Introduction
Oligodendroglioma (OG) is a diffusely infiltrating glioma representing approximately 5% of all primary intracranial tumors.[1] These tumors often involve the cortical gray and the white matter and are most commonly located in the frontal lobes. Historically, the OG was diagnosed by the histological appearance of the tumor. However, in 2016, the World Health Organization (WHO) changed the criteria for classifying central nervous system (CNS) tumors to include a phenotypic and genotypic analysis.[2] OGs are generally low-grade WHO grade II neoplasms that are slow-growing tumors and have a favorable response to treatment compared to other gliomas. The grade III anaplastic OG is a highly malignant form of the tumor that has an unfavorable prognosis and may occur de novo or as a malignant transformation from the lower grade OG.[3]
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
OGs are primary brain tumors originating from oligodendrocytes, the glial cells responsible for myelination in the CNS. Initially described by Bailey and Bucy in 1929 for their microscopic resemblance to oligodendrocytes, their exact cellular origin remains debated. The hypothesis that OGs arise either from mature oligodendrocytes or neuroprogenitor cells with glial precursors, which differentiate into oligodendroglial-type cells lacking myelinating capabilities, is supported by shared driver mutations, particularly isocitrate dehydrogenase (IDH) mutations, which are common across various diffuse glioma subtypes and serve as early events in tumorigenesis.[4][5][6][7]
The etiology of OGs is multifactorial, involving genetic, environmental, epigenetic, and cellular factors. Genetically, the hallmark codeletion of chromosomal arms 1p and 19q is diagnostic in 70% to 90% of cases and correlates with better prognosis and therapy responsiveness. Mutations in IDH1 and IDH2 further characterize these tumors, contributing to the glioma CpG island methylator phenotype (G-CIMP), which alters deoxyribonucleic acid (DNA) methylation and promotes growth. Less frequently, mutations in TP53 and ATRX help differentiate OGs from astrocytomas.
While specific environmental exposures, such as ionizing radiation or chemicals in certain industries, may play a role, definitive links remain unproven. Epigenetic changes, including altered DNA methylation and histone acetylation, with dysregulated signaling pathways like platelet-derived growth factor receptor (PDGFR) and the epidermal growth factor receptor (EGFR), further promote tumorigenesis. The tumor microenvironment, influenced by hypoxia, inflammation, and cellular interactions, also contributes to their development. Combining these factors transforms normal oligodendrocytes or their precursors into malignant cells, giving rise to OGs. Research into these mechanisms continues to inform diagnostic and therapeutic advancements, offering hope for improved patient outcomes.
Epidemiology
OGs are uncommon, with an incidence of 0.2 per 100,000 people, and are the third most common primary brain neoplasm following the glioblastoma and the diffuse astrocytoma. OGs comprise approximately 5% of all primary CNS tumors.[5][8][9] These tumors have a slight male predominance, reported from 1.1 to 2, with as low as a 0.92 male-to-female ratio, as seen from the results of a study.[10] OGs have a slight bimodal distribution but are primarily adult neoplasm, with a peak incidence occurring in the fourth and fifth decades and a smaller tumor incidence peak in children aged 6 to 12.[9] OGs in pediatric patients usually demonstrate different molecular markers than are seen in the adult form, raising the issue of whether they represent the same neoplasm.
Pathophysiology
OGs are diffuse gliomas that arise predominantly in the hemispheric cerebral white matter, with 80% to 90% being supratentorial. The frontal lobes are the most commonly affected sites, although involvement of the temporal and parietal lobes is also frequently observed. These tumors are typically located in a cortical-subcortical distribution, with diffuse infiltration into the adjacent white matter. Rarely, OGs can occur in intraventricular or subependymal locations.
Genetic Features
A hallmark of OGs is chromosomal arms 1p and 19q codeletion, which occurs in 70% to 90% of cases. This genetic alteration is diagnostic and associated with better prognosis and enhanced responsiveness to radiotherapy and chemotherapy. Nearly all OGs harbor mutations in the IDH1 or IDH2 genes, producing the oncometabolite 2-hydroxyglutarate. This molecule induces a G-CIMP methylator phenotype, promoting DNA hypermethylation, impairing cellular differentiation, and facilitating tumor growth. Other genetic alterations, such as TP53 mutations or ATRX gene alterations, are less common and help distinguish OGs from other gliomas.
Epigenetic and Cellular Mechanisms
Epigenetic modifications, including DNA hypermethylation and histone changes, further contribute to tumor development by altering gene expression and cell differentiation. Dysregulated signaling pathways, such as those involving PDGFR, EGFR, and phosphoinositide 3-kinase (PI3K), drive tumor cell proliferation, angiogenesis, and resistance to apoptosis. The tumor microenvironment, characterized by hypoxia, inflammation, and immune evasion, plays an additional role in sustaining tumor progression.
Cellular Origin
Historically, OGs were thought to originate from mature oligodendrocytes. However, recent evidence suggests that they arise from neuroprogenitor cells with glial precursor characteristics. These progenitor cells acquire mutations and signaling dysregulation, transforming into malignant cells with an oligodendroglial phenotype.
Histopathology
OGs exhibit distinctive macroscopic and microscopic characteristics that reflect their origin and biological behavior. Macroscopically, OGs are typically gelatinous gray masses with areas of cystic degeneration, calcifications, and occasionally small focal hemorrhages. These features are often observed during surgical resection or imaging studies. Microscopically, OGs are characterized by sheets of uniform, small-to-medium-sized cells with spherical nuclei surrounded by a clear perinuclear halo, creating the classic "fried egg" appearance. This artifact arises from routine histological processing with formalin and paraffin fixation. A network of thin-walled capillaries, described as a "chicken wire" vascular pattern, is a hallmark of these tumors. Scattered calcifications and psammoma bodies are commonly identified.
Occasionally, myelin-like structures are observed on the tumor surface, although they do not meet the criteria for true myelin sheaths.[6] High-grade, anaplastic OGs (WHO grade III) exhibit increased cellular density, significant nuclear atypia, and heightened mitotic activity. These tumors may also display microvascular proliferation, increased blood vessel density, and necrosis, reflecting tumor cells' accelerated growth and proliferation.
Immunohistochemistry aids in differentiating OGs from other gliomas:
- Olig2
- This transcription factor is associated with oligodendrocyte progenitor cells used in neuronal or oligodendrocytic cell differentiation consistently expressed in OGs.[11]
- Glial fibrillary acidic protein
- This is commonly expressed in OGs but to a lesser degree than in astrocytomas.[5]
- Myelin basic protein
- OGs lack basic myelin proteins found in myelin-producing oligodendrocytes and Schwann cells.
- Ki-67
- This marker for cellular proliferation is low in low-grade OGs but elevated in anaplastic forms.
Molecular features are critical for diagnosis:
- 1p/19q codeletion
- This defining genetic hallmark seen in 70% to 90% of cases is strongly associated with better prognosis and response to therapy.
- IDH mutations
- This mutation, found in nearly all OGs, contributes to tumorigenesis through the production of the oncometabolite 2-hydroxyglutarate.
- TERT promoter mutations
- This is commonly present and involved in telomere maintenance.
OGs are predominantly located in the hemispheric white matter, with 80% to 90% being supratentorial and most commonly found in the frontal lobes. They are diffusely infiltrative, involving adjacent white matter, and less frequently occur in subependymal or intraventricular locations. These histopathological and molecular features facilitate accurate diagnosis and inform prognosis and therapeutic strategies. Advanced understanding of the tumor microenvironment and genetic drivers continues to shape the management of OGs.
History and Physical
History
Patients with OGs often present with nonspecific symptoms such as headaches or more specific findings like seizures, which are among the most common presenting complaints. Seizures are reported in 35% to 91% of cases, making them a hallmark symptom of OG.[9] These seizures may range from focal to generalized and are often refractory to standard antiepileptic therapy, particularly in recurrent or progressive cases. Persistent or progressive headaches may indicate increased intracranial pressure or local mass effect. At the same time, cognitive or personality changes, especially with frontal lobe tumors, can include impaired judgment, memory deficits, or disinhibition.
Depending on the tumor's location, patients occasionally develop focal neurological deficits such as weakness, sensory disturbances, aphasia, or visual field deficits. Increased intracranial pressure symptoms, including nausea, vomiting, and papilledema, may occur in cases with significant mass effects or hydrocephalus. Any patient presenting with new-onset seizures, focal neurological deficits, or unexplained cognitive changes should undergo CNS imaging to evaluate for underlying pathologies, including OGs.
Physical Examination
Findings on physical examination depend on the tumor’s location, size, and mass effect.
- Neurological Examination
- Seizures
- May be ongoing or reported as a recurrent issue in the patient’s history
- Focal neurological deficits
- Weakness, sensory loss, or aphasia consistent with the affected cortical region
- Visual field deficits
- Homonymous hemianopia or quadrantanopia, particularly with temporal or parietal lobe involvement
- Cognitive and behavioral changes
- Impaired executive function, personality changes, or memory deficits with frontal lobe lesions
- Seizures
- Signs of increased intracranial pressure
- Papilledema on fundoscopic examination
- Sixth nerve palsy due to raised pressure
- Location-specific symptoms
- Frontal lobe
- Disinhibition, apathy, or difficulty with planning and judgment
- Temporal lobe
- Memory disturbances, auditory hallucinations, or language deficits
- Parietal lobe
- Sensory deficits, spatial neglect
- Occipital lobe
- Visual field deficits or cortical blindness
- Frontal lobe
Patients with high-grade or anaplastic OGs may present with a rapid onset of symptoms and severe neurological deficits due to the tumor’s fast growth. The history and physical examination provide critical clues for early recognition and guide the need for diagnostic imaging and further management.
Evaluation
Laboratory Tests
Laboratory investigations for OGs are generally nonspecific but may be performed to assess general health and rule out other conditions. Routine tests like a complete blood count and serum electrolytes can help identify complications such as anemia, infections, or electrolyte disturbances that may arise from the tumor or seizures. However, there are no definitive blood biomarkers for OGs in clinical practice. Genetic markers, such as glial fibrillary acidic protein and Olig2, may be used in research settings to help understand the tumor’s molecular characteristics. These markers are more useful in differentiating OGs from other gliomas, but histological and molecular analysis remains the primary diagnostic approach.
Radiographic Imaging
Computed tomography scan
Computed tomography (CT) is often the initial imaging modality used for patients presenting with nonspecific neurological symptoms, particularly to rule out acute abnormalities such as hemorrhage or ischemia quickly. OGs on CT are typically hypodense or occasionally isodense with the surrounding brain tissue and appear as peripheral masses that may expand the cortical gyri (see Image. Oligodendroglioma, Computed Tomography Scan). Calcifications, seen in up to 90% of OGs, often present in a gyriform pattern, highly suggestive of this tumor type.[12][13] The presence of peritumoral edema and hemorrhage is uncommon, and the degree of contrast enhancement is usually mild, varying by tumor grade.
Magnetic resonance imaging
Magnetic resonance imaging (MRI) offers a better characterization of the tumor appearance than CT and can help define the OG's true extent and infiltrative nature. T2 weighted sequences will demonstrate a circumscribed heterogeneously hyperintense tumor, often cortical/subcortical in location, and may demonstrate a slight peritumoral hyperintense signal. Within the tumor, there are frequently small areas of cystic change, microhemorrhage, and macroscopic calcifications demonstrated by T2 hypointense signal due to susceptibility signal loss and increased tumor heterogeneity. OGs are hypointense compared to gray matter on T1 weighted imaging, and enhancement in low-grade tumors is variable, ranging from patchy enhancement to moderate enhancement, where the presence of enhancement does not necessarily represent a higher histological grade of OG. An OG typically has facilitated rather than restricted diffusion, with hyperintensity seen on apparent diffusion coefficient maps.
More advanced MRI techniques, such as spectroscopy and perfusion, may complement anatomic imaging and help characterize and grade the tumor type. However, most OGs are low-grade, so these advanced imaging techniques may not provide significant additional information to the more standard anatomic imaging. Spectroscopy shows a mildly elevated choline level and decreased N-acetyl aspartate peaks, seen in most neoplasms, which are more prominent in high-grade tumors than low-grade gliomas due to their increased mitotic activity. Elevated myoinositol and 2-hydroxyglutarate presence may suggest isocitrate dehydrogenase mutation status.[14]
The presence of elevated lipid and lactate peaks generally suggests a high-grade tumor.[13] T2 weighted dynamic susceptibility contrast perfusion imaging can show elevated cerebral blood flow and cerebral blood volume in higher-grade tumors due to neovascularization compared to low-grade tumors. However, there is a significant degree of overlap between them.[13][15][16] T1 weighted dynamic contrast-enhanced perfusion may show elevated Ktrans, representing leaky vasculature and the diffusion of contrast outside of the capillary endothelium in higher-grade tumors compared to low-grade tumors. Still, again, there is significant overlap where most low-grade tumors do not have significant contrast enhancement.[17][18][19]
Preoperative evaluation of white matter tracts using MRI diffusion tensor imaging sequences and tractography can demonstrate their position and proximity to the tumor. This typically shows displacement of the tracts rather than destruction by the tumor. Additional evaluation with functional MRI can demonstrate the tumor's proximity to the eloquent cortex, including important language, motor, sensory, and visual areas.
Genetic Testing and Histopathology
Histopathologic examination remains essential for definitive diagnosis, as radiographic imaging alone cannot distinguish OGs from other gliomas, especially low-grade tumors. All patients with a tumor will require tissue sampling and histologic examination of that tissue for diagnosis. In 2016, the WHO changed the criteria for classifying CNS tumors to include phenotypic and genotypic analysis. The diagnosis of OG is now determined using molecular markers, and the tumor must possess both an IDH1 or IDH2 mutation and 1p/19q codeletion to be classified as an OG.[2][20] The 1p/19q mutation leads to epigenetic changes and hypermethylation of the genome, demonstrating a distinct biological phenotype associated with improved survival rates.
Tumors that cannot be tested for these genetic markers but would traditionally be diagnosed as OG on histology should be designated OG not otherwise specified (NOS). When a molecular analysis is unavailable, or the molecular diagnosis is equivocal, the diagnosis of OG NOS should be used. Pediatric OG often does not have the characteristics of IDH mutation and 1p/19q codeletion present in their adult counterparts, which may genetically represent a different disease.[5][21][22][23] Further studies on adult and pediatric molecular and genetic markers are ongoing.
Treatment / Management
Treatment of an OG is multifactorial and consists of surgical, chemotherapy, and radiation therapy (RT) treatment methods.
Surgery
Surgical treatments are considered on a case-by-case basis. Tumor location, patient comorbidities, and other surgical risk factors are used to determine the type of surgery that will benefit the optimal patient. Complete or gross total resection of the tumor is the primary treatment of choice for the OG. This approach will increase overall survival time and can occasionally be curative. Therefore, all attempts should be made to achieve this surgical result if possible.[9][24][25][26] Suppose a complete tumor resection cannot be obtained. In that case, a debulking procedure can be performed to help improve neurological symptoms and seizure control and decrease the potential for malignant transformation. This procedure has also increased overall survival time and time to tumor progression.[27] In patients with partially resected low-grade tumors that are being observed and demonstrate increased tumor growth over time, or in patients with concern for malignant transformation to a higher-grade neoplasm, repeat surgical resection is still recommended in an attempt to cure the patient potentially or to confirm the histological grade of the tumor.(A1)
Surgical resection techniques depend on the location of the OG, where the eloquent cortex and vital neurological structures must be preserved. The location of the craniotomy utilized for each approach and access will vary, and some surgeons prefer to perform the resection on an awake patient to stimulate, test, and map adjacent brain parenchyma to help determine the extent of tissue that can safely be removed. Intraoperative imaging techniques, such as intraoperative MRI or ultrasound, have also been used to assist in maximizing the extent of the tumor resection. These adjunct imaging techniques may be more efficacious than standard stereotactic navigation, increase operative time, and require special equipment and training.
Using 5-aminolevulinic acid (5-ALA) fluorescent dye can also be a helpful surgical tool. 5-ALA is a heme precursor that is shown to cause fluorescence in malignant tissue by creating fluorescent porphyrins, which can be visualized intraoperatively. This agent has also been shown to have statistically significant improvement in the extent of resection of high-grade gliomas over conventional stereotactic navigation and is overall similar in efficacy to intraoperative MRI. However, it has only been shown in small sample sizes.[9][28][29][30] Further study and evaluation may be warranted.(A1)
Radiation
RT also plays an important role in treating OG, most often after surgical resection. There does not appear to be a difference in the overall survival between early or delayed postoperative RT. Still, an increased time to progression has been shown with early treatment, in addition to better seizure control following surgery.[31](A1)
Standard focal or limited field hyperfractionated RT is typically employed. The treatment field consists of the surgical resection bed, areas of residual enhancement on T1 postcontrast imaging, and a margin of up to 3 cm, including the adjacent T2/FLAIR signal hyperintensity that may represent infiltrating disease. This margin is modified to minimize the involvement of critical neural structures or if microscopic/infiltrating disease is considered less likely to present.[32] The radiation dose administered has varying recommendation amounts of 45 to 65 Gy in 1.8 to 2 Gy fractions, without significant survival benefit seen between 45 Gy and 59.4 Gy or 54 Gy and 65 Gy treatments. Dosing and the treatment schedule are case-dependent, based on prognosis, with higher doses typically reserved for higher-grade tumors. Hypofractionated doses may be applied in older individuals with poor prognoses, giving fewer, higher single fractions for the same or lower total dose.[12][32][33][34] RT is usually withheld from children to minimize some of the negative long-term side effects of radiation, such as cognitive impairment, personality changes, hypopituitarism, motor and coordination abnormalities, and the development of other neoplasms.[9][35] Adults receiving RT may develop some of these negative effects after therapy or can experience other problems, such as radiation necrosis.(A1)
Chemotherapy
In addition to RT, chemotherapy is a commonly used adjunct, demonstrating increased survival in RT with chemotherapy groups compared to RT alone.[3] There is good evidence that a combination of procarbazine, lomustine, and vincristine (PCV) can be beneficial for treating OGs.[36][37] However, this regimen is typically limited to 6 cycles due to adverse events from the agents.[33][38] These adverse drug events, most often leukopenia and thrombocytopenia, can lead to early discontinuation of the treatment in many patients. For this reason, temozolomide (TMZ) is often the chemotherapy of choice. TMZ is an oral DNA alkylating agent that patients better tolerate than nitrosourea-containing regimens such as PCV. TMZ alone has been shown to occasionally result in a recurrent hypermutated tumor phenotype that is TMZ-resistant. At this time, the combination of RT and chemotherapy affords better overall survival and progression-free survival, but the choice of PCV or TMZ is still debated. Results from recent studies have shown that PCV could lead to better overall survival compared to TMZ in 1p/19q co-deleted patients, advocating for PCV as first-line use chemotherapy in the treatment of OG.[39][40] Currently, the ongoing CODEL study (NCT00887146) assessing outcomes of RT and PCV and RT and TMZ will give a better understanding between these 2 treatment regimens and potentially a determination of superiority. However, results may still be 7 to 10 years in the future.[12][41](A1)
Additionally, noncytotoxic treatment has been approved for recurrent glioblastoma use in the United States, Canada, and Europe. Still, it may also benefit anaplastic OGs that demonstrate increased vascularity, as evidenced by enhancement and peritumoral edema on MRI. Bevacizumab is a monoclonal antibody against vascular endothelial growth factor (VEGF), and these antibodies bind to the VEGF receptors, decreasing neovascularization and vascular permeability and helping to stabilize the blood-brain barrier.[36] This result eventually leads to less peritumoral edema and decreased need for corticosteroids, improving symptoms and progression-free survival as seen in some study results. However, this treatment overall appears limited to recurrent disease, palliative treatment, and in patients with brain radiation necrosis.[42][43]
Differential Diagnosis
Diffuse astrocytoma is the primary differential diagnosis on imaging and is virtually indistinguishable from OG. On imaging studies, diffuse astrocytoma often has less dystrophic calcification and more white matter and less cortical involvement than the OG. However, the main discriminating factors between the 2 tumor types are the genetic markers, where the lack of the 1p/19q deletion occurs in the astrocytoma.
Glioblastoma is an important diagnostic consideration from OG because this tumor carries a much poorer prognosis and is, unfortunately, much more common in adults than children. Glioblastoma will typically demonstrate more avid and heterogeneous enhancement than low-grade OG. Although glioblastoma may be hard to distinguish from the higher grade, it enhances anaplastic OG. Glioblastoma can sometimes have patchy areas of restricted diffusion and areas of central necrosis, which are both uncommon in OG. Calcification is uncommon in glioblastoma and very common in OG. Histologically, glioblastoma will have more aggressive features such as necrosis and neovascularization, and they lack the 1p/19q deletion and are often IDH mutation negative compared to OG on molecular analysis.
Dysembryoplastic neuroepithelial tumors (DNETs) are another low-grade cortical-subcortical-based neoplasm that can look similar to OG in imaging studies. However, DNETs tend to have a more “bubbly” cystic T2 hyperintense appearance and may have adjacent cortical dysplasia. Compared to OGs, which are more often diagnosed in older adults, DNETs are more often found in children and young adults. Ganglioglioma is another cortical-subcortical neoplasm that occurs more often in children and young adults and can be a seizure focus. On MRI, gangliogliomas typically present as cysts with enhancing nodules and are more commonly seen in the temporal lobe than the OG. A multinodular and vacuolating neuronal tumor is a cortical-subcortical lesion that may have imaging overlap with OG and typically appears as a cluster of small bubbly cysts that are T2/FLAIR hyperintense and do not typically enhance. The lesion is typically incidentally found and maybe a malformation/dysplastic lesion instead of a true neoplasm.
Prognosis
Low-grade OGs (WHO Grade II), which by definition have 1p/19q deletion and IDH mutant, have a better prognosis than astrocytomas without these genetic markers.[44] OGs have a median survival time of 10 to 12 years and 5-year progression-free and overall survival rates of 51% to 83%. Younger patients, patients without other comorbidities, and those with a greater extent of tumor resection tend to have a better prognosis.[8][10][24][25][26] The overall and 5-year survival rates decrease with higher-grade anaplastic OGs, where the median survival time is 3.5 years in WHO Grade III tumors.
Complications
Complications from OGs may include seizures, postsurgical complications, and thromboembolic events. Chemotherapeutic adverse events, including myelosuppression limiting treatment with PCV, as well as nausea, vomiting, and other gastrointestinal symptoms that occur with TMZ. The long-term adverse events of radiation therapy include cognitive deficits, gait abnormalities, or radiation necrosis. Residual or recurrent OGs can also undergo malignant degeneration over time.
Deterrence and Patient Education
While the exact cause of OGs remains unclear, deterrence focuses on minimizing potential risk factors where possible. Patients should avoid unnecessary exposure to ionizing radiation and limit contact with known carcinogens, particularly in occupational settings. Encouraging a healthy lifestyle, including smoking cessation and balanced nutrition, may also promote general brain health, though specific preventative strategies for OGs are not established.
Patient education is vital in improving outcomes. Clinicians should ensure patients and caregivers understand the nature of the disease, the need for regular follow-ups, and the rationale for imaging and treatment plans. Discussing symptoms of recurrence, potential adverse events from treatment, and the importance of adhering to prescribed therapies is crucial. Providing access to resources, including support groups and educational materials, can help patients confidently navigate their diagnosis and treatment journey. Additionally, multidisciplinary teams should emphasize open communication to address patient concerns, manage expectations, and enhance quality of life.
Pearls and Other Issues
The OG is a low-grade (WHO Grade II) CNS neoplasm with many imaging characteristics similar to other astrocytomas, so tissue sampling is essential for an accurate diagnosis. The OG is defined by its IDH mutation and 1p/19q deletion status and several imaging features that suggest the presence of an OG as opposed to an astrocytoma that includes course calcification and cortical-subcortical location.
The OG is chemotherapy and radiation therapy sensitive, with a good prognosis when compared to other glial neoplasms. However, the higher-grade anaplastic OG (WHO Grade III) has a worse prognosis. Current studies are ongoing to evaluate the optimal combinations of RT and chemotherapy for treating OGs, and other targeted therapies are also being evaluated.[45][46]
Enhancing Healthcare Team Outcomes
Effective oligodendroglioma (OG) management requires a multidisciplinary approach, where clinicians, nurses, pharmacists, and other healthcare professionals collaborate seamlessly to enhance patient-centered care and outcomes. Advanced clinicians, including neurologists, neurosurgeons, and oncologists, lead the diagnostic and therapeutic decision-making, using advanced imaging, histopathological, and molecular analyses to tailor treatment plans. Advanced clinicians and nurses play pivotal roles in patient education, symptom management, and care coordination, ensuring patients and their families understand the disease process and treatment options. Pharmacists provide critical input on chemotherapeutic regimens, ensuring accurate dosing, monitoring for drug interactions, and managing side effects to enhance patient safety.
Interprofessional communication and coordinated care strategies are vital in delivering comprehensive care for patients with OG. Regular tumor board discussions and collaborative treatment planning allow for the integration of diverse expertise, ensuring individualized and evidence-based care. Clear communication across teams facilitates seamless transitions between diagnostic evaluations, surgical interventions, and postoperative care. Additionally, ongoing psychosocial support from social workers and mental health professionals addresses patients' and caregivers' emotional and psychological needs. By fostering a culture of teamwork and open communication, healthcare professionals can improve outcomes, enhance patient safety, and optimize the overall quality of care for individuals with OG.
Media
(Click Image to Enlarge)
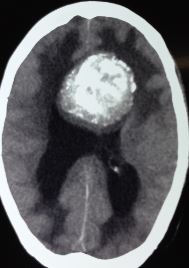
Oligodendroglioma, Computed Tomography Scan. On computed tomography scans, oligodendrogliomas are typically hypodense or occasionally isodense with the surrounding brain tissue and appear as peripheral masses that may expand the cortical gyri.
Contributed by S Munakomi, MD
References
Ostrom QT, Gittleman H, Liao P, Vecchione-Koval T, Wolinsky Y, Kruchko C, Barnholtz-Sloan JS. CBTRUS Statistical Report: Primary brain and other central nervous system tumors diagnosed in the United States in 2010-2014. Neuro-oncology. 2017 Nov 6:19(suppl_5):v1-v88. doi: 10.1093/neuonc/nox158. Epub [PubMed PMID: 29117289]
Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, Ohgaki H, Wiestler OD, Kleihues P, Ellison DW. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta neuropathologica. 2016 Jun:131(6):803-20. doi: 10.1007/s00401-016-1545-1. Epub 2016 May 9 [PubMed PMID: 27157931]
Anderson MD, Gilbert MR. Treatment recommendations for anaplastic oligodendrogliomas that are codeleted. Oncology (Williston Park, N.Y.). 2013 Apr:27(4):315-20, 322 [PubMed PMID: 23781695]
Cairncross G, Jenkins R. Gliomas with 1p/19q codeletion: a.k.a. oligodendroglioma. Cancer journal (Sudbury, Mass.). 2008 Nov-Dec:14(6):352-7. doi: 10.1097/PPO.0b013e31818d8178. Epub [PubMed PMID: 19060598]
Wesseling P, van den Bent M, Perry A. Oligodendroglioma: pathology, molecular mechanisms and markers. Acta neuropathologica. 2015 Jun:129(6):809-27. doi: 10.1007/s00401-015-1424-1. Epub 2015 May 6 [PubMed PMID: 25943885]
Komori T. Pathology of oligodendroglia: An overview. Neuropathology : official journal of the Japanese Society of Neuropathology. 2017 Oct:37(5):465-474. doi: 10.1111/neup.12389. Epub 2017 May 26 [PubMed PMID: 28548216]
Level 3 (low-level) evidenceLiu YQ, Chai RC, Wang YZ, Wang Z, Liu X, Wu F, Jiang T. Amino acid metabolism-related gene expression-based risk signature can better predict overall survival for glioma. Cancer science. 2019 Jan:110(1):321-333. doi: 10.1111/cas.13878. Epub 2018 Dec 17 [PubMed PMID: 30431206]
Van den Bent MJ, Reni M, Gatta G, Vecht C. Oligodendroglioma. Critical reviews in oncology/hematology. 2008 Jun:66(3):262-72. doi: 10.1016/j.critrevonc.2007.11.007. Epub 2008 Feb 12 [PubMed PMID: 18272388]
Engelhard HH. Current diagnosis and treatment of oligodendroglioma. Neurosurgical focus. 2002 Feb 15:12(2):E2 [PubMed PMID: 16212319]
Ohgaki H, Kleihues P. Population-based studies on incidence, survival rates, and genetic alterations in astrocytic and oligodendroglial gliomas. Journal of neuropathology and experimental neurology. 2005 Jun:64(6):479-89 [PubMed PMID: 15977639]
Sun Y, Meijer DH, Alberta JA, Mehta S, Kane MF, Tien AC, Fu H, Petryniak MA, Potter GB, Liu Z, Powers JF, Runquist IS, Rowitch DH, Stiles CD. Phosphorylation state of Olig2 regulates proliferation of neural progenitors. Neuron. 2011 Mar 10:69(5):906-17. doi: 10.1016/j.neuron.2011.02.005. Epub [PubMed PMID: 21382551]
Level 3 (low-level) evidencevan den Bent MJ, Chang SM. Grade II and III Oligodendroglioma and Astrocytoma. Neurologic clinics. 2018 Aug:36(3):467-484. doi: 10.1016/j.ncl.2018.04.005. Epub 2018 Jun 15 [PubMed PMID: 30072066]
Smits M. Imaging of oligodendroglioma. The British journal of radiology. 2016:89(1060):20150857. doi: 10.1259/bjr.20150857. Epub 2016 Feb 5 [PubMed PMID: 26849038]
Choi C, Ganji SK, DeBerardinis RJ, Hatanpaa KJ, Rakheja D, Kovacs Z, Yang XL, Mashimo T, Raisanen JM, Marin-Valencia I, Pascual JM, Madden CJ, Mickey BE, Malloy CR, Bachoo RM, Maher EA. 2-hydroxyglutarate detection by magnetic resonance spectroscopy in IDH-mutated patients with gliomas. Nature medicine. 2012 Jan 26:18(4):624-9. doi: 10.1038/nm.2682. Epub 2012 Jan 26 [PubMed PMID: 22281806]
Ellenbogen JR, Walker C, Jenkinson MD. Genetics and imaging of oligodendroglial tumors. CNS oncology. 2015:4(5):307-15. doi: 10.2217/cns.15.37. Epub 2015 Oct 19 [PubMed PMID: 26478219]
Delgado AF, Delgado AF. Discrimination between Glioma Grades II and III Using Dynamic Susceptibility Perfusion MRI: A Meta-Analysis. AJNR. American journal of neuroradiology. 2017 Jul:38(7):1348-1355. doi: 10.3174/ajnr.A5218. Epub 2017 May 18 [PubMed PMID: 28522666]
Level 1 (high-level) evidenceAnzalone N, Castellano A, Cadioli M, Conte GM, Cuccarini V, Bizzi A, Grimaldi M, Costa A, Grillea G, Vitali P, Aquino D, Terreni MR, Torri V, Erickson BJ, Caulo M. Brain Gliomas: Multicenter Standardized Assessment of Dynamic Contrast-enhanced and Dynamic Susceptibility Contrast MR Images. Radiology. 2018 Jun:287(3):933-943. doi: 10.1148/radiol.2017170362. Epub 2018 Jan 22 [PubMed PMID: 29361245]
Liang J, Liu D, Gao P, Zhang D, Chen H, Shi C, Luo L. Diagnostic Values of DCE-MRI and DSC-MRI for Differentiation Between High-grade and Low-grade Gliomas: A Comprehensive Meta-analysis. Academic radiology. 2018 Mar:25(3):338-348. doi: 10.1016/j.acra.2017.10.001. Epub 2017 Dec 6 [PubMed PMID: 29223713]
Level 1 (high-level) evidenceZhao M, Guo LL, Huang N, Wu Q, Zhou L, Zhao H, Zhang J, Fu K. Quantitative analysis of permeability for glioma grading using dynamic contrast-enhanced magnetic resonance imaging. Oncology letters. 2017 Nov:14(5):5418-5426. doi: 10.3892/ol.2017.6895. Epub 2017 Sep 6 [PubMed PMID: 29113174]
Level 3 (low-level) evidenceBai J, Varghese J, Jain R. Adult Glioma WHO Classification Update, Genomics, and Imaging: What the Radiologists Need to Know. Topics in magnetic resonance imaging : TMRI. 2020 Apr:29(2):71-82. doi: 10.1097/RMR.0000000000000234. Epub [PubMed PMID: 32271284]
Rodriguez FJ, Tihan T, Lin D, McDonald W, Nigro J, Feuerstein B, Jackson S, Cohen K, Burger PC. Clinicopathologic features of pediatric oligodendrogliomas: a series of 50 patients. The American journal of surgical pathology. 2014 Aug:38(8):1058-70. doi: 10.1097/PAS.0000000000000221. Epub [PubMed PMID: 24805856]
Richard H, Stogner-Underwood K, Fuller C. Congenital oligodendroglioma: clinicopathologic and molecular assessment with review of the literature. Case reports in pathology. 2015:2015():370234. doi: 10.1155/2015/370234. Epub 2015 Feb 10 [PubMed PMID: 25755903]
Level 3 (low-level) evidenceNauen D, Haley L, Lin MT, Perry A, Giannini C, Burger PC, Rodriguez FJ. Molecular Analysis of Pediatric Oligodendrogliomas Highlights Genetic Differences with Adult Counterparts and Other Pediatric Gliomas. Brain pathology (Zurich, Switzerland). 2016 Mar:26(2):206-14. doi: 10.1111/bpa.12291. Epub 2015 Aug 14 [PubMed PMID: 26206478]
Kinslow CJ, Garton ALA, Rae AI, Marcus LP, Adams CM, McKhann GM, Sisti MB, Connolly ES, Bruce JN, Neugut AI, Sonabend AM, Canoll P, Cheng SK, Wang TJC. Extent of resection and survival for oligodendroglioma: a U.S. population-based study. Journal of neuro-oncology. 2019 Sep:144(3):591-601. doi: 10.1007/s11060-019-03261-5. Epub 2019 Aug 12 [PubMed PMID: 31407129]
Buckner JC. Factors influencing survival in high-grade gliomas. Seminars in oncology. 2003 Dec:30(6 Suppl 19):10-4 [PubMed PMID: 14765378]
Kim YZ, Kim CY, Wee CW, Roh TH, Hong JB, Oh HJ, Kang SG, Kang SH, Kong DS, Kim SH, Kim SH, Kim SH, Kim YJ, Kim EH, Kim IA, Kim HS, Park JS, Park HJ, Song SW, Sung KS, Yang SH, Yoon WS, Yoon HI, Lee J, Lee ST, Lee SW, Lee YS, Lim J, Chang JH, Jung TY, Jung HL, Cho JH, Choi SH, Choi HS, Lim DH, Chung DS, KSNO Guideline Working Group. The Korean Society for Neuro-Oncology (KSNO) Guideline for WHO Grade II Cerebral Gliomas in Adults: Version 2019.01. Brain tumor research and treatment. 2019 Oct:7(2):74-84. doi: 10.14791/btrt.2019.7.e43. Epub [PubMed PMID: 31686437]
Jiang B, Chaichana K, Veeravagu A, Chang SD, Black KL, Patil CG. Biopsy versus resection for the management of low-grade gliomas. The Cochrane database of systematic reviews. 2017 Apr 27:4(4):CD009319. doi: 10.1002/14651858.CD009319.pub3. Epub 2017 Apr 27 [PubMed PMID: 28447767]
Level 1 (high-level) evidenceGolub D, Hyde J, Dogra S, Nicholson J, Kirkwood KA, Gohel P, Loftus S, Schwartz TH. Intraoperative MRI versus 5-ALA in high-grade glioma resection: a network meta-analysis. Journal of neurosurgery. 2021 Feb 1:134(2):484-498. doi: 10.3171/2019.12.JNS191203. Epub 2020 Feb 21 [PubMed PMID: 32084631]
Level 1 (high-level) evidenceJenkinson MD, Barone DG, Bryant A, Vale L, Bulbeck H, Lawrie TA, Hart MG, Watts C. Intraoperative imaging technology to maximise extent of resection for glioma. The Cochrane database of systematic reviews. 2018 Jan 22:1(1):CD012788. doi: 10.1002/14651858.CD012788.pub2. Epub 2018 Jan 22 [PubMed PMID: 29355914]
Level 1 (high-level) evidenceGandhi S, Tayebi Meybodi A, Belykh E, Cavallo C, Zhao X, Syed MP, Borba Moreira L, Lawton MT, Nakaji P, Preul MC. Survival Outcomes Among Patients With High-Grade Glioma Treated With 5-Aminolevulinic Acid-Guided Surgery: A Systematic Review and Meta-Analysis. Frontiers in oncology. 2019:9():620. doi: 10.3389/fonc.2019.00620. Epub 2019 Jul 17 [PubMed PMID: 31380272]
Level 1 (high-level) evidenceDhawan S, Patil CG, Chen C, Venteicher AS. Early versus delayed postoperative radiotherapy for treatment of low-grade gliomas. The Cochrane database of systematic reviews. 2020 Jan 20:1(1):CD009229. doi: 10.1002/14651858.CD009229.pub3. Epub 2020 Jan 20 [PubMed PMID: 31958162]
Level 1 (high-level) evidenceWeller M, van den Bent M, Tonn JC, Stupp R, Preusser M, Cohen-Jonathan-Moyal E, Henriksson R, Le Rhun E, Balana C, Chinot O, Bendszus M, Reijneveld JC, Dhermain F, French P, Marosi C, Watts C, Oberg I, Pilkington G, Baumert BG, Taphoorn MJB, Hegi M, Westphal M, Reifenberger G, Soffietti R, Wick W, European Association for Neuro-Oncology (EANO) Task Force on Gliomas. European Association for Neuro-Oncology (EANO) guideline on the diagnosis and treatment of adult astrocytic and oligodendroglial gliomas. The Lancet. Oncology. 2017 Jun:18(6):e315-e329. doi: 10.1016/S1470-2045(17)30194-8. Epub 2017 May 5 [PubMed PMID: 28483413]
van den Bent MJ, Smits M, Kros JM, Chang SM. Diffuse Infiltrating Oligodendroglioma and Astrocytoma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2017 Jul 20:35(21):2394-2401. doi: 10.1200/JCO.2017.72.6737. Epub 2017 Jun 22 [PubMed PMID: 28640702]
Cairncross G, Wang M, Shaw E, Jenkins R, Brachman D, Buckner J, Fink K, Souhami L, Laperriere N, Curran W, Mehta M. Phase III trial of chemoradiotherapy for anaplastic oligodendroglioma: long-term results of RTOG 9402. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2013 Jan 20:31(3):337-43. doi: 10.1200/JCO.2012.43.2674. Epub 2012 Oct 15 [PubMed PMID: 23071247]
Level 1 (high-level) evidenceCayuela N, Jaramillo-Jiménez E, Càmara E, Majós C, Vidal N, Lucas A, Gil-Gil M, Graus F, Bruna J, Simó M. Cognitive and brain structural changes in long-term oligodendroglial tumor survivors. Neuro-oncology. 2019 Nov 4:21(11):1470-1479. doi: 10.1093/neuonc/noz130. Epub [PubMed PMID: 31549152]
Bou Zerdan M, Assi HI. Oligodendroglioma: A Review of Management and Pathways. Frontiers in molecular neuroscience. 2021:14():722396. doi: 10.3389/fnmol.2021.722396. Epub 2021 Oct 5 [PubMed PMID: 34675774]
Sasaki H, Kitamura Y, Toda M, Hirose Y, Yoshida K. Oligodendroglioma, IDH-mutant and 1p/19q-codeleted-prognostic factors, standard of care and chemotherapy, and future perspectives with neoadjuvant strategy. Brain tumor pathology. 2024 Apr:41(2):43-49. doi: 10.1007/s10014-024-00480-1. Epub 2024 Apr 2 [PubMed PMID: 38564040]
Level 3 (low-level) evidenceFogh SE, Boreta L, Nakamura JL, Johnson DR, Chi AS, Kurz SC. Neuro-Oncology Practice Clinical Debate: Early treatment or observation for patients with newly diagnosed oligodendroglioma and small-volume residual disease. Neuro-oncology practice. 2021 Feb:8(1):11-17. doi: 10.1093/nop/npaa037. Epub 2020 Jun 27 [PubMed PMID: 33664965]
McDuff SGR, Dietrich J, Atkins KM, Oh KS, Loeffler JS, Shih HA. Radiation and chemotherapy for high-risk lower grade gliomas: Choosing between temozolomide and PCV. Cancer medicine. 2020 Jan:9(1):3-11. doi: 10.1002/cam4.2686. Epub 2019 Nov 7 [PubMed PMID: 31701682]
Brandner S, McAleenan A, Jones HE, Kernohan A, Robinson T, Schmidt L, Dawson S, Kelly C, Leal ES, Faulkner CL, Palmer A, Wragg C, Jefferies S, Vale L, Higgins JPT, Kurian KM. Diagnostic accuracy of 1p/19q codeletion tests in oligodendroglioma: A comprehensive meta-analysis based on a Cochrane systematic review. Neuropathology and applied neurobiology. 2022 Jun:48(4):e12790. doi: 10.1111/nan.12790. Epub 2022 Mar 3 [PubMed PMID: 34958131]
Level 1 (high-level) evidenceDrappatz J, Lieberman F. Chemotherapy of Oligodendrogliomas. Progress in neurological surgery. 2018:31():152-161. doi: 10.1159/000467376. Epub 2018 Jan 25 [PubMed PMID: 29393183]
Grimm SA, Chamberlain MC. Bevacizumab and other novel therapies for recurrent oligodendroglial tumors. CNS oncology. 2015:4(5):333-9. doi: 10.2217/cns.15.27. Epub 2015 Oct 28 [PubMed PMID: 26509217]
Zhuang H, Shi S, Yuan Z, Chang JY. Bevacizumab treatment for radiation brain necrosis: mechanism, efficacy and issues. Molecular cancer. 2019 Feb 7:18(1):21. doi: 10.1186/s12943-019-0950-1. Epub 2019 Feb 7 [PubMed PMID: 30732625]
Carstam L, Latini F, Solheim O, Bartek J Jr, Pedersen LK, Zetterling M, Beniaminov S, Sjåvik K, Ryttlefors M, Jensdottir M, Rydenhag B, Smits A, Jakola AS. Long-term follow up of patients with WHO grade 2 oligodendroglioma. Journal of neuro-oncology. 2023 Aug:164(1):65-74. doi: 10.1007/s11060-023-04368-6. Epub 2023 Aug 21 [PubMed PMID: 37603235]
Lamb YN. Correction: Vorasidenib: First Approval. Drugs. 2024 Dec:84(12):1673. doi: 10.1007/s40265-024-02127-z. Epub 2024 Dec 2 [PubMed PMID: 39617874]
Lamb YN. Vorasidenib: First Approval. Drugs. 2024 Oct:84(10):1325-1331. doi: 10.1007/s40265-024-02097-2. Epub 2024 Oct 8 [PubMed PMID: 39375303]