 Autosomal Dominant Polycystic Kidney Disease
Autosomal Dominant Polycystic Kidney Disease
Introduction
Autosomal dominant polycystic kidney disease (ADPKD) is the most common genetic cause of renal failure worldwide. ADPKD is a multisystem and progressive inherited disorder with renal cyst formation, kidney enlargement, and extrarenal organ involvement (eg, liver, pancreas, spleen, cardiac, and arachnoid membranes).
In the adult population, ADPKD occurs in all races and is responsible for 6% to 10% of all patients on dialysis in the United States. Cysts may be detected in childhood or even in utero, but clinical manifestations do not typically appear until the third or fourth decade of life.
Autosomal recessive polycystic kidney disease (ARPKD) is very rare and affects 1 in 20,000 - 40,000 live births.[1] ARPKD has a much more severe clinical course and is caused by mutations in polycystic kidney and hepatic disease 1 (PKHD1), which encodes fibrocystin.[1] ARPKD usually presents in childhood and often causes death in childhood or perinatally. Please see StatPearl's companion reference article, Autosomal Recessive Polycystic Kidney Disease.[1]
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
ADPKD involves mutations in various genes, two of which are identified. PKD1 (chromosome 16p13.3) accounts for 85% of ADPKD cases, and PKD2 (4q21) contributes 15%.
Mutations in GANAB are thought to contribute to 1% of ADPKD patients with more variable polycystic liver disease.[2]
Mutations in the PKD1 and PKD2 genes express similar phenotypes. In the PKD1 form, about 50% of patients need renal replacement therapy by 60 years. PKD2 mutations are seen in older individuals and present with a milder disease with fewer renal cysts, late-onset hypertension, and less end-stage kidney disease (ESKD) than PKD1.[3]
Epidemiology
ADPKD is a worldwide condition affecting all races, with a prevalence rate of diagnosed cases ranging from 1:400 to 1:1,000.[4] While some patients remain asymptomatic their whole lives, ADPKD is not considered a benign disorder, as about half and possibly as many as three-quarters of all patients affected develop end-stage kidney disease by the time they reach 70 years of age, constituting up to 10% of all patients with ESKD.[5][6][7]
End-stage kidney disease caused by ADPKD in African Americans is less common than in White Americans. ESKD caused by ADPKD in men and women are respectively 8.7 and 6.9 per 1 million population in the United States using data from 1998 to 2001.
Corresponding data for the same time frame are 7.8 and 6.0 per million individuals in Europe and 5.6 and 4.0 per million in Japan. The worldwide incidence is about 12.5 million individuals.[8][9]
Due to increased awareness, early detection, and hypertension treatment, the age of onset of ESKD has increased, and all-cause mortality has decreased. Renal disease is more severe in males; however, polycystic liver disease is much more prominent in females, suggesting an element of hormonal regulation.[10]
Pathophysiology
Eighty-five percent of patients with ADPKD have PKD1 mutations, 15% have the PKD2 gene mutation, and about 1% have GANAB gene mutations. Other mutations leading to PKD are unidentified, and 10% to 15% of patients with ADPKD have no known family history, suggesting a high de novo mutation rate.[10] PKD1 and PKD2 mutations have the same phenotype, but patients with PKD2 have a milder disease with fewer renal cysts, later onset of hypertension, and less ESKD than patients with PKD1. Patients with GANAB mutations also have a milder phenotype than PKD1 but have more associated hepatic disease.
- PKD1 codes for polycystin-1 (PC1), an integral membrane protein with a long, extracellular N-terminal, 11 transmembrane regions, and a short, intracellular C-terminal. PC1 is present in focal adhesions, primary cilia, tight junctions, desmosomes, and adherens junctions and plays a vital role in cell-to-cell and cell-to-matrix interactions.
- PKD2 codes for polycystin-2 (PC2), which has a short cytoplasmic N-terminal, 6 transmembrane regions, and a short cytoplasmic C-terminal. PC2 is present in the endoplasmic reticulum, plasma membrane, primary cilia, centrosomes, and mitotic spindles in dividing cells. PC2 is also involved in intracellular calcium regulation.
- Both PCI and PC2 are present in the primary cilia of renal epithelial cells and play a role in producing transmembrane calcium currents in the presence of stretch or luminal flow and increasing intracellular calcium.[11][12][13] PC1 and PC2 play a crucial role in cell proliferation, differentiation, and fluid secretion through G protein-mediated or JAK-STAT-mediated signaling pathways, which results in increased intracellular concentrations of cyclic adenosine monophosphate (cAMP), leading to an increase in chloride secretion across the luminal membrane.[14]
Chloride-rich fluid secretion is an essential component of cystogenesis, leading to the expansion of cysts, even after the detachment from their parent nephron. The accumulation of cyst fluid, rich in chloride and sodium, relies on the active luminal excretion of chloride primarily through the cystic fibrosis transmembrane conductor regulator (CFTR).[15][16][17]
Increased levels of cAMP are found in animal models of ADPKD in the kidneys, liver, and vascular smooth muscle cells, and this plays a vital role in the proliferation of different cell types.[18][19] Cyclic AMP increases the proliferative pathways in cells derived from ADPKD kidneys while at the same time inhibiting the proliferation of cells from normal human kidneys.
Each renal cyst is believed to originate from a single, genetically transformed clonal hyperproliferative epithelial cell. A somatic mutation, known as the "second hit," in either the PKD1 or PKD2 gene leads to cyst growth and development. The continuous proliferation of epithelial cells, fluid secretion, and alterations in the extracellular matrix result in focal outpouching from the parent nephron.
Cyst formation can occur in proximal and distal tubules but is most common in the distal nephron and collecting duct.[10] Cysts become separate from the parent nephron when their size exceeds 2 cm and continue to autonomously secrete fluid, leading to cyst expansion and kidney enlargement, which, in turn, results in a reduction in functional nephrons. The continuous expansion of cysts compresses renal vessels, leading to intrarenal ischemia, which activates the renin-angiotensin-aldosterone system as well as other factors.[20]
Hypertension in ADPKD is postulated to be related to local areas of kidney ischemia, which develop due to cyst expansion. This results in increased renin release and a rise in blood pressure.[21][22] Attenuated arteries in the walls of cysts and cells in the connective tissue surrounding cysts contain cells with renin, suggesting that increasing cyst size leads to worsening hypertension.[23]
The cysts also trigger an inflammatory response in the surrounding renal parenchyma and promote renal fibrosis. Progressive cyst expansion, grreater systemic vascular resistance, sodium retention, and increassing renal fibrosis ultimately lead to ESKD.[13]
Liver cysts are a common manifestation of ADPKD and are more prevalent in women. They are shown to increase in size and number in response to high estrogen states such as pregnancy and oral contraceptive use.
In the hepatic system, the absence of polycystin leads to cyst formation, increased cell proliferation and apoptosis, enhanced fluid secretion, abnormal cell–matrix interactions, and alterations in cell polarity. Proliferative and secretive activities of cystic epithelium are regulated by estrogens either directly or by increasing growth signals, such as nerve growth factor, IGF1, FSH, and VEGF.[24]
A higher cyst burden, as reflected by increased total kidney volume, is considered a risk factor for the development of hypertension, even with normal serum creatinine levels.[25][26]
Histopathology
Histologic samples show epithelial cell proliferation, abnormal fluid secretion, and abnormal extracellular matrix deposition of the cystic epithelial cells. Alterations in the pericystic blood and lymphatic microvasculature accompany these changes.
Tubular dilation, microcysts, and tubular atrophy are common. Tubular dilation is also associated with an enlarged Bowman's space. There is extensive interstitial fibrosis in the peritubular area, especially near cysts.
Cyst enlargement compresses the surrounding nephrons, interstitium, and renal vasculature. Vascular changes include fibrosclerosis and lumen narrowing. Inflammatory cells are prominent, especially fibroblasts and monocytes.[10][27]
History and Physical
Patients with ADPKD can present with a variety of clinical conditions, such as autosomal dominant polycystic kidney disease, which is a disorder affecting many organ systems. Renal function can remain normal for decades. However, once the glomerular filtration rate (GFR) starts to decline, renal impairment is usually rapid, with an average GFR loss of 4.0 to 5.0 mL/minute/year.
Male sex, early age of onset of hypertension, PKD1 genotype, increased total kidney volume, and the presence of proteinuria are worse prognostic indicators. Total kidney volume is the primary predictive biomarker of future GFR loss.
The most common clinical presentations are hypertension, anemia, liver cysts, hematuria, flank pain, abdominal masses, urinary tract infections, renal failure, nephrolithiasis, and renal cancers.
Hypertension is the most universal and earliest clinical presentation in most patients with ADPKD.[21][22][28] Microalbuminuria, proteinuria, and hematuria are also more prevalent in patients with hypertension and ADPKD. Episodes of acute flank pain are often seen due to cyst bleeding, infection, stones, and, rarely, tumors.
Gross (visible) hematuria may also be the initial presenting symptom.[29] Cyst hemorrhage is a frequent complication causing gross hematuria when the cyst communicates with the collecting system. It can manifest as fever, raising the possibility of cyst infection. Occasionally, a hemorrhagic cyst will rupture, resulting in a retroperitoneal bleed.
Urinary tract infections (UTIs) are common in ADPKD. Urinary tract infections include cystitis, acute pyelonephritis, cyst infection, and perinephric abscesses. Escherichia coli, Klebsiella, Proteus species, and other Enterobacteriaceae are the most common infectious causes identified.
Renal stone disease occurs in about 20% to 25% of patients with ADPKD.[30][31][32][33] Most stones are composed of uric acid, calcium oxalate, or both. This is due to lower urinary volumes, intrarenal stasis due to anatomical obstruction, hypocitraturia, and low urinary magnesium levels.[34][35] Renal calculi can be challenging to diagnose on imaging in ADPKD patients because of calcifications in the renal parenchyma and cyst walls.[36][37] Unenhanced CT scans of the abdomen and pelvis are the most helpful for diagnosing nephrolithiasis in ADPKD patients.[36][37]
The prevalence of hepatic cysts increases with age, and polycystic liver disease should be suspected when four or more cysts are present in the hepatic parenchyma.[38][39] Patients may remain asymptomatic or present with liver impairment or pain secondary to infection or rupture of the cyst. About 7% to 36% of patients also have pancreatic cysts, which are more common with PKD2 mutations than with PKD1.[40]
Mitral valve prolapse and aortic regurgitation are the most common cardiovascular abnormalities associated with ADPKD.[41] It is also associated with increased coronary aneurysms, asymptomatic pericardial effusions, congenital heart malformations, aortic valve stenosis, and atrial fibrillation. Gastrointestinal abnormalities are also common; up to 50% of patients with ADPKD will also have diverticulosis.[10]
Although children with ADPKD do not generally have renal impairment (as opposed to ARPKD), they have higher rates of hypertension and proteinuria than children without ADPKD, which requires close monitoring and treatment.
It is also suggested that in children, the presence of early-onset ADPKD is equivalent to the incidence of ARPKD, so differentiation between these two disorders may be difficult.[10]
Evaluation
The diagnosis of autosomal dominant polycystic kidney disease is usually made by a combination of a positive family history, imaging with ultrasonography and/or CT scans, and clinical findings like hypertension and/or renal failure. The disorder should also be suspected in patients with renal impairment associated with multiple bilateral cysts on renal imaging by CT or renal ultrasound, with or without a family history of ADPKD (see Image. Polycystic Kidneys (ADPKD) and Liver Cysts).
Ultrasound is usually sufficient for asymptomatic patients with normal renal function (see Image. Polycystic Kidney). In children, if the initial ultrasound findings are negative for kidney cysts, follow-up ultrasounds should be deferred until adolescence (between ages 15 and 18).[42]
CT or MRI can help estimate the height-adjusted total kidney volume for risk stratification of disease progression and may be beneficial for management (see Image. Autosomal Dominant Polycystic Kidney Disease).
In patients with a strong family history and palpable kidneys, a baseline CT or MRI can be helpful in the management of possible future complications, tracking the progression of the disease, and early identification of possible nephrolithiasis.
The Renal Association Clinical Practice Guidelines recommend that parents or caregivers of individuals with ADPKD should receive education regarding the risk of inheriting ADPKD. Blood pressure should be checked every two years for at-risk children older than two years and young adults.
Genetic testing for autosomal dominant polycystic kidney disease can be performed using direct DNA sequencing or by DNA gene linkage analysis (which requires samples from at least three family members, including some with and without ADPKD.) Genetic testing enjoys an accuracy rate of 99% for both PKD1 and PKD2 genes and is commercially available.
The primary indication for genetic testing is in young adults with negative ultrasonographic findings who have a family history suggestive of ADPKD and are being evaluated as potential kidney donors, considering having children or becoming pregnant.[43] Genetic testing for ADPKD is not frequently performed or required in the diagnosis or management of most cases of ADPKD, although it can be useful in prognosis, diagnostic confirmation, estimating disease progression, and family planning.[44]
Genetic testing may also be used to confirm an ADPKD diagnosis when clinical factors and imaging are inconclusive.[45]
The decision to undergo genetic testing or renal ultrasonography for asymptomatic children should be collaborative, involving caretakers, parents, and healthcare professionals.
Ultrasound Criteria for ADPKD: Original Ravine PKD1 Diagnostic Criteria [46]
- Ages 15 to 29 years: 2 or more cysts, unilateral or bilateral.
- Ages 30 to 59 years: 2 or more cysts in each kidney.
- Ages 60 years or older: 4 or more cysts in each kidney.
These criteria are also used for diagnosing PKD2 but are less accurate. Two notable characteristics of these criteria are as follows: [47]
- Three or more total cysts in those aged 15 to 39 years have a positive predictive value of 100%.
- Two or fewer cysts in those older than 40 years have a negative predictive value of 100%.
Autosomal Dominant vs Autosomal Recessive Polycystic Kidney Disease
Autosomal Dominant Polycystic Kidney Disease:
- Symptoms typically begin starting at age 30.
- Incidence of 1:400 - 1:1,000. (Relatively common.)
- Hepatic cysts may develop but do not progress to significant liver disease or failure.
- Strong family history since the condition is inherited as a dominant trait.
- Average life expectancy is roughly from 53 to 70 years.
- ESKD typically develops in patients in their 60's.
Autosomal Recessive Polycystic Kidney Disease:
- Symptoms typically begin at birth or shortly thereafter.
- Incidence of 1:20,000 - 1:40,000. (Relatively rare.)
- Hepatic cysts develop early and may be seen prenatally. Liver disease is significant and may progress to cirrhosis.
- Usually no family history of this disease as it is passed on as a recessive trait.
- Many patients (20%) will die early, within the first month, but those who survive the newborn period have a 90% chance of living to at least 20 years.
- ESKD develops in 50% of patients who reach 20 years of age.
Treatment / Management
The Mayo classification system (classes 1A, 1B, 1C, 1D, and 1E) can be ranked from lowest to highest risk for worse disease outcomes. Of these criteria, total kidney volume is the most useful and diagnostic.[48][49]
The last three stages are associated with a higher risk for ESKD, and these criteria help to identify high-risk patients who would benefit from more aggressive management.
Life Style and Dietary Modifications are recommended, although not definitively proven to prevent ADPKD progression. Patients are instructed to drink three liters of fluids daily to suppress vasopressin, thereby decreasing cAMP production and inhibiting cyst production.[50][51] (B3)
All ADPKD patients are advised to limit their sodium intake to less than 2 grams per day. The CRISP study showed a positive correlation between the increase in total kidney volume and 24-hour urine sodium excretion.[52] Restricted sodium intake can also improve blood pressure. (B2)
Flank pain from other causes that may require intervention, such as infection, stone, herpes zoster, and neoplasms, should be excluded. Tricyclic antidepressants are useful, as in other chronic pain syndromes, and are well tolerated. Cyst aspiration, under ultrasound or CT guidance, can be done if distortion of the kidney by a large cyst is considered a likely or possible cause of the pain. If multiple cysts are contributing to pain, laparoscopic or surgical cyst fenestration or unroofing may be of benefit.
Cyst hemorrhage episodes are usually self-limited, and patients respond well to conservative management with bed rest, analgesics, and increased fluid intake to prevent clots from obstructing the upper urinary tract. Rarely, bleeding is more severe, leading to hemodynamic instability, which would require hospitalization, transfusions, and supportive care.
Cyst and urinary tract infections require immediate treatment to prevent retrograde seeding of the renal parenchyma.[53] Agents of choice include trimethoprim-sulfamethoxazole, ertapenem, chloramphenicol, fluoroquinolones, sulfamethoxazole/trimethoprim, and clindamycin as they all have good cyst penetration. If fever persists after 1 to 2 weeks of appropriate antimicrobial therapy, infected cysts should be drained percutaneously or surgically. In the case of end-stage polycystic kidneys, nephrectomy should be considered.
Nephrolithiasis is much more common in patients with ADPKD than in the general population. ADPKD patients will tend to make uric acid stones preferentially (most common), followed by calcium oxalate urinary calculi.[30] Symptomatic stones are treated the same as in other patients, but since most calculi are uric acid, urinary alkalinization therapy is suggested whenever possible.[54] (A1)
Potassium citrate and higher oral fluid intake (to increase urinary volume) are generally the treatments of choice in stone-forming conditions associated with ADPKD, such as distal acidification disorders, low urinary volumes, and aciduria. 24-hour urine testing is recommended to identify all the risk factors in ADPKD patients with or at risk for urolithiasis.
Management of hypertension is essential in reducing cardiovascular mortality and slowing the progression of renal failure. As per the HALT-PKD study, the target blood pressure range is less than 120 to 125/80 mm Hg, similar to other patients with chronic kidney disease.[55] In patients with preserved or nearly preserved GFR, a lower blood pressure goal of less than 110/75 mm Hg is associated with a decreased incidence of cardiovascular events and a slower rate of cyst growth.[55](A1)
Angiotensin inhibitors are the preferred agents if there is no contraindication. ACE inhibition also protects the glomeruli by decreasing intraglomerular pressure and reducing the rate of GFR decline in those with proteinuria. Beta-blockers and calcium-channel blockers are second-line treatments. Thiazides are preferred in patients with normal renal function as a third-line and in patients with hypercalciuria, while loop diuretics are recommended in patients with impaired renal function as an alternative to thiazides+.[56][57]
Tolvaptan slows the progression of autosomal dominant polycystic kidney disease by blocking the reception of vasopressin signaling at the V2 receptor, lowering the intracellular cyclic AMP that would otherwise stimulate cystic proliferation and growth.[50][51] It is the only FDA-approved medicine for ADPKD at high risk for disease progression; however, due to its high cost and adverse effects, its use is recommended only in patients at high risk of disease progression or who demonstrate rapidly declining kidney function.(B3)
Modified guidelines have shifted from using a patient's historical GFR to total kidney volume, which precedes a decline in GFR. It is thought that in older patients, medical comorbidities contribute to their declining GFR more than the ADPKD disorder; therefore, preference for tolvaptan use is geared towards patients 55 years or younger with a rapidly declining GFR and fewer comorbidities, but there is no definite age cutoff.[58][59][60](B3)
A current guideline for tolvaptan use is a yearly decline in GFR of ≥3.0 mL/min, primarily attributed to ADPKD. Loss estimation should be derived from at least five measurements over four years. Indicators that the GFR decline could be due to factors other than ADPKD include known vascular disease, uncontrolled hypertension, diabetes mellitus, and severe proteinuria (>1 g/d).[58] Tolvaptan use has not been studied in patients younger than 18.[58](B3)
The "Tolvaptan Efficacy and Safety in Management of Autosomal Dominant Polycystic Kidney Disease (TEMPO)" and "Replicating Evidence of Preserved Renal Function: An Investigation of Tolvaptan Safety and Efficacy in ADPKD (REPRISE)" are two landmark trials showing the efficacy of tolvaptan in preventing GFR decline.[61][62][63] However, tolvaptan comes with a black box warning due to its potential for liver failure. Baseline measurements of AST, ALT, and bilirubin should be checked and repeated at two and four weeks after initiation, followed by monthly measurements for the first eighteen months and then quarterly.(A1)
Due to the risks of liver injury, tolvaptan for ADPKD is only available through a restricted distribution protocol under an FDA-approved Risk Evaluation and Mitigation Strategy (REMS) program. Tolvaptan is contraindicated in patients taking CYP3A4 inhibitors, and these patients were excluded from the TEMPO and REPRISE trials. Common adverse effects among patients taking tolvaptan include increased thirst, polyuria, nocturia, polydipsia, and hypernatremia, common causes of drug discontinuation.[58][64](B3)
Statins are used in chronic kidney disease patients as renal failure is equivalent to coronary heart disease. Evidence for slowing the disease progress in ADPKD with statins is conflicting.[65]
Mammalian targets of rapamycin (mTOR) inhibitors, sirolimus and everolimus, have been studied but have not shown any benefit on renal outcomes.[64][66] Significant adverse effects also preclude their use.[64][66](A1)
Somatostatin and somatostatin analogs such as octreotide, lanreotide, and pasireotide have not been shown to affect the disease progression of ADPKD.[67](A1)
Nephrectomy is indicated in patients with ADPKD with unbearable abdominal discomfort, anorexia, renal cell carcinoma, renal hemorrhage, kidney infection with gas-forming agents, and staghorn calculi in nonfunctioning kidneys with persistent urinary tract infections. Nephrectomy for large, cystic kidneys can be considered for ESKD patients if symptomatic or to increase the abdominal capacity for a future renal transplant.
ADPKD with ESKD requires hemodialysis, renal transplantation, or peritoneal dialysis. Survival of ADPKD patients on hemodialysis is higher (10%-15% mortality at 5 years) than patients on hemodialysis due to other causes, due to less coronary artery disease in this population.[68]
Treatments on the Horizon
There are several possible future treatments being studied for ADPKD.[64]
- Glucosylceramide synthase inhibitors are a class of drugs involved in building complex glycosphingolipids and have been shown to reduce cyst formation in animal models through an unknown mechanism of action.[64][69]
- Transepithelial chloride secretion is a key mechanism behind cystogenesis and is mediated through the cystic fibrosis transmembrane conductance regulator (CFTR).[64] CFTR inhibitors are being studied for possible inhibition of cyst formation in ADPKD, but studies are currently preclinical.[64]
- Metformin is a well-established drug that blocks the aerobic glycolysis pathway, which is linked to cell proliferation, renal cyst formation, and progression. Studies found that metformin was safe and well-tolerated in adults with ADPKD and may reduce renal function decline.[64][70]
- Curcumin, produced by the spice turmeric, activates the transcription of antioxidants, suppresses inflammation, and reduces cell proliferation.[71] It is currently under study for various diseases, and researchers are investigating its potential positive effects on reducing cell growth and improving arterial function in ADPKD.[70][71][72] (A1)
Differential Diagnosis
Renal cysts can be seen in other systemic diseases.
Bardet-Biedl syndrome is a complex genetic disorder characterized by various symptoms, including vision problems, obesity, and kidney abnormalities.[73][74] Renal cysts can be one of the kidney manifestations in this syndrome but are not the sole defining feature.
HNF1B mutations can lead to renal cysts, often associated with various other systemic disorders such as early-onset diabetes, early-onset gout, pancreatic hypoplasia, abnormal liver function, and genital tract malformations.[75]
Medullary sponge kidney disease is a rare (1:5,000 individuals) congenital disorder that can cause the formation of small cysts or renal collecting tubule dilatations.[76] While it can lead to renal cyst-like structures, it does not progress to renal failure but is highly associated with nephrocalcinosis and nephrolithiasis, often due to hypocitraturia and hypercalciuria.[76] It is also associated with a higher incidence of urinary tract infections.[76] Please see StatPearl's companion reference article, Medullary Sponge Kidney.[76]
Orofaciodigital syndrome type I is a genetic disorder characterized by various abnormalities in the face, oral cavity, and digits.[77] While kidney abnormalities, including cysts, can occur, it is not typically the primary diagnostic criterion.[77]
Renal cysts can be seen in tuberous sclerosis, but this condition is usually associated with characteristic skin lesions, facial angiofibroma, connective tissue nevi, and cardiac, renal, and pulmonary manifestations.[78][79]
Conditions in the differential diagnosis of ADPKD include: [45]
- Autosomal dominant tubulointerstitial nephropathy
- Autosomal recessive polycystic kidney disease
- Bardet-Biedl syndrome
- HANAC syndrome
- HNF1B mutations
- Medullary sponge kidney
- Multiple simple renal cysts
- Orofaciodigial syndrome type I
- Parapelvic renal cysts
- Renal spongiosis
- Tuberous sclerosis
- Von Hippel-Lindau syndrome
Prognosis
About 50% of patients with ADPKD will develop ESKD by 70 years, and possibly up to 75%.[5][6][7][10]
A Canadian study reported that 25% of patients with ADPKD had ESKD by 47 years, 50% by 59 years, and 75% by 70 years.[5][10]
A French study showed that 22% of patients with ADPKD had ESKD by 50 years, 42% by 58 years, and 72% by 73 years.[10]
Overall life expectancy for patients with ADPKD is 53 to 70 years.[80][81]
Complications
- End-stage kidney disease requiring dialysis or renal transplantation is the most common complication of ADPKD.
- A ruptured cerebral aneurysm is the most serious extrarenal complication of ADPKD and has a four times higher prevalence in ADPKD patients compared to the general population.[82] The risk for a cerebral aneurysm increases with a personal or family history of cerebral aneurysm or subarachnoid hemorrhage, female sex, and older age.
- Smoking, hypertension, and excess alcohol intake are modifiable risk factors.
- Hepatic cysts are also a known complication of ADPKD, and the incidence increases with age, with an approximate prevalence of 10% to 20% up to the age of 30 years and reaching up to 50% to 70% in those older than 60.[83] Liver cysts may be associated with hepatic pain from cyst hemorrhage or hepatic cyst infection but rarely cause hepatic function impairment.
- ADPKD is often associated with diverticular disease, nephrolithiasis, and abdominal wall or inguinal hernias.
Deterrence and Patient Education
Patients should regularly follow up with nephrologists after diagnosis. Early diagnosis, risk assessment, appropriate pharmacotherapy, and proper management of nephrolithiasis risk factors, urinary citrate levels, hypertension, and proteinuria can help slow the progression of the disease.
Lifestyle modifications like increased oral intake of water, restricted salt intake, and avoidance of NSAIDs are associated with long-term benefits.
Enhancing Healthcare Team Outcomes
Autosomal dominant polycystic kidney disease is a systemic disorder that affects many organs; hence, a multidisciplinary management approach is necessary. A nephrologist, urologist, interventional radiologist, cardiologist, social worker, and dialysis nurse are key professionals required to care for these patients.
The patient must be educated on optimal blood pressure control and regular blood testing to assess renal function. Patients also need to be made aware of complications that may include cerebral aneurysms, hypertension, kidney stones, and end-stage kidney disease.[84][85]
The PRO-PKD score has been developed to predict the prognosis of ADPKD.[86][87] As uncontrolled hypertension accelerates the decline in renal function, it is essential to manage blood pressure optimally.
Pain control and early treatment of cysts or urinary tract infections can improve the quality of life in ADPKD patients. Education of patients and families about the condition can also reduce the number of hospitalizations.[88][89]
Media
(Click Image to Enlarge)
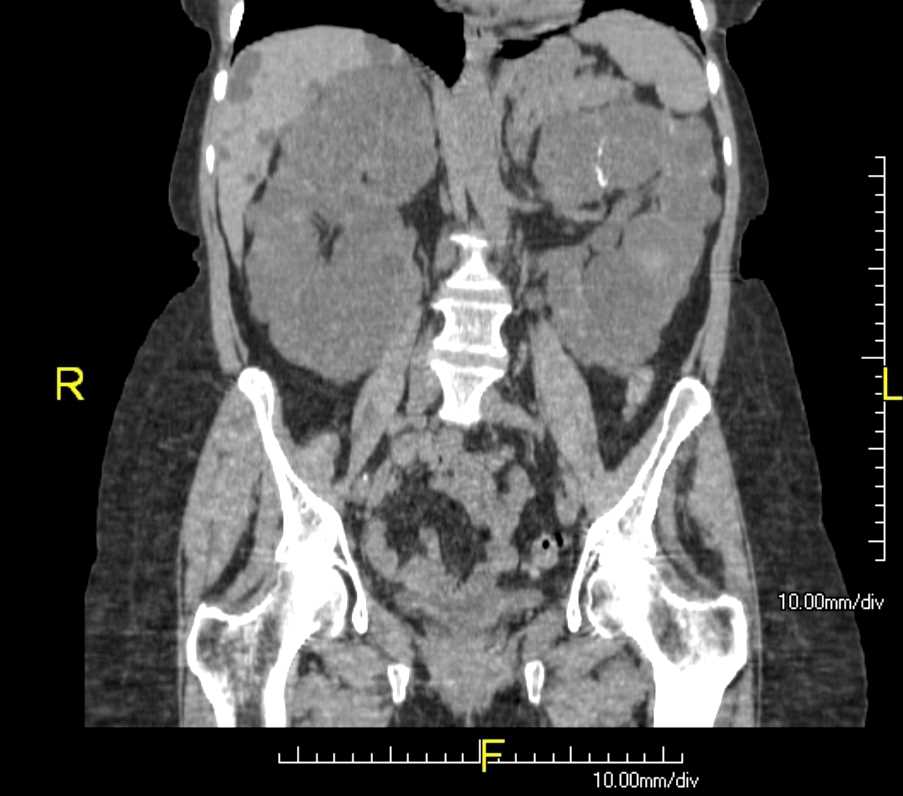
Polycystic Kidneys (ADPKD) and Liver Cysts. CT coronal view of abdomen.
Contributed by Scott Dulebohn, MD
(Click Image to Enlarge)
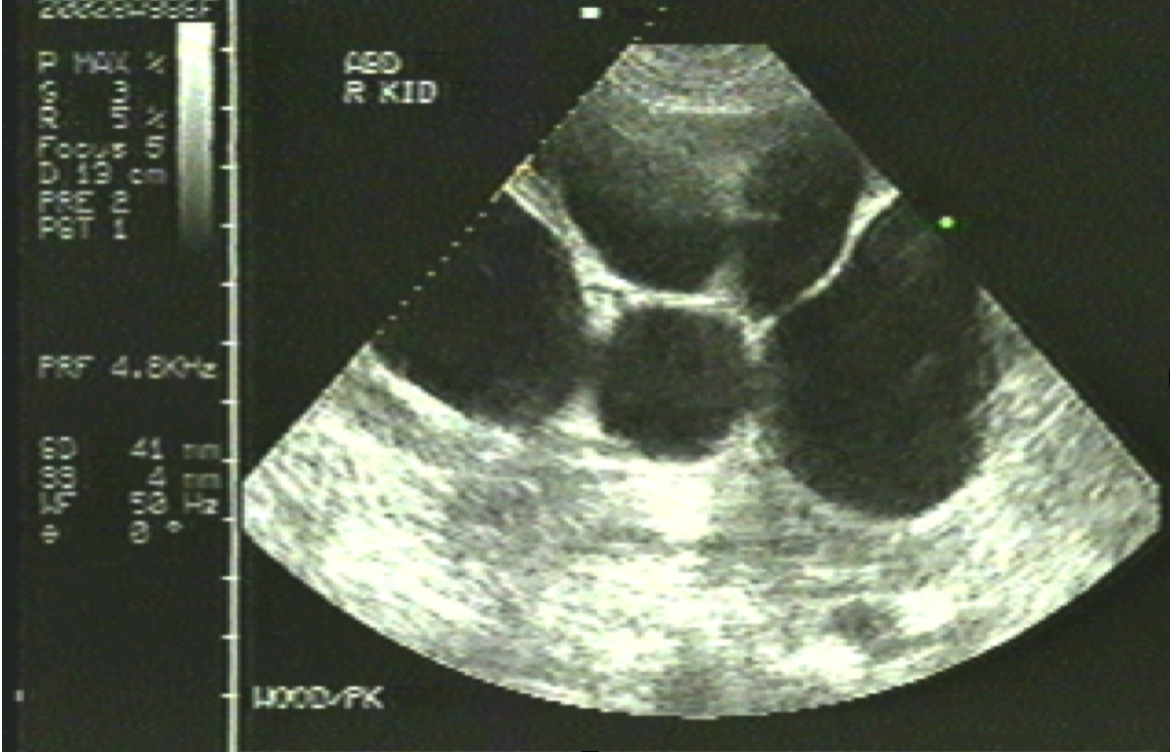
Polycystic Kidney
Contributed by Michael Lambert, MD
(Click Image to Enlarge)
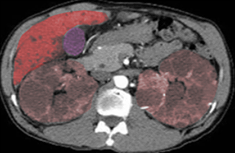
Autosomal Polycystic Kidney Disease
Image courtesy S Bhimji, MD
References
Subramanian S, Leslie SW, Ahmad T. Autosomal Recessive Polycystic Kidney Disease. StatPearls. 2024 Jan:(): [PubMed PMID: 30725822]
Porath B, Gainullin VG, Cornec-Le Gall E, Dillinger EK, Heyer CM, Hopp K, Edwards ME, Madsen CD, Mauritz SR, Banks CJ, Baheti S, Reddy B, Herrero JI, Bañales JM, Hogan MC, Tasic V, Watnick TJ, Chapman AB, Vigneau C, Lavainne F, Audrézet MP, Ferec C, Le Meur Y, Torres VE, Genkyst Study Group, HALT Progression of Polycystic Kidney Disease Group, Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease, Harris PC. Mutations in GANAB, Encoding the Glucosidase IIα Subunit, Cause Autosomal-Dominant Polycystic Kidney and Liver Disease. American journal of human genetics. 2016 Jun 2:98(6):1193-1207. doi: 10.1016/j.ajhg.2016.05.004. Epub [PubMed PMID: 27259053]
Steele C, You Z, Gitomer BY, Brosnahan GM, Abebe KZ, Braun WE, Chapman AB, Harris PC, Perrone RD, Steinman TI, Torres VE, Yu ASL, Chonchol M, Nowak KL. PKD1 Compared With PK D2 Genotype and Cardiac Hospitalizations in the Halt Progression of Polycystic Kidney Disease Studies. Kidney international reports. 2022 Jan:7(1):117-120. doi: 10.1016/j.ekir.2021.09.013. Epub 2021 Oct 7 [PubMed PMID: 35005320]
Shaw C, Simms RJ, Pitcher D, Sandford R. Epidemiology of patients in England and Wales with autosomal dominant polycystic kidney disease and end-stage renal failure. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 2014 Oct:29(10):1910-8. doi: 10.1093/ndt/gfu087. Epub 2014 Apr 15 [PubMed PMID: 24737444]
Budhram B, Akbari A, Brown P, Biyani M, Knoll G, Zimmerman D, Edwards C, McCormick B, Bugeja A, Sood MM. End-Stage Kidney Disease in Patients With Autosomal Dominant Polycystic Kidney Disease: A 12-Year Study Based on the Canadian Organ Replacement Registry. Canadian journal of kidney health and disease. 2018:5():2054358118778568. doi: 10.1177/2054358118778568. Epub 2018 Jun 11 [PubMed PMID: 29977583]
Spithoven EM, Kramer A, Meijer E, Orskov B, Wanner C, Caskey F, Collart F, Finne P, Fogarty DG, Groothoff JW, Hoitsma A, Nogier MB, Postorino M, Ravani P, Zurriaga O, Jager KJ, Gansevoort RT, ERA-EDTA Registry, EuroCYST Consortium, WGIKD, EuroCYST Consortium, WGIKD. Analysis of data from the ERA-EDTA Registry indicates that conventional treatments for chronic kidney disease do not reduce the need for renal replacement therapy in autosomal dominant polycystic kidney disease. Kidney international. 2014 Dec:86(6):1244-52. doi: 10.1038/ki.2014.120. Epub 2014 May 14 [PubMed PMID: 24827775]
Abbott KC, Agodoa LY. Polycystic kidney disease at end-stage renal disease in the United States: patient characteristics and survival. Clinical nephrology. 2002 Mar:57(3):208-14 [PubMed PMID: 11924752]
Harris PC, Torres VE. Polycystic kidney disease. Annual review of medicine. 2009:60():321-37. doi: 10.1146/annurev.med.60.101707.125712. Epub [PubMed PMID: 18947299]
Level 3 (low-level) evidenceLiebau MC, Mekahli D, Perrone R, Soyfer B, Fedeles S. Polycystic Kidney Disease Drug Development: A Conference Report. Kidney medicine. 2023 Mar:5(3):100596. doi: 10.1016/j.xkme.2022.100596. Epub 2022 Dec 27 [PubMed PMID: 36698747]
Bergmann C, Guay-Woodford LM, Harris PC, Horie S, Peters DJM, Torres VE. Polycystic kidney disease. Nature reviews. Disease primers. 2018 Dec 6:4(1):50. doi: 10.1038/s41572-018-0047-y. Epub 2018 Dec 6 [PubMed PMID: 30523303]
Gallagher AR, Germino GG, Somlo S. Molecular advances in autosomal dominant polycystic kidney disease. Advances in chronic kidney disease. 2010 Mar:17(2):118-30. doi: 10.1053/j.ackd.2010.01.002. Epub [PubMed PMID: 20219615]
Level 3 (low-level) evidenceHanaoka K, Qian F, Boletta A, Bhunia AK, Piontek K, Tsiokas L, Sukhatme VP, Guggino WB, Germino GG. Co-assembly of polycystin-1 and -2 produces unique cation-permeable currents. Nature. 2000 Dec 21-28:408(6815):990-4 [PubMed PMID: 11140688]
Level 3 (low-level) evidenceWatnick T, Germino G. From cilia to cyst. Nature genetics. 2003 Aug:34(4):355-6 [PubMed PMID: 12923538]
Olsan EE, Mukherjee S, Wulkersdorfer B, Shillingford JM, Giovannone AJ, Todorov G, Song X, Pei Y, Weimbs T. Signal transducer and activator of transcription-6 (STAT6) inhibition suppresses renal cyst growth in polycystic kidney disease. Proceedings of the National Academy of Sciences of the United States of America. 2011 Nov 1:108(44):18067-72. doi: 10.1073/pnas.1111966108. Epub 2011 Oct 24 [PubMed PMID: 22025716]
Level 3 (low-level) evidenceGrantham JJ, Ye M, Davidow C, Holub B, Sharma M. Evidence for a potent lipid secretagogue in the cyst fluids of patients with autosomal dominant polycystic kidney disease. Journal of the American Society of Nephrology : JASN. 1995 Oct:6(4):1242-9 [PubMed PMID: 8589292]
Level 3 (low-level) evidenceDavidow CJ, Maser RL, Rome LA, Calvet JP, Grantham JJ. The cystic fibrosis transmembrane conductance regulator mediates transepithelial fluid secretion by human autosomal dominant polycystic kidney disease epithelium in vitro. Kidney international. 1996 Jul:50(1):208-18 [PubMed PMID: 8807590]
Hanaoka K, Devuyst O, Schwiebert EM, Wilson PD, Guggino WB. A role for CFTR in human autosomal dominant polycystic kidney disease. The American journal of physiology. 1996 Jan:270(1 Pt 1):C389-99 [PubMed PMID: 8772467]
Grantham JJ, Ye M, Gattone VH 2nd, Sullivan LP. In vitro fluid secretion by epithelium from polycystic kidneys. The Journal of clinical investigation. 1995 Jan:95(1):195-202 [PubMed PMID: 7814614]
Grantham JJ. Lillian Jean Kaplan International Prize for advancement in the understanding of polycystic kidney disease. Understanding polycystic kidney disease: a systems biology approach. Kidney international. 2003 Oct:64(4):1157-62 [PubMed PMID: 12969132]
Level 3 (low-level) evidenceSagar PS, Rangan GK. Cardiovascular Manifestations and Management in ADPKD. Kidney international reports. 2023 Oct:8(10):1924-1940. doi: 10.1016/j.ekir.2023.07.017. Epub 2023 Aug 4 [PubMed PMID: 37850017]
Bell PE, Hossack KF, Gabow PA, Durr JA, Johnson AM, Schrier RW. Hypertension in autosomal dominant polycystic kidney disease. Kidney international. 1988 Nov:34(5):683-90 [PubMed PMID: 2974094]
Chapman AB, Johnson A, Gabow PA, Schrier RW. The renin-angiotensin-aldosterone system and autosomal dominant polycystic kidney disease. The New England journal of medicine. 1990 Oct 18:323(16):1091-6 [PubMed PMID: 2215576]
Torres VE, Donovan KA, Scicli G, Holley KE, Thibodeau SN, Carretero OA, Inagami T, McAteer JA, Johnson CM. Synthesis of renin by tubulocystic epithelium in autosomal-dominant polycystic kidney disease. Kidney international. 1992 Aug:42(2):364-73 [PubMed PMID: 1405319]
Onori P, Franchitto A, Mancinelli R, Carpino G, Alvaro D, Francis H, Alpini G, Gaudio E. Polycystic liver diseases. Digestive and liver disease : official journal of the Italian Society of Gastroenterology and the Italian Association for the Study of the Liver. 2010 Apr:42(4):261-71. doi: 10.1016/j.dld.2010.01.006. Epub 2010 Feb 6 [PubMed PMID: 20138815]
Gabow PA, Chapman AB, Johnson AM, Tangel DJ, Duley IT, Kaehny WD, Manco-Johnson M, Schrier RW. Renal structure and hypertension in autosomal dominant polycystic kidney disease. Kidney international. 1990 Dec:38(6):1177-80 [PubMed PMID: 2074659]
Perrone RD, Oberdhan D, Ouyang J, Bichet DG, Budde K, Chapman AB, Gitomer BY, Horie S, Ong ACM, Torres VE, Turner AN, Krasa H. OVERTURE: A Worldwide, Prospective, Observational Study of Disease Characteristics in Patients With ADPKD. Kidney international reports. 2023 May:8(5):989-1001. doi: 10.1016/j.ekir.2023.02.1073. Epub 2023 Feb 13 [PubMed PMID: 37180499]
Level 2 (mid-level) evidenceCaroli A, Antiga L, Conti S, Sonzogni A, Fasolini G, Ondei P, Perico N, Remuzzi G, Remuzzi A. Intermediate volume on computed tomography imaging defines a fibrotic compartment that predicts glomerular filtration rate decline in autosomal dominant polycystic kidney disease patients. The American journal of pathology. 2011 Aug:179(2):619-27. doi: 10.1016/j.ajpath.2011.04.036. Epub 2011 Jun 17 [PubMed PMID: 21683674]
Level 2 (mid-level) evidenceTorres VE, Harris PC, Pirson Y. Autosomal dominant polycystic kidney disease. Lancet (London, England). 2007 Apr 14:369(9569):1287-1301. doi: 10.1016/S0140-6736(07)60601-1. Epub [PubMed PMID: 17434405]
Level 3 (low-level) evidenceGalliani M, Chicca S, Vitaliano E, Di Lullo L, Giannakakis K, Paone A. [Renal manifestation of Autosomal Dominant Polycystic Kidney Disease]. Giornale italiano di nefrologia : organo ufficiale della Societa italiana di nefrologia. 2018 Mar:35(2):. pii: 2018-vol2. Epub [PubMed PMID: 29582957]
Kalatharan V, Grewal G, Nash DM, Welk B, Sarma S, Pei Y, Garg AX. Stone Prevalence in Autosomal Dominant Polycystic Kidney Disease: A Systematic Review and Meta-Analysis. Canadian journal of kidney health and disease. 2020:7():2054358120934628. doi: 10.1177/2054358120934628. Epub 2020 Jul 4 [PubMed PMID: 35186303]
Level 1 (high-level) evidenceMallett A, Patel M, Tunnicliffe DJ, Rangan GK. KHA-CARI Autosomal Dominant Polycystic Kidney Disease Guideline: Management of Renal Stone Disease. Seminars in nephrology. 2015 Nov:35(6):603-606.e3. doi: 10.1016/j.semnephrol.2015.10.012. Epub [PubMed PMID: 26718165]
Gabow PA. Autosomal dominant polycystic kidney disease--more than a renal disease. American journal of kidney diseases : the official journal of the National Kidney Foundation. 1990 Nov:16(5):403-13 [PubMed PMID: 2239929]
Torres VE, Wilson DM, Hattery RR, Segura JW. Renal stone disease in autosomal dominant polycystic kidney disease. American journal of kidney diseases : the official journal of the National Kidney Foundation. 1993 Oct:22(4):513-9 [PubMed PMID: 8213789]
Grampsas SA, Chandhoke PS, Fan J, Glass MA, Townsend R, Johnson AM, Gabow P. Anatomic and metabolic risk factors for nephrolithiasis in patients with autosomal dominant polycystic kidney disease. American journal of kidney diseases : the official journal of the National Kidney Foundation. 2000 Jul:36(1):53-7 [PubMed PMID: 10873872]
Chasan O, Mirioglu S, Artan AS, Gursu M, Kazancioglu R, Elcioglu OC. Assessment of metabolic risk factors for nephrolithiasis in patients with autosomal dominant polycystic kidney disease: a cross-sectional study. Clinical and experimental nephrology. 2023 Nov:27(11):912-918. doi: 10.1007/s10157-023-02378-2. Epub 2023 Jul 26 [PubMed PMID: 37493903]
Level 2 (mid-level) evidenceNishiura JL, Neves RF, Eloi SR, Cintra SM, Ajzen SA, Heilberg IP. Evaluation of nephrolithiasis in autosomal dominant polycystic kidney disease patients. Clinical journal of the American Society of Nephrology : CJASN. 2009 Apr:4(4):838-44. doi: 10.2215/CJN.03100608. Epub 2009 Apr 1 [PubMed PMID: 19339428]
Levine E, Grantham JJ. Calcified renal stones and cyst calcifications in autosomal dominant polycystic kidney disease: clinical and CT study in 84 patients. AJR. American journal of roentgenology. 1992 Jul:159(1):77-81 [PubMed PMID: 1609726]
Gabow PA, Johnson AM, Kaehny WD, Manco-Johnson ML, Duley IT, Everson GT. Risk factors for the development of hepatic cysts in autosomal dominant polycystic kidney disease. Hepatology (Baltimore, Md.). 1990 Jun:11(6):1033-7 [PubMed PMID: 2365280]
Everson GT. Hepatic cysts in autosomal dominant polycystic kidney disease. American journal of kidney diseases : the official journal of the National Kidney Foundation. 1993 Oct:22(4):520-5 [PubMed PMID: 8213790]
Level 3 (low-level) evidenceKim JA, Blumenfeld JD, Chhabra S, Dutruel SP, Thimmappa ND, Bobb WO, Donahue S, Rennert HE, Tan AY, Giambrone AE, Prince MR. Pancreatic Cysts in Autosomal Dominant Polycystic Kidney Disease: Prevalence and Association with PKD2 Gene Mutations. Radiology. 2016 Sep:280(3):762-70. doi: 10.1148/radiol.2016151650. Epub 2016 Apr 5 [PubMed PMID: 27046073]
Fick GM, Gabow PA. Hereditary and acquired cystic disease of the kidney. Kidney international. 1994 Oct:46(4):951-64 [PubMed PMID: 7861721]
Dudley J, Winyard P, Marlais M, Cuthell O, Harris T, Chong J, Sayer J, Gale DP, Moore L, Turner K, Burrows S, Sandford R. Clinical practice guideline monitoring children and young people with, or at risk of developing autosomal dominant polycystic kidney disease (ADPKD). BMC nephrology. 2019 Apr 30:20(1):148. doi: 10.1186/s12882-019-1285-2. Epub 2019 Apr 30 [PubMed PMID: 31039757]
Level 1 (high-level) evidenceAl Sayyab M, Chapman A. Pregnancy in Autosomal Dominant Polycystic Kidney Disease. Advances in kidney disease and health. 2023 Sep:30(5):454-460. doi: 10.1053/j.akdh.2023.10.006. Epub [PubMed PMID: 38032583]
Level 3 (low-level) evidenceNoce EM. Considerations for genetic testing in individuals with autosomal dominant polycystic kidney disease. Journal of the American Association of Nurse Practitioners. 2022 Dec 1:34(12):1249-1251. doi: 10.1097/JXX.0000000000000787. Epub 2022 Dec 1 [PubMed PMID: 36469907]
Ars E, Bernis C, Fraga G, Furlano M, Martínez V, Martins J, Ortiz A, Pérez-Gómez MV, Rodríguez-Pérez JC, Sans L, Torra R, en nombre del grupo de trabajo de Enfermedades Renales Hereditarias de la Sociedad Española de Nefrología. Consensus document on autosomal dominant polycystic kindey disease from the Spanish Working Group on Inherited Kindey Diseases. Review 2020. Nefrologia. 2022 Jul-Aug:42(4):367-389. doi: 10.1016/j.nefroe.2022.11.011. Epub 2022 Nov 17 [PubMed PMID: 36404270]
Level 3 (low-level) evidencePetrucci I, Clementi A, Sessa C, Torrisi I, Meola M. Ultrasound and color Doppler applications in chronic kidney disease. Journal of nephrology. 2018 Dec:31(6):863-879. doi: 10.1007/s40620-018-0531-1. Epub 2018 Sep 6 [PubMed PMID: 30191413]
Pei Y, Obaji J, Dupuis A, Paterson AD, Magistroni R, Dicks E, Parfrey P, Cramer B, Coto E, Torra R, San Millan JL, Gibson R, Breuning M, Peters D, Ravine D. Unified criteria for ultrasonographic diagnosis of ADPKD. Journal of the American Society of Nephrology : JASN. 2009 Jan:20(1):205-12. doi: 10.1681/ASN.2008050507. Epub 2008 Oct 22 [PubMed PMID: 18945943]
Irazabal MV, Rangel LJ, Bergstralh EJ, Osborn SL, Harmon AJ, Sundsbak JL, Bae KT, Chapman AB, Grantham JJ, Mrug M, Hogan MC, El-Zoghby ZM, Harris PC, Erickson BJ, King BF, Torres VE, CRISP Investigators. Imaging classification of autosomal dominant polycystic kidney disease: a simple model for selecting patients for clinical trials. Journal of the American Society of Nephrology : JASN. 2015 Jan:26(1):160-72. doi: 10.1681/ASN.2013101138. Epub 2014 Jun 5 [PubMed PMID: 24904092]
Bae KT, Sun H, Lee JG, Bae K, Wang J, Tao C, Chapman AB, Torres VE, Grantham JJ, Mrug M, Bennett WM, Flessner MF, Landsittel DP, Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease. Novel methodology to evaluate renal cysts in polycystic kidney disease. American journal of nephrology. 2014:39(3):210-7. doi: 10.1159/000358604. Epub 2014 Feb 22 [PubMed PMID: 24576800]
Torres VE, Bankir L, Grantham JJ. A case for water in the treatment of polycystic kidney disease. Clinical journal of the American Society of Nephrology : CJASN. 2009 Jun:4(6):1140-50. doi: 10.2215/CJN.00790209. Epub 2009 May 14 [PubMed PMID: 19443627]
Level 3 (low-level) evidenceBarash I, Ponda MP, Goldfarb DS, Skolnik EY. A pilot clinical study to evaluate changes in urine osmolality and urine cAMP in response to acute and chronic water loading in autosomal dominant polycystic kidney disease. Clinical journal of the American Society of Nephrology : CJASN. 2010 Apr:5(4):693-7. doi: 10.2215/CJN.04180609. Epub 2010 Feb 18 [PubMed PMID: 20167686]
Level 3 (low-level) evidenceChapman AB, Guay-Woodford LM, Grantham JJ, Torres VE, Bae KT, Baumgarten DA, Kenney PJ, King BF Jr, Glockner JF, Wetzel LH, Brummer ME, O'Neill WC, Robbin ML, Bennett WM, Klahr S, Hirschman GH, Kimmel PL, Thompson PA, Miller JP, Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease cohort. Renal structure in early autosomal-dominant polycystic kidney disease (ADPKD): The Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease (CRISP) cohort. Kidney international. 2003 Sep:64(3):1035-45 [PubMed PMID: 12911554]
Level 2 (mid-level) evidenceVikrant S, Parashar A. Autosomal dominant polycystic kidney disease: Study of clinical characteristics in an Indian population. Saudi journal of kidney diseases and transplantation : an official publication of the Saudi Center for Organ Transplantation, Saudi Arabia. 2017 Jan-Feb:28(1):115-124. doi: 10.4103/1319-2442.198163. Epub [PubMed PMID: 28098112]
KC M, Leslie SW. Uric Acid Nephrolithiasis. StatPearls. 2024 Jan:(): [PubMed PMID: 32809561]
Schrier RW, Abebe KZ, Perrone RD, Torres VE, Braun WE, Steinman TI, Winklhofer FT, Brosnahan G, Czarnecki PG, Hogan MC, Miskulin DC, Rahbari-Oskoui FF, Grantham JJ, Harris PC, Flessner MF, Bae KT, Moore CG, Chapman AB, HALT-PKD Trial Investigators. Blood pressure in early autosomal dominant polycystic kidney disease. The New England journal of medicine. 2014 Dec 11:371(24):2255-66. doi: 10.1056/NEJMoa1402685. Epub 2014 Nov 15 [PubMed PMID: 25399733]
Level 1 (high-level) evidenceArogundade FA, Akinbodewa AA, Sanusi AA, Okunola O, Hassan MO, Akinsola A. Clinical presentation and outcome of autosomal dominant polycystic kidney disease in Nigeria. African health sciences. 2018 Sep:18(3):671-680. doi: 10.4314/ahs.v18i3.25. Epub [PubMed PMID: 30603000]
Müller RU, Benzing T. Management of autosomal-dominant polycystic kidney disease-state-of-the-art. Clinical kidney journal. 2018 Dec:11(Suppl 1):i2-i13. doi: 10.1093/ckj/sfy103. Epub 2018 Dec 17 [PubMed PMID: 30581561]
Müller RU, Messchendorp AL, Birn H, Capasso G, Cornec-Le Gall E, Devuyst O, van Eerde A, Guirchoun P, Harris T, Hoorn EJ, Knoers NVAM, Korst U, Mekahli D, Le Meur Y, Nijenhuis T, Ong ACM, Sayer JA, Schaefer F, Servais A, Tesar V, Torra R, Walsh SB, Gansevoort RT. An update on the use of tolvaptan for autosomal dominant polycystic kidney disease: consensus statement on behalf of the ERA Working Group on Inherited Kidney Disorders, the European Rare Kidney Disease Reference Network and Polycystic Kidney Disease International. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 2022 Apr 25:37(5):825-839. doi: 10.1093/ndt/gfab312. Epub [PubMed PMID: 35134221]
Level 3 (low-level) evidencePatel SJ, Sadowski CK. An update on treatments for autosomal dominant polycystic kidney disease. JAAPA : official journal of the American Academy of Physician Assistants. 2023 Jun 1:36(6):11-16. doi: 10.1097/01.JAA.0000931420.46207.82. Epub [PubMed PMID: 37163712]
Kim Y, Han S. Recent updates in therapeutic approach using tolvaptan for autosomal dominant polycystic kidney disease. The Korean journal of internal medicine. 2023 May:38(3):322-331. doi: 10.3904/kjim.2022.376. Epub 2023 Apr 25 [PubMed PMID: 37089056]
Lacquaniti A. [Tolvaptan and autosomal dominant polycystic kidney disease in the adult: let's give time to the "TEMPO" trial (Tolvaptan Efficacy and Safety in Management of Autosomal Dominant Polycystic Kidney Disease and Its Outcomes)]. Giornale italiano di nefrologia : organo ufficiale della Societa italiana di nefrologia. 2013 Jan-Feb:30(1):. pii: gin/30.1.10. Epub [PubMed PMID: 25083527]
Torres VE, Higashihara E, Devuyst O, Chapman AB, Gansevoort RT, Grantham JJ, Perrone RD, Ouyang J, Blais JD, Czerwiec FS, TEMPO 3:4 Trial Investigators. Effect of Tolvaptan in Autosomal Dominant Polycystic Kidney Disease by CKD Stage: Results from the TEMPO 3:4 Trial. Clinical journal of the American Society of Nephrology : CJASN. 2016 May 6:11(5):803-811. doi: 10.2215/CJN.06300615. Epub 2016 Feb 23 [PubMed PMID: 26912543]
Chebib FT, Zhou X, Garbinsky D, Davenport E, Nunna S, Oberdhan D, Fernandes A. Tolvaptan and Kidney Function Decline in Older Individuals With Autosomal Dominant Polycystic Kidney Disease: A Pooled Analysis of Randomized Clinical Trials and Observational Studies. Kidney medicine. 2023 Jun:5(6):100639. doi: 10.1016/j.xkme.2023.100639. Epub 2023 Apr 14 [PubMed PMID: 37250503]
Level 1 (high-level) evidenceBais T, Gansevoort RT, Meijer E. Drugs in Clinical Development to Treat Autosomal Dominant Polycystic Kidney Disease. Drugs. 2022 Jul:82(10):1095-1115. doi: 10.1007/s40265-022-01745-9. Epub 2022 Jul 19 [PubMed PMID: 35852784]
Shoaf SE, Ouyang J, Sergeyeva O, Estilo A, Li H, Leung D. A Post Hoc Analysis of Statin Use in Tolvaptan Autosomal Dominant Polycystic Kidney Disease Pivotal Trials. Clinical journal of the American Society of Nephrology : CJASN. 2020 May 7:15(5):643-650. doi: 10.2215/CJN.08170719. Epub 2020 Apr 2 [PubMed PMID: 32241780]
Lin CH, Chao CT, Wu MY, Lo WC, Lin TC, Wu MS. Use of mammalian target of rapamycin inhibitors in patient with autosomal dominant polycystic kidney disease: an updated meta-analysis. International urology and nephrology. 2019 Nov:51(11):2015-2025. doi: 10.1007/s11255-019-02292-1. Epub 2019 Oct 1 [PubMed PMID: 31578673]
Level 1 (high-level) evidenceMeijer E, Visser FW, van Aerts RMM, Blijdorp CJ, Casteleijn NF, D'Agnolo HMA, Dekker SEI, Drenth JPH, de Fijter JW, van Gastel MDA, Gevers TJ, Lantinga MA, Losekoot M, Messchendorp AL, Neijenhuis MK, Pena MJ, Peters DJM, Salih M, Soonawala D, Spithoven EM, Wetzels JF, Zietse R, Gansevoort RT, DIPAK-1 Investigators. Effect of Lanreotide on Kidney Function in Patients With Autosomal Dominant Polycystic Kidney Disease: The DIPAK 1 Randomized Clinical Trial. JAMA. 2018 Nov 20:320(19):2010-2019. doi: 10.1001/jama.2018.15870. Epub [PubMed PMID: 30422235]
Level 1 (high-level) evidencePirson Y, Christophe JL, Goffin E. Outcome of renal replacement therapy in autosomal dominant polycystic kidney disease. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 1996:11 Suppl 6():24-8 [PubMed PMID: 9044324]
Natoli TA, Modur V, Ibraghimov-Beskrovnaya O. Glycosphingolipid metabolism and polycystic kidney disease. Cellular signalling. 2020 May:69():109526. doi: 10.1016/j.cellsig.2020.109526. Epub 2020 Jan 10 [PubMed PMID: 31911181]
Sieben CJ, Harris PC. Experimental Models of Polycystic Kidney Disease: Applications and Therapeutic Testing. Kidney360. 2023 Aug 1:4(8):1155-1173. doi: 10.34067/KID.0000000000000209. Epub 2023 Jul 7 [PubMed PMID: 37418622]
Nowak KL, Farmer-Bailey H, Wang W, You Z, Steele C, Cadnapaphornchai MA, Klawitter J, Patel N, George D, Jovanovich A, Soranno DE, Gitomer B, Chonchol M. Curcumin Therapy to Treat Vascular Dysfunction in Children and Young Adults with ADPKD: A Randomized Controlled Trial. Clinical journal of the American Society of Nephrology : CJASN. 2022 Feb:17(2):240-250. doi: 10.2215/CJN.08950621. Epub 2021 Dec 14 [PubMed PMID: 34907021]
Level 1 (high-level) evidenceGhafouri-Fard S, Shoorei H, Bahroudi Z, Hussen BM, Talebi SF, Taheri M, Ayatollahi SA. Nrf2-Related Therapeutic Effects of Curcumin in Different Disorders. Biomolecules. 2022 Jan 5:12(1):. doi: 10.3390/biom12010082. Epub 2022 Jan 5 [PubMed PMID: 35053230]
Tomlinson JW. Bardet-Biedl syndrome: A focus on genetics, mechanisms and metabolic dysfunction. Diabetes, obesity & metabolism. 2024 Apr:26 Suppl 2():13-24. doi: 10.1111/dom.15480. Epub 2024 Feb 1 [PubMed PMID: 38302651]
Melluso A, Secondulfo F, Capolongo G, Capasso G, Zacchia M. Bardet-Biedl Syndrome: Current Perspectives and Clinical Outlook. Therapeutics and clinical risk management. 2023:19():115-132. doi: 10.2147/TCRM.S338653. Epub 2023 Jan 30 [PubMed PMID: 36741589]
Level 3 (low-level) evidenceClissold RL, Hamilton AJ, Hattersley AT, Ellard S, Bingham C. HNF1B-associated renal and extra-renal disease-an expanding clinical spectrum. Nature reviews. Nephrology. 2015 Feb:11(2):102-12. doi: 10.1038/nrneph.2014.232. Epub 2014 Dec 23 [PubMed PMID: 25536396]
Garfield K, Leslie SW. Medullary Sponge Kidney. StatPearls. 2024 Jan:(): [PubMed PMID: 29262095]
Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, Franco B, Bruel AL, Thauvin-Robinet C. Oral-Facial-Digital Syndrome Type I. GeneReviews(®). 1993:(): [PubMed PMID: 20301367]
Portocarrero LKL, Quental KN, Samorano LP, Oliveira ZNP, Rivitti-Machado MCDM. Tuberous sclerosis complex: review based on new diagnostic criteria. Anais brasileiros de dermatologia. 2018 Jun:93(3):323-331. doi: 10.1590/abd1806-4841.20186972. Epub [PubMed PMID: 29924239]
Garfield K, Leslie SW. Simple Renal Cyst. StatPearls. 2024 Jan:(): [PubMed PMID: 29763075]
Wilson PD. Polycystic kidney disease. The New England journal of medicine. 2004 Jan 8:350(2):151-64 [PubMed PMID: 14711914]
Rozenfeld MN, Ansari SA, Mohan P, Shaibani A, Russell EJ, Hurley MC. Autosomal Dominant Polycystic Kidney Disease and Intracranial Aneurysms: Is There an Increased Risk of Treatment? AJNR. American journal of neuroradiology. 2016 Feb:37(2):290-3. doi: 10.3174/ajnr.A4490. Epub 2015 Sep 3 [PubMed PMID: 26338918]
Ushio Y, Kataoka H, Akagawa H, Sato M, Manabe S, Kawachi K, Makabe S, Akihisa T, Seki M, Teraoka A, Iwasa N, Yoshida R, Tsuchiya K, Nitta K, Hoshino J, Mochizuki T. Factors associated with early-onset intracranial aneurysms in patients with autosomal dominant polycystic kidney disease. Journal of nephrology. 2024 Feb 5:():. doi: 10.1007/s40620-023-01866-8. Epub 2024 Feb 5 [PubMed PMID: 38315279]
Bugazia S, Hogan MC. Extrarenal Manifestations: Polycystic Liver Disease and Its Complications. Advances in kidney disease and health. 2023 Sep:30(5):440-453. doi: 10.1053/j.akdh.2023.10.004. Epub [PubMed PMID: 37943238]
Level 3 (low-level) evidenceKönig JC, Titieni A, Konrad M, NEOCYST Consortium. Network for Early Onset Cystic Kidney Diseases-A Comprehensive Multidisciplinary Approach to Hereditary Cystic Kidney Diseases in Childhood. Frontiers in pediatrics. 2018:6():24. doi: 10.3389/fped.2018.00024. Epub 2018 Feb 13 [PubMed PMID: 29497606]
Chebib FT, Perrone RD, Chapman AB, Dahl NK, Harris PC, Mrug M, Mustafa RA, Rastogi A, Watnick T, Yu ASL, Torres VE. A Practical Guide for Treatment of Rapidly Progressive ADPKD with Tolvaptan. Journal of the American Society of Nephrology : JASN. 2018 Oct:29(10):2458-2470. doi: 10.1681/ASN.2018060590. Epub 2018 Sep 18 [PubMed PMID: 30228150]
Corradi V, Gastaldon F, Caprara C, Giuliani A, Martino F, Ferrari F, Ronco C. Predictors of rapid disease progression in autosomal dominant polycystic kidney disease. Minerva medica. 2017 Feb:108(1):43-56. doi: 10.23736/S0026-4806.16.04830-8. Epub 2016 Oct 4 [PubMed PMID: 27701376]
Ryu H, Park HC, Oh YK, Sangadi I, Wong A, Mei C, Ecder T, Wang AY, Kao TW, Huang JW, Rangan GK, Ahn C. RAPID-ADPKD (Retrospective epidemiological study of Asia-Pacific patients with rapId Disease progression of Autosomal Dominant Polycystic Kidney Disease): study protocol for a multinational, retrospective cohort study. BMJ open. 2020 Feb 6:10(2):e034103. doi: 10.1136/bmjopen-2019-034103. Epub 2020 Feb 6 [PubMed PMID: 32034027]
Level 2 (mid-level) evidencede Chickera S, Akbari A, Levin A, Tang M, Brown P, Djurdev O, Biyani M, Clark EG, Sood MM. The Risk of Adverse Events in Patients With Polycystic Kidney Disease With Advanced Chronic Kidney Disease. Canadian journal of kidney health and disease. 2018:5():2054358118774537. doi: 10.1177/2054358118774537. Epub 2018 Jun 8 [PubMed PMID: 30186614]
Wilkinson DA, Heung M, Deol A, Chaudhary N, Gemmete JJ, Thompson BG, Pandey AS. Cerebral Aneurysms in Autosomal Dominant Polycystic Kidney Disease: A Comparison of Management Approaches. Neurosurgery. 2019 Jun 1:84(6):E352-E361. doi: 10.1093/neuros/nyy336. Epub [PubMed PMID: 30060240]