Introduction
The pituitary gland is located within the sella turcica, a bony depression in the sphenoid bone. The pituitary gland is divided into anterior and posterior segments. The anterior pituitary (adenohypophysis) is comprised of glandular tissue, while the posterior pituitary (neurohypophysis) is comprised of neural tissue. The pituitary gland is connected to the hypothalamus through the pituitary stalk. The pituitary stalk has a portal circulation for the anterior pituitary and neuronal connection for the posterior pituitary. The anterior pituitary gland is regulated by hormones secreted by the hypothalamus, while the posterior pituitary hormones are produced by the hypothalamus itself and are subsequently stored in the posterior pituitary for release.[1]
Pituitary tumors arising from the anterior pituitary gland account for approximately 15% of intracranial neoplasms and are usually benign tumors.[2][3] Rarely, pituitary tumors may metastasize and are then termed pituitary carcinoma.[4] Pituitary carcinoma differs from systemic metastasis to the pituitary gland, which may originate from the lung or breast, and less commonly from the prostate, renal, and colon, among others.[5]
Here we present an overview of pituitary cancer, including its epidemiology, clinical presentation, evaluation, available predictive markers, current, and emerging treatment options, as well as prognosis.
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
The exact underlying etiology of pituitary gland tumors is not yet known but most likely occurs due to genetic abnormalities and alterations. There are ongoing studies focusing on the underlying molecular mechanisms of pituitary tumor behavior. Transcriptional and post-transcriptional regulation by way of methylation, histone regulation[6][7], and modification of the expression of long non-coding ribonucleic acids (RNAs) or specific microRNAs have been found to correlate with malignancy in a few studies.[8] Pituitary tumors associated with specific germline mutations (MEN1, AIP, GPR101) have been shown to be more frequently resistant or invasive.[9]
Some hereditary conditions are also related to an increased risk of pituitary tumors. For example, multiple endocrine neoplasias (MEN)-1 syndrome is often associated with pituitary adenomas. No environmental or lifestyle changes have been directly linked to the underlying etiology of pituitary tumors, and therefore there are no preventable recommendations at this time.[10][11]
Epidemiology
The prevalence of pituitary carcinoma is extremely low and estimated at 1/1,000,000, which is <.01% of all detected anterior pituitary gland tumors.[12] Pituitary carcinomas usually originate from functioning pituitary gland tumors and are mostly represented by lactotroph and corticotroph carcinomas, with each representing around one-third of cases.[13][14]
The median age at diagnosis of pituitary carcinoma is 44 to 45.[13][15] The median latency period from the time of diagnosis to the occurrence of the first metastasis is approximately 5 to 7.5 years.[15][13] A switch from a non-functioning pituitary tumor to a functioning tumor has been documented in cases where pituitary adenomas have progressed to pituitary carcinomas.[16][17][15]
Pathophysiology
The pathophysiology of pituitary cancer is genetically related, but most mutations are unknown and actively being studied. There are ongoing efforts to determine the association with specific gene mutations.
There are six hormones produced and secreted by the anterior pituitary, which include luteinizing hormone (LH), follicle-stimulating hormone (FSH), adrenocorticotrophic hormone (ACTH), prolactin, thyroid-stimulating hormone (TSH), and growth hormone (GH). The hormones stored in the posterior pituitary gland are oxytocin and anti-diuretic hormone (ADH).[1]
The release of LH and FSH is controlled by gonadotropin-releasing hormone (GnRH) from the hypothalamus, while ACTH hormone secretion is controlled by the release of corticotropin-releasing hormone (CRH) from the hypothalamus. TSH is released after being stimulated by the thyrotropin-releasing hormone (TRH) from the hypothalamus. The hypothalamic hormones that control the release of GH are regulated by two hormones, including growth hormone-releasing hormone (GHRH), which stimulates the release, and somatostatin, which inhibits the release of GH. The release of prolactin is inhibited by dopamine release from the hypothalamus.[18]
Pituitary gland tumors are rarely cancerous and mostly benign. Pituitary tumors may function with hormone production or non-functioning without producing hormones. Functioning tumors cause different types of signs and symptoms depending on the specific hormone they secrete.[19]
Histopathology
The most common subtypes of pituitary carcinoma are corticotroph and lactotroph carcinomas.[16] Some carcinomas may appear similar to benign tumors of the pituitary gland and lack features such as cellular atypia and cellular dedifferentiation. To date, there is no single pathologic marker for malignancy of the pituitary gland. The concept of 'high-risk adenomas' has been proposed, which includes rapid growth, a high Ki67 index, and radiologic invasion.[20]
It has been proposed that tumors that are invasive, clinically aggressive, and highly proliferative (mitotic count >2, Ki67 index ≥10%, and p53 positive) should be considered pituitary tumors with malignant potential.[21] There are numerous prior and active studies focusing on the molecular mechanisms of pituitary tumor behavior. Corresponding markers have, unfortunately, been poorly validated to date.
History and Physical
The clinical presentation of pituitary tumors is classically divided into functional (secreting) and non-functional (nonsecretory) tumors. In general, functional tumors present earlier as a result of symptoms related to the physiologic effects of the excess hormones they secrete.[22] Unless incidentally discovered on radiographic imaging for work-up of a different indication, non-functional pituitary tumors typically do not present until they are large enough to result in neurologic deficits due to mass effect on adjacent structures. Pituitary carcinoma is suspected if neck or back pain is present, suggesting possible spine metastases.
Structures that are commonly compressed by pituitary tumors resulting in symptoms include the optic chiasm, the cavernous sinus, and the third ventricle. Mass effect on the optic chiasm due to tumor growth superiorly through the diaphragma sella commonly presents as bitemporal hemianopsia and may also cause a decrease in visual acuity. Cranial nerves within the cavernous sinus include the oculomotor nerve (III), trochlear (IV), first and second divisions of the trigeminal nerve (V1, V2), as well as abducens nerve (VI). If a pituitary tumor exerts a mass effect on the cranial nerves in the cavernous sinus, it may result in ptosis, diplopia, or facial pain. Occlusion of the cavernous sinus itself may result in chemosis and ptosis. If the third ventricle is involved, the mass effect may result in obstructive hydrocephalus. Larger pituitary tumors, such as macroadenomas, may produce non-specific symptoms, such as headaches, potentially due to increased intrasellar pressure. Invasive pituitary tumors may infrequently present with cerebrospinal fluid (CSF) rhinorrhea.[23]
Symptoms Related to Hormone Oversecretion
- Prolactin: Oversecretion of prolactin may cause impotence in men. In females, over-secretion of prolactin can cause amenorrhea-galactorrhea syndrome.
- Growth hormone (GH): Produces gigantism in prepubertal children. In adults, it presents as acromegaly.
- Adrenocorticotropic hormone (ACTH): Overproduction of ACTH results in Cushing's disease, which presents with a constellation of findings that is not limited to but includes hypertension, cutaneous purple striae, reduced libido in men and women, atrophic skin with easy bruising, depression, generalized muscle wasting, and weight gain, which is characterized by centripetal fat distribution in addition to fat deposition in the upper thoracic spine, supraclavicular fat pad with a round face and slender extremities.
- Thyrotropin (TSH): Secondary hyperthyroidism, which may present with symptoms of hyperthyroidism such as sweating, intolerance to heat, diarrhea, and weight loss, among others.
- Follicle-stimulating hormone (FSH) and/or luteinizing hormone (LH): Oversecretion of the gonadotropins typically does not produce a clinical syndrome. Oversecretion of FSH may produce ovarian hyperstimulation in women of reproductive age, causing ovarian cysts in addition to amenorrhea and galactorrhea.[24]
Symptoms related to the underproduction of pituitary hormones may be caused by compression of the normal pituitary gland by large pituitary tumors. The order of sensitivity of compression in which pituitary hormones become depressed is GH, gonadotropins (FSH & LH), TSH, ACTH, and prolactin. The order may be remembered by the mnemonic Go Look For The Adenoma Please.
Pituitary apoplexy can occur due to the rapid expansion of a pituitary tumor from necrosis or hemorrhage; any characteristically presents with headache, neurologic deficit, and/or endocrinologic deficit. In contrast to the typical situation with pituitary tumors, pituitary apoplexy more commonly presents with ophthalmoplegia rather than visual pathway deficit.[25]
Evaluation
Laboratory Tests
Laboratory testing involves both blood and urine testing. Specific hormonal level testing is usually dictated by correlating symptoms; however, more commonly, a panel of hormone testing is performed when incidental or nonfunctioning tumors are involved. An endocrinologic workup is performed primarily to rule out hypersecretory tumors. Routinely tested levels include prolactin and insulin-like growth factor (IGF-1). Early morning cortisol levels are reserved for patients with symptomatic manifestations of Cushing disease.[26]
Hyposecretory adenomas are tumors that cause the hyposecretion of hormones from the pituitary gland and can involve one or more hormones. Hyposecreting tumors require measuring levels of multiple hormones such as thyroid-stimulating hormone (TSH), follicle-stimulating hormone (FSH), luteinizing hormone (LH), free T4, total testosterone (in men), and cortisol. Hypogonadotropic hypogonadism requires magnetic resonance imaging (MRI) of the brain at the time of diagnosis to rule out a pituitary tumor as the underlying etiology. Confirmatory testing with stimulatory testing follows basic laboratory testing, and these tests are specific to the hormone of interest.[27][28][29]
Imaging
A sellar MRI is the recommended radiographic imaging technique for evaluating the pituitary gland. On imaging or gross pathology, they can be classified according to their size as macroadenomas when greater than 1 centimeter or microadenomas when less than 1 centimeter.[30] A computed tomography angiography (CTA) of the head may also be useful for pre-operative planning to evaluate the relationships of adjacent vascular structures, such as the internal carotid artery. In the absence of histopathologic features of malignancy, pituitary carcinoma is confirmed when systemic and/or craniospinal metastasis is identified radiographically, typically with spinal MRIs if suspected.[31]
Treatment / Management
Surgery
It is estimated that approximately 80.5% of pituitary carcinomas have been treated at least twice with surgery (including 27.9% of patients who underwent at least four surgeries).[16] Surgical debulking can be beneficial in patients with mass-effect on adjacent structures such as the optic pathway, third ventricle, or in advanced cases, the brainstem. (B3)
Radiotherapy
In one survey, it was estimated that approximately 42.9% of patients with pituitary carcinoma were treated with at least two courses of radiotherapy.[16] The optimal technique(s) should be discussed with specialists in radiotherapy and radiosurgery on a case-by-case basis. Conformal radiotherapy can be considered and aims to provide antitumoral and antisecretory objectives. Data on the utility of conformal radiotherapy is limited to aggressive, rapidly growing tumors and pituitary carcinoma. There are a few case reports of using two different radiation modalities to treat aggressive pituitary tumors with suboptimal efficacy.[32] Radiotherapy should be considered as a part of a multimodal treatment approach as its efficacy is often delayed.(B2)
Chemotherapy
Temozolomide is an oral alkylating agent which exerts its pharmacologic effect by deoxyribonucleic acid (DNA) methylation, resulting in irreversible DNA damage. Temozolomide is the recommended first-line chemotherapy. Temozolomide has been shown to improve progression-free and overall 5-year survival rates in responders.[33][34][35] Despite temozolomide being the first-line chemotherapeutic agent, only 6% of patients with pituitary carcinoma showed a complete radiologic response (mass absent radiographically), 31% showed partial response (tumor size decreased by >30%), stable disease was seen in 33% (tumor size increased by < 10% or decreased by < 30%), and progressive disease (tumor size >10% or new metastasis) was seen in 30%.[16] Improved response to temozolomide was observed in three specific situations: concomitant administration of radiotherapy, clinically functioning pituitary tumor, and low O6-Methylguanine-DNA Methyltransferase (MGMT) immunohistochemical expression.[16](B3)
For tumors that are progressing on or after the first course of temozolomide, there are, unfortunately, no other evidence-based treatments available at this time; therefore, maximizing and prolonging temozolomide efficacy is critical. Despite limited experience, options include long-term treatment with temozolomide as long as it is effective and clinically tolerated.[36][37] Additionally, using temozolomide earlier in the management of pituitary carcinoma as a part of the Stupp protocol instead of as “rescue therapy” is an option.[36][38][39]
Antisecretory Medications
Controlling pituitary hormone hypersecretion is as mandatory in patients with pituitary cancer as in any patient with a functional pituitary tumor. For patients with lactotroph tumors, dopamine agonists, such as cabergoline, can be given at high doses.[40] In patients with hypercortisolism, steroidogenesis inhibitors, such as metyrapone or ketoconazole (11-beta-hydroxylase inhibitor, and antifungal agent, respectively), are crucial to avoid morbidity and mortality from excess cortisol.[41] Finally, for somatotrophic tumors, somatostatin analogs (octreotide), growth hormone receptor antagonists (pegvisomant), or dopamine agonists can be continued at maximally tolerated doses. (A1)
Future Therapies
Potential future therapeutic options that are currently being studied include immunotherapy as well as molecularly targeted therapies such as bevacizumab (anti-VEGF antibody), everolimus (mTOR inhibitor), and several tyrosine kinase inhibitors (TKIs).[42]
Differential Diagnosis
Pituitary cancer arises from the pituitary gland. A helpful mnemonic for the radiographic differential diagnosis of lesions within the pituitary region is SATCHMO. Satchmo is best known as the nickname for the famous musician Louis Armstrong.
- S: Sarcoidosis, sellar tumor (pituitary adenoma, pituitary carcinoma)
- A: Aneurysm (typically superior hypophyseal)
- T: Teratoma, tuberculosis (other granulomatous diseases)
- C: Craniopharyngioma, chordoma, cleft cyst (Rathke's)
- H: Hypothalamic glioma, hamartoma, histiocytosis
- M: Meningioma, metastasis
- O: Optic nerve glioma
Radiation Oncology
It has been estimated that approximately 42.9% of patients with pituitary carcinoma are treated with at least two courses of radiotherapy.[16] The optimal technique(s) should be discussed with specialists in radiotherapy and radiosurgery on a case-by-case basis. Conformal radiotherapy can be considered and is aimed at providing antitumoral and antisecretory objectives. Data on the utility of conformal radiotherapy is limited to aggressive, rapidly growing tumors and pituitary carcinoma. There are a few case reports of using two different radiation modalities to treat aggressive pituitary tumors with suboptimal efficacy.[32] Radiotherapy should be considered as part of a multimodal treatment approach as its efficacy is often delayed.
Medical Oncology
Temozolomide is an oral alkylating agent which exerts its pharmacologic effect by deoxyribonucleic acid (DNA) methylation, resulting in irreversible DNA damage. Temozolomide is the recommended first-line chemotherapy. Temozolomide has been shown to improve progression-free and overall 5-year survival rates in responders.[33][34][35] Despite temozolomide being the first-line chemotherapeutic agent, only 6% of patients with pituitary carcinoma showed a complete radiologic response (mass absent radiographically), 31% showed partial response (tumor size decreased by >30%), stable disease was seen in 33% (tumor size increased by <10% or decreased by <30%), and progressive disease (tumor size >10% or new metastasis) was seen in 30%.[16] Improved response to temozolomide was observed in three specific situations: concomitant administration of radiotherapy, clinically functioning pituitary tumor, and low O6-methylguanine-DNA methyltransferase (MGMT) immunohistochemical expression.[16]
For tumors that are progressing on or after the first course of temozolomide, there are, unfortunately, no other evidence-based treatments available at this time; therefore, maximizing and prolonging temozolomide efficacy is critical. Despite limited experience, options include long-term treatment with temozolomide as long as it is effective and clinically tolerated.[36][37] Additionally, using temozolomide earlier in the management of pituitary carcinoma as a part of the Stupp protocol instead of as “rescue therapy” is an option.[36][38][39]
Controlling pituitary hormone hypersecretion is as mandatory in patients with pituitary cancer as in any patient with a functional pituitary tumor. For patients with lactotroph tumors, dopamine agonists, such as cabergoline, can be given at high doses.[40] In patients with hypercortisolism, steroidogenesis inhibitors, such as metyrapone or ketoconazole (11-beta-hydroxylase inhibitor, and antifungal agent, respectively), are crucial to avoid morbidity and mortality from excess cortisol.[41] Finally, for somatotrophic tumors, somatostatin analogs (octreotide), growth hormone receptor antagonists (pegvisomant), or dopamine agonists can be continued at maximally tolerated doses.
Potential future therapeutic options that are currently being studied include immunotherapy as well as molecularly targeted therapies such as bevacizumab (anti-VEGF antibody), everolimus (mTOR inhibitor), and several tyrosine kinase inhibitors (TKIs).[42]
Prognosis
The overall prognosis of benign pituitary tumors remains fair in terms of mortality compared to other neurohormonal tumors. The survival rate is approximately 82% five years after diagnosis.[43] The prognosis is much less favorable for pituitary carcinoma. The mean survival time for pituitary carcinoma is <4 years once metastases have been identified.[14]
Complications
One of the most notorious and severe complications of any pituitary tumor is blindness. The encroachment of the tumor on the chiasm or nerves can cause partial or complete blindness. Some tumors that do not present with visual defects can progress rapidly to cause vision loss.[44]
Diabetes insipidus can occur in macroadenomas, which causes vasopressin deficiency and can, in turn, cause increased thirst and polyuria.[45]
Pituitary apoplexy, or hemorrhage of the pituitary gland, requires emergent attention and can cause a severe headache, ophthalmoplegia, and visual disturbances immediately at onset.
Due in part because of need for repeat surgery, as well as the severity of the underlying disease, an increased number of surgical complications, including hydrocephalus requiring placement of a ventricular shunt, is observed.[46][47] To minimize the risk of major adverse events and complications, surgery should be performed by a neurosurgeon with a high volume of experience in treating pituitary masses.[4] Cases of sellar fibrosarcomas, osteosarcomas, and undifferentiated pleomorphic sarcomas have been reported in patients treated with radiotherapy for pituitary adenoma, developing after a mean of 10.5 years.[48] There is some controversy surrounding radiotherapy’s pathogenic role in transforming benign into malignant pituitary tumors. In a large patient series including 641 benign pituitary tumors, the rate of suspected malignant transformation when patients were followed over eight years was not statistically different from that of the general population.[49]
Additional peri-operative complications which may occur include infection (meningitis or pituitary abscess), CSF leak, entry into the cavernous sinus with possible injury to structures contained within the sinus, and rarely internal carotid artery rupture, which may occur intraoperatively or in a delayed fashion due to a pseudoaneurysm created during surgery.[50][51][52][51]
Deterrence and Patient Education
The pituitary gland is located in the base of the brain and is involved in hormonal regulation. Pituitary gland tumors may cause symptoms from mass effect on adjacent structures, typically the optic pathway involved in vision, or via glandular involvement with increased or decreased gland secretion. The signs/symptoms that are evaluated include vision changes if due to mass effect, hormonally may present with irregular menstrual cycles, milky discharge from breasts in females, and can cause erection problems or low sex drive in men. If there is an excess of steroid hormones, patients will typically have symptoms that will include weight gain, weak bones, and high blood pressure.
Several other related conditions or symptoms caused by different types of hormones may present with excess growth of the hands or head. Patients may additionally present with non-specific symptoms, such as headaches. Pituitary cancer comprises a very small proportion of pituitary tumors and may rarely arise as a metastatic lesion from elsewhere in the body or, more commonly, from the pituitary gland itself, referred to as pituitary carcinoma. Pituitary carcinoma may present with neck and/or back pain if there are metastatic lesions involving the spine.
There are varying modalities of diagnosis that typically involve a combination of laboratory testing and radiographic imaging. This will be determined by the physician on a case-by-case basis. Treatment typically includes surgical intervention followed by possibly adjuvant radiosurgery and/or chemotherapy/immunotherapies.
Enhancing Healthcare Team Outcomes
Pituitary cancer is rare, and improving outcomes requires coordinated interprofessional care and communication between healthcare team members. An interprofessional team approach is recommended, including primary care clinicians, endocrinology, ophthalmology, neurosurgery, otolaryngology, radiology, radiation oncology, and oncology. Surgical intervention is necessary, in addition to possibly adjuvant radiosurgery and/or chemotherapy, as well as emerging immunotherapies. Chemotherapy can benefit from a consultation with an oncology specialized pharmacist. Patients should have ongoing discussions with specialists to discuss the risks and benefits of the various treatment options. The interprofessional model will drive the best patient outcomes in pituitary cancer. [Level 5]
Media
(Click Image to Enlarge)
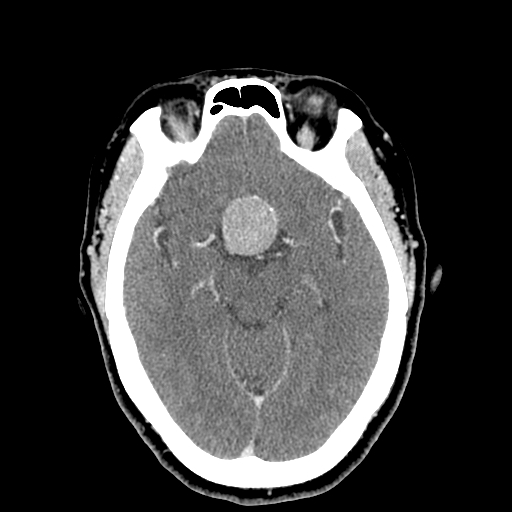
Pituitary Macroadenoma on Head Computed Tomography. The tumor appears as a round, hyperintense mass in the frontal region.
Contributed by Scott Dulebohn, MD
References
Molitch ME. Diagnosis and Treatment of Pituitary Adenomas: A Review. JAMA. 2017 Feb 7:317(5):516-524. doi: 10.1001/jama.2016.19699. Epub [PubMed PMID: 28170483]
Melmed S. Pituitary-Tumor Endocrinopathies. The New England journal of medicine. 2020 Mar 5:382(10):937-950. doi: 10.1056/NEJMra1810772. Epub [PubMed PMID: 32130815]
Saeger W, Lüdecke DK, Buchfelder M, Fahlbusch R, Quabbe HJ, Petersenn S. Pathohistological classification of pituitary tumors: 10 years of experience with the German Pituitary Tumor Registry. European journal of endocrinology. 2007 Feb:156(2):203-16 [PubMed PMID: 17287410]
Raverot G, Burman P, McCormack A, Heaney A, Petersenn S, Popovic V, Trouillas J, Dekkers OM, European Society of Endocrinology. European Society of Endocrinology Clinical Practice Guidelines for the management of aggressive pituitary tumours and carcinomas. European journal of endocrinology. 2018 Jan:178(1):G1-G24. doi: 10.1530/EJE-17-0796. Epub 2017 Oct 18 [PubMed PMID: 29046323]
Level 1 (high-level) evidenceShimon I. Metastatic Spread to the Pituitary. Neuroendocrinology. 2020:110(9-10):805-808. doi: 10.1159/000506810. Epub 2020 Feb 27 [PubMed PMID: 32101869]
Miyake Y, Adachi JI, Suzuki T, Mishima K, Araki R, Mizuno R, Nishikawa R. TERT promoter methylation is significantly associated with TERT upregulation and disease progression in pituitary adenomas. Journal of neuro-oncology. 2019 Jan:141(1):131-138. doi: 10.1007/s11060-018-03016-8. Epub 2018 Nov 3 [PubMed PMID: 30392088]
Pease M, Ling C, Mack WJ, Wang K, Zada G. The role of epigenetic modification in tumorigenesis and progression of pituitary adenomas: a systematic review of the literature. PloS one. 2013:8(12):e82619. doi: 10.1371/journal.pone.0082619. Epub 2013 Dec 18 [PubMed PMID: 24367530]
Level 1 (high-level) evidenceStilling G, Sun Z, Zhang S, Jin L, Righi A, Kovācs G, Korbonits M, Scheithauer BW, Kovacs K, Lloyd RV. MicroRNA expression in ACTH-producing pituitary tumors: up-regulation of microRNA-122 and -493 in pituitary carcinomas. Endocrine. 2010 Aug:38(1):67-75. doi: 10.1007/s12020-010-9346-0. Epub 2010 May 27 [PubMed PMID: 20960104]
Srirangam Nadhamuni V, Korbonits M. Novel Insights into Pituitary Tumorigenesis: Genetic and Epigenetic Mechanisms. Endocrine reviews. 2020 Dec 1:41(6):821-46. doi: 10.1210/endrev/bnaa006. Epub [PubMed PMID: 32201880]
Bassett JH, Forbes SA, Pannett AA, Lloyd SE, Christie PT, Wooding C, Harding B, Besser GM, Edwards CR, Monson JP, Sampson J, Wass JA, Wheeler MH, Thakker RV. Characterization of mutations in patients with multiple endocrine neoplasia type 1. American journal of human genetics. 1998 Feb:62(2):232-44 [PubMed PMID: 9463336]
Crabtree JS, Scacheri PC, Ward JM, Garrett-Beal L, Emmert-Buck MR, Edgemon KA, Lorang D, Libutti SK, Chandrasekharappa SC, Marx SJ, Spiegel AM, Collins FS. A mouse model of multiple endocrine neoplasia, type 1, develops multiple endocrine tumors. Proceedings of the National Academy of Sciences of the United States of America. 2001 Jan 30:98(3):1118-23 [PubMed PMID: 11158604]
Level 3 (low-level) evidenceDekkers OM, Karavitaki N, Pereira AM. The epidemiology of aggressive pituitary tumors (and its challenges). Reviews in endocrine & metabolic disorders. 2020 Jun:21(2):209-212. doi: 10.1007/s11154-020-09556-7. Epub [PubMed PMID: 32361816]
Yoo F, Kuan EC, Heaney AP, Bergsneider M, Wang MB. Corticotrophic pituitary carcinoma with cervical metastases: case series and literature review. Pituitary. 2018 Jun:21(3):290-301. doi: 10.1007/s11102-018-0872-8. Epub [PubMed PMID: 29404894]
Level 2 (mid-level) evidenceDudziak K, Honegger J, Bornemann A, Horger M, Müssig K. Pituitary carcinoma with malignant growth from first presentation and fulminant clinical course--case report and review of the literature. The Journal of clinical endocrinology and metabolism. 2011 Sep:96(9):2665-9. doi: 10.1210/jc.2011-1166. Epub 2011 Jun 29 [PubMed PMID: 21715538]
Level 3 (low-level) evidenceSantos-Pinheiro F, Penas-Prado M, Kamiya-Matsuoka C, Waguespack SG, Mahajan A, Brown PD, Shah KB, Fuller GN, McCutcheon IE. Treatment and long-term outcomes in pituitary carcinoma: a cohort study. European journal of endocrinology. 2019 Oct:181(4):397-407. doi: 10.1530/EJE-18-0795. Epub [PubMed PMID: 31349217]
McCormack A, Dekkers OM, Petersenn S, Popovic V, Trouillas J, Raverot G, Burman P, ESE survey collaborators. Treatment of aggressive pituitary tumours and carcinomas: results of a European Society of Endocrinology (ESE) survey 2016. European journal of endocrinology. 2018 Mar:178(3):265-276. doi: 10.1530/EJE-17-0933. Epub 2018 Jan 12 [PubMed PMID: 29330228]
Level 3 (low-level) evidenceKasuki L, Raverot G. Definition and diagnosis of aggressive pituitary tumors. Reviews in endocrine & metabolic disorders. 2020 Jun:21(2):203-208. doi: 10.1007/s11154-019-09531-x. Epub [PubMed PMID: 31808044]
Alkhani AM, Cusimano M, Kovacs K, Bilbao JM, Horvath E, Singer W. Cytology of pituitary thyrotroph hyperplasia in protracted primary hypothyroidism. Pituitary. 1999 May:1(3-4):291-5 [PubMed PMID: 11081211]
Level 3 (low-level) evidenceShimono T, Hatabu H, Kasagi K, Miki Y, Nishizawa S, Misaki T, Hiraga A, Konishi J. Rapid progression of pituitary hyperplasia in humans with primary hypothyroidism: demonstration with MR imaging. Radiology. 1999 Nov:213(2):383-8 [PubMed PMID: 10551216]
Mete O, Lopes MB. Overview of the 2017 WHO Classification of Pituitary Tumors. Endocrine pathology. 2017 Sep:28(3):228-243. doi: 10.1007/s12022-017-9498-z. Epub [PubMed PMID: 28766057]
Level 3 (low-level) evidenceTrouillas J, Jaffrain-Rea ML, Vasiljevic A, Dekkers O, Popovic V, Wierinckx A, McCormack A, Petersenn S, Burman P, Raverot G, Villa C. Are aggressive pituitary tumors and carcinomas two sides of the same coin? Pathologists reply to clinician's questions. Reviews in endocrine & metabolic disorders. 2020 Jun:21(2):243-251. doi: 10.1007/s11154-020-09562-9. Epub [PubMed PMID: 32504268]
Ebersold MJ, Quast LM, Laws ER Jr, Scheithauer B, Randall RV. Long-term results in transsphenoidal removal of nonfunctioning pituitary adenomas. Journal of neurosurgery. 1986 May:64(5):713-9 [PubMed PMID: 3701419]
Nutkiewicz A, DeFeo DR, Kohut RI, Fierstein S. Cerebrospinal fluid rhinorrhea as a presentation of pituitary adenoma. Neurosurgery. 1980 Feb:6(2):195-7 [PubMed PMID: 7366811]
Level 3 (low-level) evidenceKawaguchi T, Ogawa Y, Ito K, Watanabe M, Tominaga T. Follicle-stimulating hormone-secreting pituitary adenoma manifesting as recurrent ovarian cysts in a young woman--latent risk of unidentified ovarian hyperstimulation: a case report. BMC research notes. 2013 Oct 11:6():408. doi: 10.1186/1756-0500-6-408. Epub 2013 Oct 11 [PubMed PMID: 24119690]
Level 3 (low-level) evidenceGlezer A, Bronstein MD. Pituitary apoplexy: pathophysiology, diagnosis and management. Archives of endocrinology and metabolism. 2015 Jun:59(3):259-64. doi: 10.1590/2359-3997000000047. Epub [PubMed PMID: 26154095]
St-Jean E, Blain F, Comtois R. High prolactin levels may be missed by immunoradiometric assay in patients with macroprolactinomas. Clinical endocrinology. 1996 Mar:44(3):305-9 [PubMed PMID: 8729527]
Ónnestam L, Berinder K, Burman P, Dahlqvist P, Engström BE, Wahlberg J, Nyström HF. National incidence and prevalence of TSH-secreting pituitary adenomas in Sweden. The Journal of clinical endocrinology and metabolism. 2013 Feb:98(2):626-35. doi: 10.1210/jc.2012-3362. Epub 2013 Jan 7 [PubMed PMID: 23295463]
Petakov MS, Damjanović SS, Nikolić-Durović MM, Dragojlović ZL, Obradović S, Gligorović MS, Simić MZ, Popović VP. Pituitary adenomas secreting large amounts of prolactin may give false low values in immunoradiometric assays. The hook effect. Journal of endocrinological investigation. 1998 Mar:21(3):184-8 [PubMed PMID: 9591215]
Level 3 (low-level) evidenceBarkan AL, Chandler WF. Giant pituitary prolactinoma with falsely low serum prolactin: the pitfall of the "high-dose hook effect": case report. Neurosurgery. 1998 Apr:42(4):913-5; discussion 915-6 [PubMed PMID: 9574657]
Level 3 (low-level) evidenceMarniemi J, Parkki MG. Radiochemical assay of glutathione S-epoxide transferase and its enhancement by phenobarbital in rat liver in vivo. Biochemical pharmacology. 1975 Sep 1:24(17):1569-72 [PubMed PMID: 9]
Level 3 (low-level) evidenceAsa SL, Mete O, Perry A, Osamura RY. Overview of the 2022 WHO Classification of Pituitary Tumors. Endocrine pathology. 2022 Mar:33(1):6-26. doi: 10.1007/s12022-022-09703-7. Epub 2022 Mar 15 [PubMed PMID: 35291028]
Level 3 (low-level) evidenceVerma J, McCutcheon IE, Waguespack SG, Mahajan A. Feasibility and outcome of re-irradiation in the treatment of multiply recurrent pituitary adenomas. Pituitary. 2014 Dec:17(6):539-45. doi: 10.1007/s11102-013-0541-x. Epub [PubMed PMID: 24272035]
Level 2 (mid-level) evidenceLasolle H, Cortet C, Castinetti F, Cloix L, Caron P, Delemer B, Desailloud R, Jublanc C, Lebrun-Frenay C, Sadoul JL, Taillandier L, Batisse-Lignier M, Bonnet F, Bourcigaux N, Bresson D, Chabre O, Chanson P, Garcia C, Haissaguerre M, Reznik Y, Borot S, Villa C, Vasiljevic A, Gaillard S, Jouanneau E, Assié G, Raverot G. Temozolomide treatment can improve overall survival in aggressive pituitary tumors and pituitary carcinomas. European journal of endocrinology. 2017 Jun:176(6):769-777. doi: 10.1530/EJE-16-0979. Epub [PubMed PMID: 28432119]
Ji Y, Vogel RI, Lou E. Temozolomide treatment of pituitary carcinomas and atypical adenomas: systematic review of case reports. Neuro-oncology practice. 2016 Sep:3(3):188-195 [PubMed PMID: 27551432]
Level 3 (low-level) evidenceElbelt U, Schlaffer SM, Buchfelder M, Knappe UJ, Vila G, Micko A, Deutschbein T, Unger N, Lammert A, Topuzoglu-Müller T, Bojunga J, Droste M, Johanssen S, Kolenda H, Ritzel K, Buslei R, Strasburger CJ, Petersenn S, Honegger J. Efficacy of Temozolomide Therapy in Patients With Aggressive Pituitary Adenomas and Carcinomas-A German Survey. The Journal of clinical endocrinology and metabolism. 2020 Mar 1:105(3):. pii: dgz211. doi: 10.1210/clinem/dgz211. Epub [PubMed PMID: 31746334]
Level 3 (low-level) evidenceIlie MD, Jouanneau E, Raverot G. Aggressive Pituitary Adenomas and Carcinomas. Endocrinology and metabolism clinics of North America. 2020 Sep:49(3):505-515. doi: 10.1016/j.ecl.2020.05.008. Epub 2020 Jul 8 [PubMed PMID: 32741485]
Lizzul L, Lombardi G, Barbot M, Ceccato F, Gardiman MP, Regazzo D, Bellu L, Mazza E, Losa M, Scaroni C. Long-course temozolomide in aggressive pituitary adenoma: real-life experience in two tertiary care centers and review of the literature. Pituitary. 2020 Aug:23(4):359-366. doi: 10.1007/s11102-020-01040-4. Epub [PubMed PMID: 32232709]
Whitelaw BC. How and when to use temozolomide to treat aggressive pituitary tumours. Endocrine-related cancer. 2019 Aug:26(9):R545-R552. doi: 10.1530/ERC-19-0083. Epub [PubMed PMID: 31311005]
Lin AL, Sum MW, DeAngelis LM. Is there a role for early chemotherapy in the management of pituitary adenomas? Neuro-oncology. 2016 Oct:18(10):1350-6. doi: 10.1093/neuonc/now059. Epub 2016 Apr 21 [PubMed PMID: 27106409]
Ono M, Miki N, Kawamata T, Makino R, Amano K, Seki T, Kubo O, Hori T, Takano K. Prospective study of high-dose cabergoline treatment of prolactinomas in 150 patients. The Journal of clinical endocrinology and metabolism. 2008 Dec:93(12):4721-7. doi: 10.1210/jc.2007-2758. Epub 2008 Sep 23 [PubMed PMID: 18812485]
Nieman LK, Biller BM, Findling JW, Murad MH, Newell-Price J, Savage MO, Tabarin A, Endocrine Society. Treatment of Cushing's Syndrome: An Endocrine Society Clinical Practice Guideline. The Journal of clinical endocrinology and metabolism. 2015 Aug:100(8):2807-31. doi: 10.1210/jc.2015-1818. Epub 2015 Jul 29 [PubMed PMID: 26222757]
Level 1 (high-level) evidenceRaverot G, Ilie MD, Lasolle H, Amodru V, Trouillas J, Castinetti F, Brue T. Aggressive pituitary tumours and pituitary carcinomas. Nature reviews. Endocrinology. 2021 Nov:17(11):671-684. doi: 10.1038/s41574-021-00550-w. Epub 2021 Sep 7 [PubMed PMID: 34493834]
Gsponer J, De Tribolet N, Déruaz JP, Janzer R, Uské A, Mirimanoff RO, Reymond MJ, Rey F, Temler E, Gaillard RC, Gomez F. Diagnosis, treatment, and outcome of pituitary tumors and other abnormal intrasellar masses. Retrospective analysis of 353 patients. Medicine. 1999 Jul:78(4):236-69 [PubMed PMID: 10424206]
Level 2 (mid-level) evidenceTaylor SL, Barakos JA, Harsh GR 4th, Wilson CB. Magnetic resonance imaging of tuberculum sellae meningiomas: preventing preoperative misdiagnosis as pituitary macroadenoma. Neurosurgery. 1992 Oct:31(4):621-7; discussion 627 [PubMed PMID: 1407446]
Pivonello R, Colao A, Di Somma C, Facciolli G, Klain M, Faggiano A, Salvatore M, Lombardi G. Impairment of bone status in patients with central diabetes insipidus. The Journal of clinical endocrinology and metabolism. 1998 Jul:83(7):2275-80 [PubMed PMID: 9661594]
Level 2 (mid-level) evidenceBakhsheshian J, Wheeler S, Strickland BA, Pham MH, Rennert RC, Carmichael J, Weiss M, Zada G. Surgical Outcomes Following Repeat Transsphenoidal Surgery for Nonfunctional Pituitary Adenomas: A Retrospective Comparative Study. Operative neurosurgery (Hagerstown, Md.). 2019 Feb 1:16(2):127-135. doi: 10.1093/ons/opy078. Epub [PubMed PMID: 29767762]
Level 2 (mid-level) evidenceGraillon T, Castinetti F, Fuentes S, Gras R, Brue T, Dufour H. Transcranial approach in giant pituitary adenomas: results and outcome in a modern series. Journal of neurosurgical sciences. 2020 Feb:64(1):25-36. doi: 10.23736/S0390-5616.16.03889-3. Epub 2017 Jan 12 [PubMed PMID: 28079350]
Guerrero-Pérez F,Vidal N,López-Vázquez M,Sánchez-Barrera R,Sánchez-Fernández JJ,Torres-Díaz A,Vilarrasa N,Villabona C, Sarcomas of the sellar region: a systematic review. Pituitary. 2021 Feb [PubMed PMID: 32785833]
Level 1 (high-level) evidenceWolf A, Naylor K, Tam M, Habibi A, Novotny J, Liščák R, Martinez-Moreno N, Martinez-Alvarez R, Sisterson N, Golfinos JG, Silverman J, Kano H, Sheehan J, Lunsford LD, Kondziolka D. Risk of radiation-associated intracranial malignancy after stereotactic radiosurgery: a retrospective, multicentre, cohort study. The Lancet. Oncology. 2019 Jan:20(1):159-164. doi: 10.1016/S1470-2045(18)30659-4. Epub 2018 Nov 22 [PubMed PMID: 30473468]
Level 2 (mid-level) evidenceDomingue JN, Wilson CB. Pituitary abscesses. Report of seven cases and review of the literature. Journal of neurosurgery. 1977 May:46(5):601-8 [PubMed PMID: 845649]
Level 3 (low-level) evidenceRobinson B. Intrasellar abscess after transsphenoidal pituitary adenomectomy. Neurosurgery. 1983 Jun:12(6):684-6 [PubMed PMID: 6348578]
Level 3 (low-level) evidenceCiric I,Mikhael M,Stafford T,Lawson L,Garces R, Transsphenoidal microsurgery of pituitary macroadenomas with long-term follow-up results. Journal of neurosurgery. 1983 Sep [PubMed PMID: 6886753]