati FG, Stocchi F. The Lee Silverman Voice Treatment (LSVT®) speech therapy in progressive supranuclear palsy. European journal of physical and rehabilitation medicine. 2015 Oct:51(5):569-74 [PubMed PMID: 26138088]
Scelzo E, Lozano AM, Hamani C, Poon YY, Aldakheel A, Zadikoff C, Lang AE, Moro E. Peduncolopontine nucleus stimulation in progressive supranuclear palsy: a randomised trial. Journal of neurology, neurosurgery, and psychiatry. 2017 Jul:88(7):613-616. doi: 10.1136/jnnp-2016-315192. Epub 2017 Feb 18 [PubMed PMID: 28214797]
Level 1 (high-level) evidenceBensimon G, Ludolph A, Agid Y, Vidailhet M, Payan C, Leigh PN, NNIPPS Study Group. Riluzole treatment, survival and diagnostic criteria in Parkinson plus disorders: the NNIPPS study. Brain : a journal of neurology. 2009 Jan:132(Pt 1):156-71. doi: 10.1093/brain/awn291. Epub 2008 Nov 23 [PubMed PMID: 19029129]
Level 2 (mid-level) evidenceBoxer AL, Lang AE, Grossman M, Knopman DS, Miller BL, Schneider LS, Doody RS, Lees A, Golbe LI, Williams DR, Corvol JC, Ludolph A, Burn D, Lorenzl S, Litvan I, Roberson ED, Höglinger GU, Koestler M, Jack CR Jr, Van Deerlin V, Randolph C, Lobach IV, Heuer HW, Gozes I, Parker L, Whitaker S, Hirman J, Stewart AJ, Gold M, Morimoto BH, AL-108-231 Investigators. Davunetide in patients with progressive supranuclear palsy: a randomised, double-blind, placebo-controlled phase 2/3 trial. The Lancet. Neurology. 2014 Jul:13(7):676-85. doi: 10.1016/S1474-4422(14)70088-2. Epub 2014 May 27 [PubMed PMID: 24873720]
Level 1 (high-level) evidenceTolosa E, Litvan I, Höglinger GU, Burn D, Lees A, Andrés MV, Gómez-Carrillo B, León T, Del Ser T, TAUROS Investigators. A phase 2 trial of the GSK-3 inhibitor tideglusib in progressive supranuclear palsy. Movement disorders : official journal of the Movement Disorder Society. 2014 Apr:29(4):470-8. doi: 10.1002/mds.25824. Epub 2014 Feb 14 [PubMed PMID: 24532007]
Level 1 (high-level) evidenceLeclair-Visonneau L, Rouaud T, Debilly B, Durif F, Houeto JL, Kreisler A, Defebvre L, Lamy E, Volteau C, Nguyen JM, Dily SL, Damier P, Boutoleau-Bretonnière C, Lejeune P, Derkinderen P. Randomized placebo-controlled trial of sodium valproate in progressive supranuclear palsy. Clinical neurology and neurosurgery. 2016 Jul:146():35-9. doi: 10.1016/j.clineuro.2016.04.021. Epub 2016 Apr 27 [PubMed PMID: 27136096]
Level 1 (high-level) evidenceNuebling G, Hensler M, Paul S, Zwergal A, Crispin A, Lorenzl S. PROSPERA: a randomized, controlled trial evaluating rasagiline in progressive supranuclear palsy. Journal of neurology. 2016 Aug:263(8):1565-74. doi: 10.1007/s00415-016-8169-1. Epub 2016 May 26 [PubMed PMID: 27230855]
Level 1 (high-level) evidenceApetauerova D, Scala SA, Hamill RW, Simon DK, Pathak S, Ruthazer R, Standaert DG, Yacoubian TA. CoQ10 in progressive supranuclear palsy: A randomized, placebo-controlled, double-blind trial. Neurology(R) neuroimmunology & neuroinflammation. 2016 Oct:3(5):e266. doi: 10.1212/NXI.0000000000000266. Epub 2016 Aug 2 [PubMed PMID: 27583276]
Level 1 (high-level) evidenceStamelou M, Reuss A, Pilatus U, Magerkurth J, Niklowitz P, Eggert KM, Krisp A, Menke T, Schade-Brittinger C, Oertel WH, Höglinger GU. Short-term effects of coenzyme Q10 in progressive supranuclear palsy: a randomized, placebo-controlled trial. Movement disorders : official journal of the Movement Disorder Society. 2008 May 15:23(7):942-949. doi: 10.1002/mds.22023. Epub [PubMed PMID: 18464278]
Level 1 (high-level) evidenceO'Sullivan SS, Massey LA, Williams DR, Silveira-Moriyama L, Kempster PA, Holton JL, Revesz T, Lees AJ. Clinical outcomes of progressive supranuclear palsy and multiple system atrophy. Brain : a journal of neurology. 2008 May:131(Pt 5):1362-72. doi: 10.1093/brain/awn065. Epub 2008 Apr 2 [PubMed PMID: 18385183]
Level 2 (mid-level) evidenceTesta D, Monza D, Ferrarini M, Soliveri P, Girotti F, Filippini G. Comparison of natural histories of progressive supranuclear palsy and multiple system atrophy. Neurological sciences : official journal of the Italian Neurological Society and of the Italian Society of Clinical Neurophysiology. 2001 Jun:22(3):247-51 [PubMed PMID: 11731878]
dell'Aquila C, Zoccolella S, Cardinali V, de Mari M, Iliceto G, Tartaglione B, Lamberti P, Logroscino G. Predictors of survival in a series of clinically diagnosed progressive supranuclear palsy patients. Parkinsonism & related disorders. 2013 Nov:19(11):980-5. doi: 10.1016/j.parkreldis.2013.06.014. Epub 2013 Aug 19 [PubMed PMID: 23968651]
Level 2 (mid-level) evidencePekmezović T, Ječmenica-Lukić M, Petrović I, Špica V, Tomić A, Kostić VS. Quality of life in patients with progressive supranuclear palsy: one-year follow-up. Journal of neurology. 2015 Sep:262(9):2042-8. doi: 10.1007/s00415-015-7815-3. Epub 2015 Jun 13 [PubMed PMID: 26070289]
Level 2 (mid-level) evidenceGlasmacher SA, Leigh PN, Saha RA. Predictors of survival in progressive supranuclear palsy and multiple system atrophy: a systematic review and meta-analysis. Journal of neurology, neurosurgery, and psychiatry. 2017 May:88(5):402-411. doi: 10.1136/jnnp-2016-314956. Epub 2017 Mar 1 [PubMed PMID: 28250027]
Level 1 (high-level) evidenceClerici I, Ferrazzoli D, Maestri R, Bossio F, Zivi I, Canesi M, Pezzoli G, Frazzitta G. Rehabilitation in progressive supranuclear palsy: Effectiveness of two multidisciplinary treatments. PloS one. 2017:12(2):e0170927. doi: 10.1371/journal.pone.0170927. Epub 2017 Feb 3 [PubMed PMID: 28158197]