Introduction
Lymphoma of the central nervous system (CNS), both primary and secondary, represents a rare but highly aggressive subset of non-Hodgkin lymphoma (NHL).[1] Despite its reputation for aggressiveness and poor prognosis, primary central nervous system lymphoma (PCNSL) often responds well to therapy and has the possibility of curative treatment. Increasingly, high-dose myeloablative therapy in combination with autologous stem cell transplantation and the advent of newer therapeutic strategies (eg, ibrutinib), offer the possibility of improved outcomes.[2] PCNSL is a rare variant of extra-nodal NHL that can impact sites anywhere along the entire neuraxis, including the orbits, leptomeninges, brain, and spinal cord.[1] On the other hand, secondary CNS lymphoma refers to systemic NHL that has disseminated to the CNS.[3]
Historically, the prognosis of PCNSL has been dismal, with overall survival of 1.5 months if untreated and a 5-year survival rate of 30%.[4][5] Diffuse B-cell lymphoma is the most common form of PCNSL, usually localized to the parenchyma, but other locations are also possible, as previously described. The underlying histology and disease localization determine the neurologic presentation, treatment options, and prognosis.[2] Patients with aggressive systemic NHL have a 2% to 27% risk of developing secondary CNS dissemination, with a median survival of 2.2 months after diagnosis.[6] The introduction of high-dose methotrexate-based chemotherapy regimens has led to substantial progress in CNS lymphoma treatment and prognosis, including improved survival.
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
Immunodeficiency, both primary and acquired, is an important risk factor for PCNSL.[6][7] PCNSL is an acquired immunodeficiency syndrome (AIDS)-defining illness, usually occurring when CD4 counts are below 50 cells/µL and in patients not on highly active antiretroviral therapy (HAART).[8] Among patients who have undergone solid organ transplantation, up to 2% of renal transplant patients and up to 7% of patients who have undergone cardiac, lung, or liver transplantation ultimately develop lymphoma in the CNS. The incidence is highest during the first year after heart and lung transplant.[7] The Epstein-Barr virus (EBV) correlates highly with CNS lymphoma in T-cell immunodeficient states, such as patients on immunosuppressants post-organ transplantation. EBV is also associated with 100% of PCNSL in patients living with AIDS.[6]
Epidemiology
CNS lymphoma is decreasing in persons living with AIDS due to the advent of HAART, but its incidence is rising in older adults.[9] The disease is rare in pediatric populations. PCNSL has an annual incidence of approximately 1700 cases in the United States or approximately 0.5 per 100,000 per year.[1][8][10][11] PNCSL comprises 3% of all primary brain tumors and 2% to 3% of all cases of NHL. Immunocompetent patients are often diagnosed between ages 50 and 70, whereas immunocompromised patients present earlier, typically in their 30s and 40s.[7] Men are affected more than women in both immunocompetent and immunocompromised groups, though there is no sex-based predilection in patients who develop CNS lymphoma after solid organ transplantation.[7][12] Other patient populations at risk for PCNSL include those with ataxia-telangiectasia, Wiskott-Aldrich syndrome, and other immunodeficiency syndromes.
The vast majority—more than 90%—of PCNSL cases are diffuse large B-cell lymphoma; primary forms of T-cell and Burkitt lymphomas and lower-grade lymphoproliferative conditions have also been reported.[2] Within the CNS, PCNSL most commonly arises in the frontal lobe and the basal ganglia, with the brainstem, cerebellum, and spinal cord less commonly affected. Most immunocompetent patients have a solitary brain mass, with multiple lesions observed in 20% to 40% of cases.[11] (See Image. Orbito-Frontal Mass, Central Nervous System Lymphoma and Image. Central Nervous System Lymphoma, Hyperdense Calvarial Based Lesion.)
Up to 25% of patients with PCNSL develop intraocular lymphoma, and primary intraocular lymphoma ultimately disseminates to the CNS more than 80% of the time.[13] Concurrent cerebrospinal fluid (CSF) and orbit involvement occur in up to 20% of cases.[11] Primary intraocular lymphoma predominates in the vitreous fluid and the retina.[12] Beyond the orbit, PCNSL rarely disseminates systemically. Roughly 40% of systemic lymphomas arise in or near the CNS, including the paranasal sinuses and the previously mentioned localizations. Secondary CNS lymphoma preferentially affects the dura and leptomeninges; leptomeningeal metastases occur in 4% to 11% of patients with systemic lymphoma.
Pathophysiology
Whether cells of PCNSL first arise from within the CNS itself or if they travel to the CNS via peripheral blood, further accumulating other mutations along the way is still unclear.[5] Diffuse large B-cell lymphoma, by far the most common PCNSL histology, can be subcategorized into 1 of 2 subtypes: the germinal center or the activated B-cell-like/nongerminal center, according to the expression of cellular markers.[2]
Several mutations in tumor suppressor and proto-oncogenes involved in B-cell activation, differentiation, and apoptosis likely contribute to the development of PCNSL. Somatic hypermutations in proto-oncogenes, such as MYC, PAX5, Rho/TTF, PIM1, and tumor suppressor genes, such as PRDM1, have been demonstrated in PCNSL cases. Nuclear factor kappa B (NF-κB) signaling is believed to play a role in disease pathogenesis. The up-regulation of activators within the NF-κB pathway, such as myeloid differentiation primary response 88 (MYD88), aspase recruitment domain family member 11 (CADR11), and cluster of differentiation 79 (CD79), and suppression of NF-κB inhibitors, such as tumor necrosis factor, alpha-induced protein 3 (TNFAIP3), have been noted in individuals with PCNSL.[5] Aberrant regulation of the Janus kinases (JAKs), and signal transducer and activator of transcription proteins (STATs) (eg, JAK/STAT) pathway is also implicated. Increased levels of interleukin (IL)-10, A JAK/STAT mediator, are seen in CSF analysis of patients with PCNSL and are often associated with a worse prognosis. The expression of MYD88 and CD79B mutations in secondary CNS lymphoma is considerably lower than in primary CNS lymphoma, suggesting a different pathogenesis mechanism.[14]
Histopathology
About 95% of PCNSL belong to the diffuse large B-cell category, with low-grade B-cell lymphoma, T-cell lymphoma, and Burkitt lymphoma accounting for the remaining cases.[5][11][15][16] There are 2 histological variants of diffuse large B-cell lymphoma: the germinal center subtype, often CD10 and BCL6 positive, and the activated B-cell-like subtype, usually expressing MUM1. More than half of PCNSL express both BCL6 and MUM1, suggesting that the tumor derives from B-cells in the process of exiting the germinal center that have not yet reached the post-germinal center stage.
PCNSL usually expresses the pan B-cell markers CD19, CD20, CD22, and CD79a, but the plasma cell markers CD38 and CD138 are often absent. The predictive value of these B-cell differentiation markers is well elucidated in systemic diffuse large B-cell lymphoma, but their significance in PCNSL remains to be determined. Similarly, overexpression of the cell cycle regulator protein MYC and the anti-apoptosis protein B-cell leukemia/lymphoma 2 protein (BCL2) have prognostic implications in systemic diffuse large B-cell lymphoma but uncertain significance in PCNSL.[5] PCNSL is highly cellular and infiltrative. Histology often displays a perivascular growth pattern known as angiotropism.[16][17] If present, a reactive perivascular infiltrate of T-cells portends a better outcome, though occurring less commonly in PCNSL than in secondary CNS lymphoma, which may help to explain PCNSL’s comparatively poorer prognosis.[5]
History and Physical
The varied clinical presentations possible in CNS lymphoma—primary or secondary in etiology—necessitate high clinical suspicion.[2] Most patients with PCNSL present with focal neurologic deficits arising from direct nerve invasion (neurolymphomatosis) or leptomeningeal involvement. However, nonspecific presentations are also possible, oftentimes of cognitive decline or behavioral disruption, particularly in older patients.[6][12]
When the initial presentation is focal, brain imaging usually takes place promptly, facilitating rapid diagnosis; cognitive/behavioral and other nonspecific presentations often cause developmental delays ranging from weeks to months. Though common in systemic lymphoma, systemic symptoms—such as fever of unknown origin, night sweats, and weight loss—rarely occur in PCNSL.[18] Focal peripheral nerve deficits suggest the presence of leptomeningeal or direct nerve invasion. Though sometimes asymptomatic, leptomeningeal disease can also cause cranial neuropathies or symptoms localizing to the cauda equina, such as urinary retention or saddle anesthesia. Direct nerve invasion can cause a painful radiculopathy. Hemiparesis, hemisensory loss, and ataxia are also possible, depending on the underlying tumor locus.[19] As many as a third of patients with PCNSL present with non-localizing signs of increased intracranial pressure, such as nausea, vomiting, and headache. Reports of frank visual deficits occur rarely, but ocular involvement occurs in as many as 25% of patients.[2]
Evaluation
Magnetic resonance imaging (MRI) brain with gadolinium contrast is the recommended first test in the diagnostic workup for patients in whom PCNSL is suspected.[2]
- Lesions are often located centrally within the cerebral white matter and periventricular regions.
- Lesions are often iso– or hypointense on unenhanced T1 weighted imaging and iso– or hyperintense on T2 sequences (see Image. Central Nervous System Lymphoma, Magnetic Resonance Image).[11]
- Surrounding edema is often present but relatively less dramatically than in malignant glioma or metastatic disease.
- Lesions in immunocompetent patients are usually solitary, with homogenous enhancement, but 20% to 40% of cases have multiple lesions, and ring-like enhancement occurs in up to 13% of cases.
- Some 30% to 80% of those who are immunodeficient present with multiple necrotic lesions in an irregular ring-enhancing pattern post-contrast.[6][12]
- Lesions typically show homogeneous enhancement (in contrast to human immunodeficienty virus [HIV]-associated PCNSL, where ring enhancement is the norm).[20]
- Micro-hemorrhages are often present in high-grade gliomas, but hemorrhage and calcifications are uncommon in primary CNS lymphoma lesions (except in AIDS-related cases).
- Conventional MRI alone cannot reliably differentiate CNS lymphoma from other neoplastic lesions in the brain, so other sequences and imaging modalities are often helpful.
- Diffusion-weighted sequencing usually demonstrates restriction due to PCNSL's high cellularity, which can then aid in differentiation from metastatic lesions and high-grade gliomas.
- MR spectroscopy (MRS) can be used to differentiate PCNSL from toxoplasmosis based on biochemical properties: the PCNSL profile reflects increased choline, lipid, and lactate peaks, along with moderately decreased N-acetyl aspartate and slightly decreased creatine peaks.[21]
MRI has low sensitivity for detecting primary vitreoretinal lymphoma (aka intraocular lymphoma), requiring thin cuts to reveal any nodular enhancing lesions on the macula or uvea.[20] Vitreous aspiration and chorioretinal biopsy are the preferred modalities for definitive diagnosis, but slit-lamp examination of both eyes is often done earlier in the workup process.[4][12][20] Flow cytometry can be used to analyze vitreous aspirates; it also has utility in CSF analysis in primary dural lymphoma, posterior fossa lesions, or secondary CNS lymphoma.[1][6] Lumbar puncture for CSF cytomorphology and flow cytometric analysis has a 6% to 13.3% diagnostic yield.[11]
Positron emission tomography (PET) scanning facilitates discrimination of PCNSL from glioma and infection. CNS lymphomas are typically more hypermetabolic than gliomas. Additionally, the uptake area on PET imaging is often more extensive than the corresponding areas on conventional imaging, reflecting the tumor's infiltration beyond the regions depicted on MR imaging. Conversely, infectious lesions are generally hypometabolic, corresponding to lower thallium-201 uptake on single-photon emission computed tomography. Further, (SPECT) and lower radio-labeled glucose uptake on PET imaging. This aids in the differentiation of PCNSLs from infectious etiologies in immunocompromised individuals.
Stereotactic (frameless) brain biopsy constitutes the mainstay of confirmatory PCNSL diagnosis,[1] yielding diagnostic confirmation in more than 91% of cases; such biopsy can be further aided by frozen section and 5-aminolevulinic acid (ALA) fluorescence.[11] Radiomics and liquid biopsy are emerging new modalities for PCNSL diagnosis.[1][11][22] CSF analysis for CD79B and MYD88 or diagnostic markers like chemokine (C-X-C motif) ligand 13 (CXCL-13), beta-2-microglobulin (B2M), and neopterin has shown promising results.[11] Newer biomarkers, such as micro ribonucleaic acid (miRNA)-21 miR-21, soluble interleukin-2 receptor (sIL-2R) in blood or CXCL-13, B2M, and neopterin in CSF, are undergoing exploration as diagnostic biomarkers.[3]
The International PCNSL Collaborative Group recommends the following tests in a comprehensive diagnostic evaluation for PCNSL:
- Contrast-enhanced MRI of the complete neuraxis (CT scans should be reserved for use only in the setting of an absolute contraindication to magnetic resonance imaging)
- Lumbar puncture
- Ophthalmologic examination
- Testicular ultrasonography in older men [1][11][23]
- Staging via a whole-body PET scan and bone marrow biopsy; if PET scan unavailable, contrast CT of the chest, abdomen, and pelvis)
- HIV and hepatitis serologies
- Performance status
- Hepatic and renal function analysis
- Lactate dehydrogenase level testing
Treatment / Management
PCNSL treatment should be initiated as soon as possible after the diagnosis,[11] usually with induction followed by consolidation/maintenance chemotherapy. Induction therapy reduces or debulks gross disease; consolidation therapy targets residual microscopic disease with the goal of cure or at least durable remission.[2](B3)
Avoid Corticosteroids
Corticosteroids should be withheld prior to diagnostic biopsy owing to their lymphocytolytic effect.[1] Inconclusive biopsies are three times more likely in patients treated with corticosteroids than in steroid-naïve patients, with non-diagnostic yields ranging from 33% to 57% in the population treated with steroids. Cell arrest and apoptosis secondary to corticosteroids can cause “ghost” or “vanishing” tumors in up to 50% of cases. Thus, corticosteroids hinder histopathological diagnosis.[1] Furthermore, steroid responsiveness is diagnostically inconclusive: neurosarcoidosis, vasculitis, neoplasms such as glioblastoma, and demyelinating conditions like multiple sclerosis all show responsiveness to steroids. When patients are treated with corticosteroids, close clinical follow-up and serial re-imaging with the provision of urgent biopsy in the case of tumor re-growth are necessary.
Historical Treatment Approaches
Due to its high radiation sensitivity, patients with newly diagnosed CNS lymphoma traditionally received treatment with whole-brain radiotherapy (WBRT). Early relapse and radiation-associated neurotoxicity were common among survivors despite initially favorable responsiveness.[4] Moreover, when treatment was limited to WBRT, median survival was a mere 12 to 18 months.[6] CNS lymphoma is not amenable to complete surgical resection due to its infiltrative nature, multifocality, and microscopic seedlings protected by the intact blood-brain barrier.[1] There is no clear evidence that gross resection offers any outcome advantage.[24] Extensive resection of PCNS is advocated only to avoid herniation.[1]
Multimodal Chemotherapeutic Approaches
The primary treatment approach for individuals with CNS lymphoma is induction chemotherapy followed by consolidation therapy. Induction therapy aims for a complete radiographic response; consolidation therapy aims to eradicate residual disease and improve overall survival.[4] Induction chemotherapy usually involves a combination of high-dose methotrexate (HD-MTX), a folate antagonist, with other chemotherapy agents, including temozolomide, cytarabine, etoposide, vincristine, carmustine, ifosfamide, thiotepa, and cyclophosphamide. The superiority of one MTX-based regimen over another has not yet been established, nor has the optimal dose for MTX itself been determined.[2] Owing to MTX’s limited CNS penetration, high doses (≥3.5 g/m) are usually necessary.[1] Good hydration is mandatory to protect systemic tissue from MTX adverse effects, and leucovorin, a folic acid analog, is typically used concomitantly.[11] Treatment regimens targeting B-cell lymphoma often incorporate rituximab, an anti-CD20 monoclonal antibody, although survival benefits have been mixed in phase 2 and 3 clinical trials.[25][26](A1)
Options for consolidation therapy include high-dose radiation (45 Gy), low-dose radiation (23.4 Gy), and dose-intensive chemotherapy with agents such as carmustine, thiotepa, cyclophosphamide, busulfan, cytarabine, and etoposide. Importantly, no survival compromise was seen in the rtesutls from studies omitting WBRT from consolidation therapy, and patients had better neurological outcomes.[4][6] Results from studies with reduced-dose WBRT in consolidation therapy have shown good progression-free and overall survival rates. Still, longer follow-up periods will be required to determine the long-term effects of the radiation, particularly on neuropsychological function. Myeloablative therapy and autologous stem cell transplant are also options for consolidation, though careful patient selection is important as the regimen can be hard to tolerate. This approach to consolidation is best tolerated by patients younger than 70 years old who are also functionally independent and have few comorbid medical conditions.[2]
Unfortunately, more than half of patients with CNS lymphoma experience eventual relapse, with an average survival of 2 months at that time.[4][17] Relapse can occur as much as 10 years after initial treatment; most occur within 5 years. While the optimal salvage regimen remains investigational, additional HD-MTX, if previously sensitive, along with other CNS agent-penetrants, such as thiotepa, cytarabine, cytarabine liposome injection, etoposide, and ifosfamide, have shown promising results.[6] Additionally, myeloablative therapy followed by stem cell transplant is a good option in younger patients. WBRT, if not previously used, remains an effective salvage option with a median overall survival rate of 11 to 19 months.[4] Several new therapeutic agents, such as lenalidomide, ibrutinib, buparlisib, nivolumab, pemetrexed, pomalidomide, temsirolimus, and pembrolizumab, are currently under investigation. Prophylaxis with HD-MTX can be considered in patients with high-risk systemic non-Hodgkin lymphoma to prevent CNS dissemination. High-risk features in these patients include a high international prognostic index score and extranodal disease, particularly in the testes.[6]
Posttreatment Monitoring for Relapse
Upon completion of multimodal chemotherapy, close and ongoing monitoring for evidence of relapse is imperative. For the first 2 years after completing initial therapy, patients should be monitored every 3 months, then every 6 months for the following 3 to 5 years, and annually until the 10-year survival milestone is reached.[11] Comprehensive monitoring includes a thorough medical history, general and neurological examination, brain imaging (MRI is the preferred modality), ophthalmologic examination, and possibly lumbar puncture. Relapsed PCNSL should undergo complete re-staging. Re-staging is not necessary for lesions refractory to first-line therapy.[1] (B3)
Differential Diagnosis
The differential diagnosis for this condition includes the following:
- The clinical presentation must guide thinking about the differential diagnosis.
- When facing known intraparenchymal disease, the chief differentials are other primary or secondary CNS neoplasms, such as high-grade gliomas and metastatic lesions.
- Whenever diffusion restriction is noted on MRI, cerebral infarct must be considered along with tumefactive demyelination or other causes of inflammatory lesions.
- When ring-enhancing lesions are noted in the setting of immunocompromise, toxoplasmosis, and fungal abscesses must be excluded.[1][15]
- Tolosa Hunt syndrome, a painful ophthalmoplegia caused by nonspecific inflammation of the cavernous sinus or superior orbital fissure, exhibits clinical and radiographic features similar to PCNSL, including steroid responsiveness.[27]
Toxicity and Adverse Effect Management
Up to 5% of patients treated with HD-MTX develop dose-related nephropathy. Adequate hydration, urinary alkalinization, avoidance of penicillin, and other drugs that interact with methotrexate are ways to mitigate this toxicity. The recommendation is to observe a 2-day gap between iodinated contrast use for imaging and HD-MTX administration. Leucovorin rescue with escalated dosing strategies, as well as using the enzyme carboxypeptidase G2 to facilitate methotrexate clearance via the kidneys, are other effective options.[6] Leukoencephalopathy following WBRT is due to the involvement of neural progenitor cells and is observed in up to 24% of cases. This condition can present as subcortical dementia, gait ataxia, and incontinence.[16]
Staging
Suspicion of CNS lymphoma necessitates staging to establish and clarify the extent of disease. This is particularly important when CNS lymphoma is possibly secondary to systemic lymphoma. Full body PET scanning or computed tomography of the chest, abdomen, and pelvis, supplemented by bone marrow biopsy and, in male patients, by dedicated testicular ultrasound.[19] Systemic disease is found in approximately 4% to 12% of patients originally thought to have PCNSL.[4][6]
Complete staging of CNS lymphoma requires gadolinium-enhanced magnetic resonance imaging of the brain and complete spinal cord, lumbar puncture, and slit lamp examination. CSF studies should include cytology and flow cytometry in addition to the standard cell counts and indices for protein and glucose.[19] Lactate dehydrogenase levels and serologies for HIV, hepatitis B, and hepatitis C are often obtained.[6] Optical coherence tomography and fluorescein angiography angiography are additional options in the diagnosis and staging of intraocular lymphoma.
Prognosis
Aggressive treatment is the standard of care in CNS lymphoma; even patients with heavy symptomatology or disease-related poor performance status are likely to benefit from the lesion’s chemo- and radiosensitivity.[2] The 5- and 10-year survival rate for PCNSL is reportedly 30% and 20%.[11] WBRT monotherapy shows a median survival of 12 to 18 months and a 5-year survival of 18% to 35%, albeit with a high risk of neurotoxicity.[16] The Radiation Therapy Oncology Group (RTOG) demonstrated that irradiation alone conferred a median survival of only 12 to 18 months, with a 5-year survival of only 3% to 4%. The RTOG studied pre-radiation CHOP—cyclophosphamide, doxorubicin hydrochloride (hydroxydaunorubicin), vincristine sulfate (Oncovin), and prednisone, a standard NHL chemotherapeutic regimen—followed by whole-brain radiotherapy, which showed no survival benefits and had significant chemotherapy-related toxicities.
When HD-MTX was used concurrently, median survival increased to 40 months, with 5-year survival rates rising by almost 22%. Patients with WBRT are vulnerable to leukoencephalopathy, which develops in 50% to 100% of older patients and up to 30% of patients younger than 60 and often presents as subcortical dementia, gait ataxia, and incontinence.[16] Periventricular white matter changes, ventricular enlargement, and cortical atrophy typically accompany leukoencephalopathy.[11] Demyelination and hippocampal neuronal loss have been postulated as the probable mechanisms.[11] For these reasons, WBRT is now only considered in patients with contraindications to chemotherapy or as a salvage therapy in refractory or relapsed cases.[11]
Current attention focuses on the long-term outcomes of consolidation therapies, and recent studies have highlighted the excellent disease control afforded by high-dose chemotherapy in combination with stem cell transplantation.[28] Recently published long-term analysis of the randomized phase 2 PRECIS study (ClinicalTrials.gov identifier NCT00863460) that compared WBRT to autologous stem cell transplantation (ASCT) reported median 8-year event-free survival rate of 67% in the ASCT arm and 39% in the WBRT arms (p = 0.03), with a significantly lower risk of relapse after ASCT (hazard ratio, 0.13; p < 0.001).[29] Neurocognitive performance and balance were notably better with ASCT than after WBRT. These recent data support using ASCT, preceded by high-dose chemotherapy (specifically, thiotepa, busulfan, and cyclophosphamide), as a first-line approach to consolidation in eligible patients.
Despite advances in chemotherapy and immunomodulatory treatments, overall survival remains challenging, with 5-year survival rates not yet attaining 40%.[30] Barriers to improved survival include:
- Limited drug access to the CNS due to the blood-brain barrier
- Performance status limits tolerability of intensive therapeutic regimens, especially among older adults
- PCNSL tumor cells may possess heretofore unidentified inherent resistance to chemotherapeutic agents [5]
Two well-known prognostication models are commonly used: the International Extra-nodal Lymphoma Study Group model includes age, performance status, lactate dehydrogenase levels, CSF protein concentration, and whether or not deep brain structures are involved; the Memorial Sloan Kettering Cancer Center model includes age and performance status.[11]
Complications
Several complications, such as gait impairment, memory loss, and incontinence, can arise in patients treated with WBRT. These most commonly occur in individuals older than 60.[6] Reports also exist of post-traumatic stress disorders in individuals after treatment. The risk of developing a second malignancy increases in long-term CNS lymphoma survivors, particularly in younger age groups. Patients with secondary CNS lymphoma would be at an increased risk for radiation-associated cardiovascular disease if applying radiation to the chest. Using anthracyclines, such as doxorubicin, has demonstrated cardiotoxicity, and rituximab is associated with an increased risk of progressive multifocal leukoencephalopathy. In post-transplant indivisuals with CNS lymphoma, allograft failure is a risk, particularly if the immunosuppressant is reduced or halted to reconstitute immune function.
Deterrence and Patient Education
Patients require education regarding their disease process and potential pitfalls to avoid while undergoing therapy. When given in high doses, MTX can precipitate in renal tubules and can cause direct tubular injury. This risk is increased when urine is acidic and in volume-depleted states, making adequate hydration paramount. Certain drugs such as nonsteriod anti-inflammatory drugs, penicillins, probenecid, phenytoin, ciprofloxacin, proton pump inhibitors, and levetiracetam interfere with renal clearance of MTX and should be avoided whenever possible.
Enhancing Healthcare Team Outcomes
Prompt diagnosis and treatment are crucial.[11] Definitive treatment should ideally be initiated at an established center within 14 days of diagnosis.[1] The involvement of an interdisciplinary team is a critical dimension in the appropriate management of CNS cancers.[31] Successful requires a collaborative and interprofessional team approach to enhance the patient’s quality of life before, during, and after treatment. Team members often include clinicians with various expertise, such as:
- Neurologists
- Ophthalmologists
- Neuroradiologists
- Neurosurgeons
- Neuropathologists
- Psychiatrists
- Medical oncologists
- Radiation oncologists
- Palliative care specialists.
- Pain management specialists
- Physical and occupational therapists
- Social workers
- Spiritual care leaders
- Oncology nurses
- Oncologic pharmacists
CNS lymphoma has a guarded prognosis; every treatment has significant adverse effects, which add to the morbidity. Relapse is common, even in patients who do respond to treatment. Involving palliative care medicine early in the disease course can be helpful. Comfort care and quality of life should not be sacrificed with exhaustive tests and procedures that do not change the prognosis.
Media
(Click Image to Enlarge)
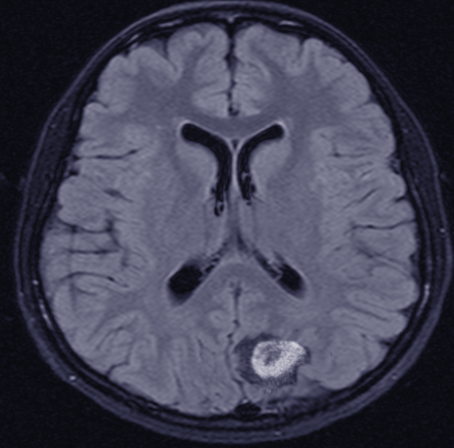
Central Nervous System Lymphoma, MRI. A magnetic resonance image of the brain with gadolinium contrast is the recommended first test in the diagnostic workup for patients with suspected primary central nervous lymphoma.
Contributed by O Chaigasame, MD
(Click Image to Enlarge)
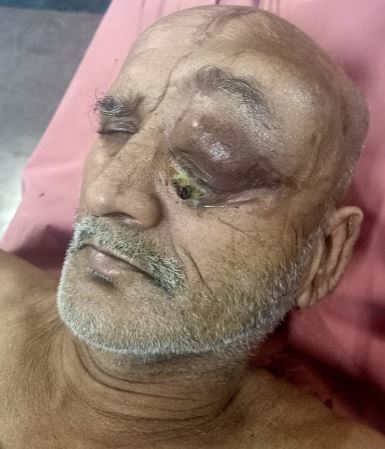
Orbito-Frontal Mass, Central Nervous System Lymphoma. This lymphoma is recurrent. These lesions (with cranial vault involvement, soft-tissue scalp swelling, and underlying intracranial mass) can mimic a meningioma.
Contributed by S Munakomi, MD
(Click Image to Enlarge)
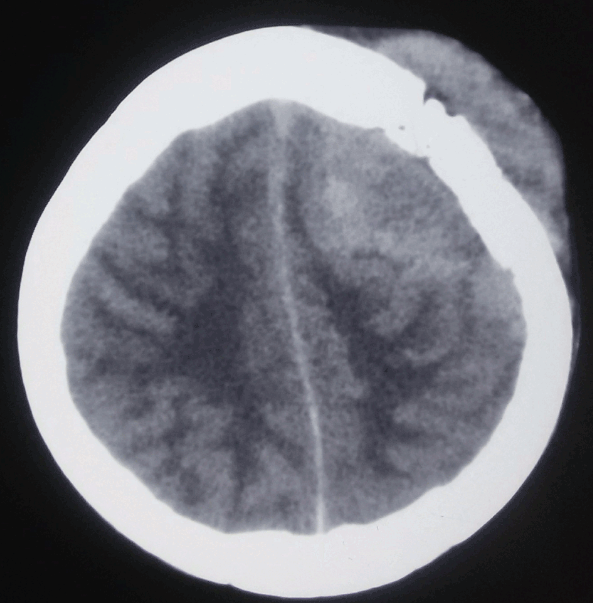
Central Nervous System Lymphoma, Hyperdense Calvarial Based Lesion. This is a primary lymphoma, referring to isolated involvement of the craniospinal axis in the absence of primary tumor elsewhere in the body.
Contributed by S Munakomi, MD
References
Fox CP, Phillips EH, Smith J, Linton K, Gallop-Evans E, Hemmaway C, Auer DP, Fuller C, Davies AJ, McKay P, Cwynarski K, British Society for Haematology. Guidelines for the diagnosis and management of primary central nervous system diffuse large B-cell lymphoma. British journal of haematology. 2019 Feb:184(3):348-363. doi: 10.1111/bjh.15661. Epub 2018 Nov 23 [PubMed PMID: 30467845]
Schaff L. Central Nervous System Lymphoma. Continuum (Minneapolis, Minn.). 2023 Dec 1:29(6):1710-1726. doi: 10.1212/CON.0000000000001356. Epub [PubMed PMID: 38085895]
van Westrhenen A, Smidt LCA, Seute T, Nierkens S, Stork ACJ, Minnema MC, Snijders TJ. Diagnostic markers for CNS lymphoma in blood and cerebrospinal fluid: a systematic review. British journal of haematology. 2018 Aug:182(3):384-403. doi: 10.1111/bjh.15410. Epub 2018 May 29 [PubMed PMID: 29808930]
Level 1 (high-level) evidenceHan CH, Batchelor TT. Diagnosis and management of primary central nervous system lymphoma. Cancer. 2017 Nov 15:123(22):4314-4324. doi: 10.1002/cncr.30965. Epub 2017 Sep 26 [PubMed PMID: 28950405]
Yang XL, Liu YB. Advances in Pathobiology of Primary Central Nervous System Lymphoma. Chinese medical journal. 2017 Aug 20:130(16):1973-1979. doi: 10.4103/0366-6999.211879. Epub [PubMed PMID: 28776551]
Level 3 (low-level) evidenceWang CC, Carnevale J, Rubenstein JL. Progress in central nervous system lymphomas. British journal of haematology. 2014 Aug:166(3):311-25. doi: 10.1111/bjh.12938. Epub 2014 May 16 [PubMed PMID: 24837460]
Bathla G, Hegde A. Lymphomatous involvement of the central nervous system. Clinical radiology. 2016 Jun:71(6):602-9. doi: 10.1016/j.crad.2016.02.006. Epub 2016 Mar 30 [PubMed PMID: 27038652]
Lv C, Wang J, Zhou M, Xu JY, Chen B, Wan Y. Primary central nervous system lymphoma in the United States, 1975-2017. Therapeutic advances in hematology. 2022:13():20406207211066166. doi: 10.1177/20406207211066166. Epub 2022 Jan 23 [PubMed PMID: 35096360]
Level 3 (low-level) evidenceOstrom QT, Price M, Neff C, Cioffi G, Waite KA, Kruchko C, Barnholtz-Sloan JS. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2015-2019. Neuro-oncology. 2022 Oct 5:24(Suppl 5):v1-v95. doi: 10.1093/neuonc/noac202. Epub [PubMed PMID: 36196752]
Lukas RV, Stupp R, Gondi V, Raizer JJ. Primary Central Nervous System Lymphoma-PART 1: Epidemiology, Diagnosis, Staging, and Prognosis. Oncology (Williston Park, N.Y.). 2018 Jan 15:32(1):17-22 [PubMed PMID: 29447417]
Löw S, Han CH, Batchelor TT. Primary central nervous system lymphoma. Therapeutic advances in neurological disorders. 2018:11():1756286418793562. doi: 10.1177/1756286418793562. Epub 2018 Oct 5 [PubMed PMID: 30305848]
Level 3 (low-level) evidenceGiannini C, Dogan A, Salomão DR. CNS lymphoma: a practical diagnostic approach. Journal of neuropathology and experimental neurology. 2014 Jun:73(6):478-94. doi: 10.1097/NEN.0000000000000076. Epub [PubMed PMID: 24806301]
Kaburaki T, Taoka K. Diagnosis and management of vitreoretinal lymphoma: present and future treatment perspectives. Japanese journal of ophthalmology. 2023 Jul:67(4):363-381. doi: 10.1007/s10384-023-00997-6. Epub 2023 May 20 [PubMed PMID: 37209195]
Level 3 (low-level) evidenceRubenstein JL, Gupta NK, Mannis GN, Lamarre AK, Treseler P. How I treat CNS lymphomas. Blood. 2013 Oct 3:122(14):2318-30. doi: 10.1182/blood-2013-06-453084. Epub 2013 Aug 20 [PubMed PMID: 23963042]
Chiavazza C, Pellerino A, Ferrio F, Cistaro A, Soffietti R, Rudà R. Primary CNS Lymphomas: Challenges in Diagnosis and Monitoring. BioMed research international. 2018:2018():3606970. doi: 10.1155/2018/3606970. Epub 2018 Jun 21 [PubMed PMID: 30035121]
Gerstner ER, Batchelor TT. Primary central nervous system lymphoma. Archives of neurology. 2010 Mar:67(3):291-7. doi: 10.1001/archneurol.2010.3. Epub [PubMed PMID: 20212226]
Grommes C, DeAngelis LM. Primary CNS Lymphoma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2017 Jul 20:35(21):2410-2418. doi: 10.1200/JCO.2017.72.7602. Epub 2017 Jun 22 [PubMed PMID: 28640701]
Ferreri AJM, Calimeri T, Cwynarski K, Dietrich J, Grommes C, Hoang-Xuan K, Hu LS, Illerhaus G, Nayak L, Ponzoni M, Batchelor TT. Primary central nervous system lymphoma. Nature reviews. Disease primers. 2023 Jun 15:9(1):29. doi: 10.1038/s41572-023-00439-0. Epub 2023 Jun 15 [PubMed PMID: 37322012]
Shah T,Venur VA, Central Nervous System Lymphoma. Seminars in neurology. 2023 Dec [PubMed PMID: 37995744]
Haldorsen IS, Espeland A, Larsson EM. Central nervous system lymphoma: characteristic findings on traditional and advanced imaging. AJNR. American journal of neuroradiology. 2011 Jun-Jul:32(6):984-92. doi: 10.3174/ajnr.A2171. Epub 2010 Jul 8 [PubMed PMID: 20616176]
Cheng G, Zhang J. Imaging features (CT, MRI, MRS, and PET/CT) of primary central nervous system lymphoma in immunocompetent patients. Neurological sciences : official journal of the Italian Neurological Society and of the Italian Society of Clinical Neurophysiology. 2019 Mar:40(3):535-542. doi: 10.1007/s10072-018-3669-7. Epub 2018 Dec 22 [PubMed PMID: 30580380]
Cao L, Zhang M, Zhang Y, Ji B, Wang X, Wang X. Progress of radiological‑pathological workflows in the differential diagnosis between primary central nervous system lymphoma and high‑grade glioma (Review). Oncology reports. 2023 Jan:49(1):. pii: 20. doi: 10.3892/or.2022.8457. Epub 2022 Dec 9 [PubMed PMID: 36484403]
Schaff LR, Grommes C. Primary central nervous system lymphoma. Blood. 2022 Sep 1:140(9):971-979. doi: 10.1182/blood.2020008377. Epub [PubMed PMID: 34699590]
Scheichel F, Pinggera D, Popadic B, Sherif C, Marhold F, Freyschlag CF. An Update on Neurosurgical Management of Primary CNS Lymphoma in Immunocompetent Patients. Frontiers in oncology. 2022:12():884724. doi: 10.3389/fonc.2022.884724. Epub 2022 Apr 20 [PubMed PMID: 35515113]
Ferreri AJ, Cwynarski K, Pulczynski E, Ponzoni M, Deckert M, Politi LS, Torri V, Fox CP, Rosée PL, Schorb E, Ambrosetti A, Roth A, Hemmaway C, Ferrari A, Linton KM, Rudà R, Binder M, Pukrop T, Balzarotti M, Fabbri A, Johnson P, Gørløv JS, Hess G, Panse J, Pisani F, Tucci A, Stilgenbauer S, Hertenstein B, Keller U, Krause SW, Levis A, Schmoll HJ, Cavalli F, Finke J, Reni M, Zucca E, Illerhaus G, International Extranodal Lymphoma Study Group (IELSG). Chemoimmunotherapy with methotrexate, cytarabine, thiotepa, and rituximab (MATRix regimen) in patients with primary CNS lymphoma: results of the first randomisation of the International Extranodal Lymphoma Study Group-32 (IELSG32) phase 2 trial. The Lancet. Haematology. 2016 May:3(5):e217-27. doi: 10.1016/S2352-3026(16)00036-3. Epub 2016 Apr 6 [PubMed PMID: 27132696]
Bromberg JEC, Issa S, Bakunina K, Minnema MC, Seute T, Durian M, Cull G, Schouten HC, Stevens WBC, Zijlstra JM, Baars JW, Nijland M, Mason KD, Beeker A, van den Bent MJ, Beijert M, Gonzales M, de Jong D, Doorduijn JK. Rituximab in patients with primary CNS lymphoma (HOVON 105/ALLG NHL 24): a randomised, open-label, phase 3 intergroup study. The Lancet. Oncology. 2019 Feb:20(2):216-228. doi: 10.1016/S1470-2045(18)30747-2. Epub 2019 Jan 7 [PubMed PMID: 30630772]
Level 1 (high-level) evidenceShazly TA, Mitchell EB, Bonhomme GR, Schuman JS. Lymphoma of the orbit masquerading as Tolosa-Hunt syndrome. BMC ophthalmology. 2015 May 15:15():51. doi: 10.1186/s12886-015-0037-8. Epub 2015 May 15 [PubMed PMID: 25971316]
Barden MM, Omuro AM. Top advances of the year: Neuro-oncology. Cancer. 2023 May 15:129(10):1467-1472. doi: 10.1002/cncr.34711. Epub 2023 Feb 24 [PubMed PMID: 36825454]
Level 3 (low-level) evidenceHouillier C, Dureau S, Taillandier L, Houot R, Chinot O, Moluçon-Chabrot C, Schmitt A, Gressin R, Choquet S, Damaj G, Peyrade F, Abraham J, Delwail V, Gyan E, Sanhes L, Cornillon J, Garidi R, Delmer A, Al Jijakli A, Morel P, Waultier A, Paillassa J, Chauchet A, Gastinne T, Laadhari M, Plissonnier AS, Feuvret L, Cassoux N, Touitou V, Ricard D, Hoang-Xuan K, Soussain C, LOC Network for CNS Lymphoma. Radiotherapy or Autologous Stem-Cell Transplantation for Primary CNS Lymphoma in Patients Age 60 Years and Younger: Long-Term Results of the Randomized Phase II PRECIS Study. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2022 Nov 10:40(32):3692-3698. doi: 10.1200/JCO.22.00491. Epub 2022 Jul 14 [PubMed PMID: 35834762]
Level 1 (high-level) evidenceHouillier C, Soussain C, Ghesquières H, Soubeyran P, Chinot O, Taillandier L, Lamy T, Choquet S, Ahle G, Damaj G, Agapé P, Moluçon-Chabrot C, Amiel A, Delwail V, Fabbro M, Jardin F, Chauchet A, Moles-Moreau MP, Morschhauser F, Casasnovas O, Gressin R, Fornecker LM, Abraham J, Marolleau JP, Tempescul A, Campello C, Colin P, Tamburini J, Laribi K, Serrier C, Haioun C, Chebrek S, Schmitt A, Blonski M, Houot R, Boyle E, Bay JO, Oberic L, Tabouret E, Waultier A, Martin-Duverneuil N, Touitou V, Cassoux N, Kas A, Mokhtari K, Charlotte F, Alentorn A, Feuvret L, Le Garff-Tavernier M, Costopoulos M, Mathon B, Peyre M, Delgadillo D, Douzane H, Genet D, Aidaoui B, Hoang-Xuan K, Gyan E. Management and outcome of primary CNS lymphoma in the modern era: An LOC network study. Neurology. 2020 Mar 10:94(10):e1027-e1039. doi: 10.1212/WNL.0000000000008900. Epub 2020 Jan 6 [PubMed PMID: 31907289]
Nabors LB,Portnow J,Ahluwalia M,Baehring J,Brem H,Brem S,Butowski N,Campian JL,Clark SW,Fabiano AJ,Forsyth P,Hattangadi-Gluth J,Holdhoff M,Horbinski C,Junck L,Kaley T,Kumthekar P,Loeffler JS,Mrugala MM,Nagpal S,Pandey M,Parney I,Peters K,Puduvalli VK,Robins I,Rockhill J,Rusthoven C,Shonka N,Shrieve DC,Swinnen LJ,Weiss S,Wen PY,Willmarth NE,Bergman MA,Darlow SD, Central Nervous System Cancers, Version 3.2020, NCCN Clinical Practice Guidelines in Oncology. Journal of the National Comprehensive Cancer Network : JNCCN. 2020 Nov 2 [PubMed PMID: 33152694]
Level 1 (high-level) evidence