Introduction
Neutrophils are the most common type of leukocytes, known colloquially as white blood cells, that normally circulate as granulocytes along the vascular endothelium in peripheral blood vessels. Neutrophils play an essential role in immune defenses because they ingest, kill, and digest invading microorganisms, including fungi and bacteria after extravasating through blood vessels, a process better known as transendothelial migration.[1] Failure to carry out this role leads to immunodeficiency, mainly characterized by recurrent infections.[2] Defects in neutrophil function can be quantitative, as seen in neutropenia, or qualitative, as seen in neutrophil dysfunction. The standard circulating neutrophil count should be greater than 1.5 x 109/L. Neutropenia can be classified as mild, moderate, severe, and agranulocytosis.[3] Please see StatPearls' associated reference, "Agranulocytosis," for further information on this topic. Neutropenia classification, per circulating neutrophil count, includes:
- Mild: 1.0 to 1.5 x 109/L
- Moderate: 0.5 to 1.0 x 109/L
- Severe: less than 0.5 x 109/L
- Agranulocytosis: less than 0.2 x 109/L with absence of neutrophil precursors in bone marrow
Neutropenia, or a state of low neutrophils, in itself has a wide differential that includes a primary disorder of production in the bone marrow, genetic deficiency, and cyclical deficiency. Additional causes of primary neutropenia include malignant disorders of marrow failure, such as leukopenia, or infiltrative hematologic neoplasms, such as high-grade lymphoma. Neutropenia can also be secondary to marrow extrinsic causes, including autoimmune diseases, cytotoxic chemotherapy, or suppressive illnesses or infections; even antibiotics can be toxic to neutrophils. Multifactorial neutropenia is also not uncommon, and is seen in patients who are on systemic chemotherapy and multiple broad-spectrum antibiotics., for instance.
Primary neutropenia, with decreased production and marrow hypoplasia, can be due to chronic benign neutropenia, cyclical neutropenia, or other congenital and genetic neutropenias. Acquired neutropenia can be secondary to cytotoxic drugs, leukemia, drug reactions, or infections. Congenital forms are rare and vary in severity. Some life-threatening neutropenias include leukocyte adhesion deficiency syndromes, Chediak-Higashi syndrome, Shwachman-Diamond syndrome, and chronic granulomatous disease.[4][5]
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
Neutrophils play a primary role in the immune defense against extracellular bacteria, including Staphylococci, Streptococci, and Escherichia coli, among others. They also protect against fungal infections, including those produced by Candida albicans. Once the neutrophil count is below 1.5 x 109/L, recurrent infections become more common. The monocyte count may increase as a compensatory mechanism.
Neutropenia can be considered acute (<3 months) or chronic (>3-6 months).[6] Neutropenia should be evaluated in conjunction with the bone marrow response which also helps clarify the pathophysiology. Circulating neutrophils account for only 3% to 5% of the total, with the remainder sequestered until activated by infection or other stimulus. The clinical significance of this is that even in cases of peripheral neutropenia if bone marrow reserves are adequate, the infectious risk may be mitigated. Neutropenia can be classified by decreased bone marrow reserves (primary) or normal bone marrow reserves (secondary).[3]
Decreased Bone Marrow Reserves
Decreased bone marrow reserves cause an initial lack of availability of neutrophils and precursor cells. This can be related to intrinsic or extrinsic factors.
- Intrinsic factors include genetic mutations (eg, ELANE, HAX1, WAS mutations) or acquired myelodysplastic syndrome, and causes vary by age.
- Extrinsic factors limiting neutrophil production can be related to 2 main causes:
- physical bone marrow infiltration/replacement/restriction—eg, leukemia, neuroblastoma, or osteopetrosis
- external limiting factors
- viral or bacterial infections, radiation, drug-induced, alcoholism
- nutritional deficiencies of folate, vitamin B12, and copper [3]
Normal Bone Marrow Reserves
Normal bone marrow reserves are common and provide the availability of neutrophils, which are then either destroyed, sequestered, consumed, or ineffectively transported/non-functional.
Genetic causes of neutropenia usually affect neutrophil chemotaxis or function. The pathophysiology section will describe the most common genetic syndromes.
Ineffective neutrophil trafficking and function can be affected at many steps. The primary causes of defective neutrophil function include failure of the following mechanisms:
- Migration into inflammation sites (abnormal chemotaxis)
- Adherence to endothelial cells
- Ingestion of bacteria
- Production of microbicidal compounds and toxic reactive oxygen species to kill pathogens
- Formation of phagolysosomes [5][7]
Drugs can cause both hypoplastic neutropenia from bone marrow suppression or autoimmune neutropenia.[8] Chemotherapeutic agents are commonly known to cause neutropenia. Levamisole is a veterinary antiparasitic drug that has been frequently used as an adulterant in cocaine and is known to cause agranulocytosis.[9] Other common drugs known to cause neutropenia include the following:[10]
- Aminopyrine
- Cephalosporins
- Dapsone
- Heavy metals (eg, arsenic, aluminum phosphate)[11]
- Hydroxychloroquine
- Hydralazine
- Lamotrigine
- Methimazole
- Oxacillin/penicillins
- Phenothiazine
- Procainamide
- Propylthiouracil
- Quinidine/quinine
- Rituximab
- Sulfasalazine
- Sulfonamides
- Trimethoprim-sulfamethoxazole
- Vancomycin
- Clozapine (see below)[10][12]
Clozapine is an antipsychotic used for treatment-resistant psychosis and has been underutilized in part due to adverse effects, including severe neutropenia and agranulocytosis (occurring in 3.8% and 0.9% of patients, respectively), requiring stringent hematologic monitoring.[13][14] The significance of neutropenia is that usage is often avoided despite clozapine being the current most effective treatment for treatment-resistant schizophrenia. A genetic predisposition to developing agranulocytosis is likely involved, and this is an area needing further study.[15][16] Rechallenge with clozapine is possible in selected cases.[14] Please see StatPearls' associated review on "Clozapine" for further details.
Autoimmune or anti-neutrophil neutropenia is a common cause of neutropenia. However, autoantibody levels do not closely correlate with the degree of neutropenia. A positive antinuclear antibody (ANA) is suggestive, but not definitive, of autoimmune neutropenia.[8] Both immunoglobulin (B-cell) and cell-mediated (T-cell, natural killer cell) immunity can be involved. Autoimmune destruction can occur in the bone marrow or peripherally, primarily through monocyte/macrophage elimination via the spleen.[6]
Autoimmune neutropenia in infancy occurs due to antineutrophil antibodies and generally has a benign course; this condition spontaneously resolves about 90% of the time within 2 to 3 years of onset.[17] Autoimmune neutropenias are more common in adult women, especially those who are between 25 and 40. Symptoms of connective tissue diseases like lupus and Sjögren syndrome are common. Felty syndrome is a classic example of autoimmune neutropenia characterized by chronic rheumatoid arthritis, splenomegaly, and activated large granular lymphocytes.[6]
Autoimmune destruction of neutrophils has also been observed in these conditions:
- Thyroid disease
- Inflammatory bowel disease
- Chronic autoimmune hepatitis
- Granulomatosis with polyangiitis
- Hodgkin lymphoma
- Infections
- Hepatitis B and C (although these can acutely cause neutropenia, they are more commonly associated with a chronic immune-mediated form)
Increased consumption of neutrophils is common in sepsis and bacterial infections. Neonates are especially prone to neutropenia from sepsis.[3]
Idiopathic neutropenia, where no cause can be found despite extensive investigation, is common. Results from one study of patients with neutropenia found no identifiable cause in 48% of patients.[8] Cell-mediated immunity may be an underlying pathology of chronic idiopathic neutropenia through mediation by myeloid-derived suppressor cells, which is an emerging area of study.[6]
Please see Table. Causes of Neutropenia for further information.
Epidemiology
Results from a study reported that in the United States, the prevalence of neutropenia was 0.38% among Mexican-Americans, 0.79% among Whites, and 4.5% among Black participants.[18] Another study reported that the risk of febrile neutropenia during the chemotherapy regimen course for treating solid tumors was 17%.[19] Chronic neutropenia is thought to be between 0.12% and 1.4% in the European population.[6] Men are affected primarily by inherited neutropenias more often than women.[20] However, autoimmune neutropenia is more common in women than men.[3]
Pathophysiology
Mutations in the gene ELANE, which encodes neutrophil elastase, have been found to be the most common cause of congenital and cyclic neutropenia and are autosomal dominant. The gene HAX1 codes for an activator of granulocyte-colony stimulating factor (G-CSF) and is the most common autosomal recessive mutation.[21][22] Severe congenital neutropenia is used to describe a heterogeneous array of gene mutations leading to variable clinical presentations. Mutations in ELANE, HAX1, and many other genes can cause maturation arrest of the neutrophils within the bone marrow, usually at the promyelocyte stage.[22][23] Severe congenital neutropenia is associated with acute myelogenous leukemia and myelodysplastic syndrome.[21][22]
Cyclic neutropenia is an autosomal dominant congenital neutropenia characterized by oscillating levels of neutrophils, monocytes, lymphocytes, platelets, and reticulocytes, most commonly on a 21-day cycle. Myeloid precursors can be seen in the peripheral blood. This condition is associated with ELANE and other mutations. Cyclic neutropenia is generally considered a benign condition but infections can occur during nadirs. G-CSF may be used in these scenarios to hasten neutrophil recovery.[22]
Chronic granulomatous disease is one of the classic primary immunodeficiencies of childhood. The clinical presentation of these diseases is recurrent multisystemic infections since childhood. An inherited X-linked recessive disorder, the most commonly observed mutation is in the CYBB gene, manifesting in a deficiency in NADPH oxidase function and failure to produce toxic reactive oxygen species. In brief, neutrophils can ingest microorganisms but cannot kill them, resulting in opportunistic infections with various microbes that include Burkholderia, Staphylococcus, Pseudomonas, and Serratia species.[20] A significant consequence is that granulomas can obstruct organs such as the stomach, esophagus, or bladder. [20]
Leukocyte adhesion deficiency disorders (LAD types 1, 2, and 3) exhibit autosomal recessive inheritance; the functional defect is the inability of neutrophils to adhere to endothelial cells so that they cannot traverse into tissues to ingest and kill bacteria. Three subtypes of LAD syndrome have been identified: In LAD I, a mutation in the beta-integrin 2 gene decreases endothelial adhesion; LAD II results from a transport mutation in the Golgi apparatus; LAD III affects the activation of beta-integrins 1, 2, and 3.[24]
Chediak-Higashi syndrome is also an autosomal recessive condition associated with neutropenia, as well as decreased natural killer and cytotoxic T-cell degranulation. This syndrome is characterized by abnormal chemotaxis preventing neutrophils from reaching bacteria and severely reduced microbicidal activity as lysosomes fail to fuse with phagosomes.[25] The mutations affect the gene LYST/CHS1, which regulates vesicle movement and the lysosomes/organelle size.[26] Oculocutaneous albinism is a commonly observed clinical feature in patients with Chediak-Higashi syndrome.[27]
WAS-related disorders: WAS is a gene encoding the protein WASp (Wiskott-Aldrich protein) in which mutations cause defective polymerization of the intracellular actin cytoskeleton. Mutations causing abnormal WASp lead to 3 main genetic syndromes:
- Wiskott-Aldrich syndrome—a genetic disorder characterized by severe immunodeficiency, thrombocytopenia, and eczema—is characterized by inactive WASp. Please see StatPearls' companion reference, "Wiskott-Aldrich Syndrome," for further details.
- X-linked thrombocytopenia characterized by bleeding, slightly increased risk of infection, and autoimmunity—is also characterized by inactive WASp.[28]
- On the other hand, gain-of-function WAS mutations, as seen in X-linked neutropenia, cause decreased neutrophil quantity but increased neutrophil activity, which shields patients against the infectious complications seen in loss-of-function WAS mutations.[29]
Shwachman-Diamond syndrome is a rare autosomal recessive disease usually caused by mutations in SBDS. This multisystemic disease has variable presentations, but over 90% of patients with this syndrome have neutropenia. About one-third have chronic neutropenia, while the other two-thirds have neutropenia intermittently. Other cell lines (red cells, platelets) are also impacted. One of the hallmarks of this multisystemic disease is exocrine pancreatic insufficiency caused by the absence of acinar cells, which can spontaneously improve over time. Hepatomegaly and skeletal deformities are also common. Please see Statpearls' companion review, Shwachman-Diamond Syndrome, for more information.[30][31]
Another unique syndrome falling under immunodeficiency syndromes is the WHIM syndrome, named after its diagnostic tetrad of warts, hypogammaglobulinemia, infections, and myelokathexis (the retention of mature neutrophils in the bone marrow). This syndrome is associated with CXCR4 mutations and results in panleukopenia. Characteristic findings are hypoglobulinemia and mild thrombocytopenia without anemia. Due to the neutrophilic excess in bone marrow, infections can actually lead to increased numbers of mobilized neutrophils in the blood, and patients often respond well to infections.[32] This phenomenon in WHIM syndrome is likely owed to the unique genetic and autoimmune mechanisms causing neutropenia, as the abnormal neutrophils can also be tagged for apoptosis in the bone marrow.[6]
Another recognized form of hereditary neutropenia is Duffy antigen null neutropenia (formerly termed benign ethnic neutropenia), which is associated with low neutrophil count (<1,500 cells/μL) without an increased infectious risk. This is a diagnosis of exclusion but is also one of the most common causes of neutropenia in people of African, Arabic, Yeminite Jew, and West Caribbean descent.[15] One theory proposes that those with the condition have a greater concentration of neutrophils within the organ compartment instead of the blood; therefore, the total neutrophil count is not low.[3][33]
Histopathology
In Chediak-Higashi syndrome, the neutrophils present with giant peroxidase-positive lysosomal granules on peripheral blood smears and in bone marrow precursors.[25][34] Chronic granulomatous disease is characterized by the presence of granulomas, which are composed of histiocytes that can fuse to form multinucleated giant cells that might be surrounded by other immune cells and covered with fibrotic tissue. The granulomas are often found in hollow viscera (stomach, colon, bladder).[20]
History and Physical
With neutropenia, there is often a history of these conditions:
- Recurrent infections
- Infections caused by rare bacteria and fungi
- Opportunistic infections
- Frequent use of antibiotics and antifungals [25][35][36]
Febrile neutropenia is common, particularly in patients recei ving systemic chemotherapy, and is covered in a different activity. For further information, please see StatPearls' review, "Febrile Neutropenia."
Patients with autoimmune neutropenia may demonstrate hepatosplenomegaly or systemic symptoms and signs of thyroid or connective tissue disease. Signs of connective tissue disease include arthralgias, skin rashes, and Raynaud phenomena. Patients with underlying malignancy may complain of various constitutional symptoms that include fatigue, malaise, bone pain, night sweats, or weight loss. Patients with WHIM syndrome will demonstrate warts, and patients with Wiskott-Aldrich syndrome characteristically demonstrate eczema. Chediak-Higashi syndrome is a hyperinflammatory syndrome described above. Physical characteristics include altered skin and eye pigmentation, specifically albinism.[37] Patients with Schwachman-Diamond syndrome often present with malabsorption as infants (but with normal sweat chloride as opposed to infants with cystic fibrosis) and malabsorption secondary to pancreatic deficiency. Other physical findings suggestive of either an inherited or acquired neutropenia may include the following:[35][38][39][40]
- Delayed separation of the umbilical cord (seen with alloimmune neutropenia) [41]
- Fever
- Meningitis
- Septicemia
- Bacteremia
- Skin infections (eg, pyodermas and abscesses)
- Malaise
- Gingivitis
- Purulent conjunctivitis
- Recurrent tonsillitis
- Sore throat
- Aphthous stomatitis
- Otitis media
- Coarse facial features
- Mucocutaneous candidiasis
- Cough
- Lung abscesses
- Pneumatoceles (air-filled invitations, usually in the lung, after infection, trauma, or with cystic lung disease)
- Sinus and lung infections
- Bronchiectasis
- Deep abscesses
- Granulomas with catalase-positive organisms
- Osteomyelitis
- Arthritis
- Peritonitis
- Intestinal malabsorption
- Splenomegaly
- Diarrhea
- Urinary sepsis
- Vasculitis
- Poor wound healing
Evaluation
The immunological investigation of a patient with neutropenia includes the assessment of immunoglobulins, the complement system, phagocytes, and autoantibodies.[42][43]
Quantitative Serum Immunoglobulins
IgG, IgM, IgA, and IgE levels can be measured. Elevated IgG and IgM levels may suggest an autoimmune etiology, especially in the presence of other systemic signs.
Blood Lymphocyte Subpopulations
- B lymphocytes (CD19 and CD20) can be measured to assess B-cell levels. Low B-cell levels and hypogammaglobulinemia are seen in some disorders like WHIM syndrome.
Phagocytic Function
The nitroblue tetrazolium test before and after stimulation with endotoxin shows a positive blue color indicative of NADPH oxidase activity and superoxide generation in response to phagocytized bacteria. This is used for screening, and tests of patients with chronic granulomatous disease will not demonstrate the characteristic positive blue color seen in patients with intact NADPH oxidase activity.[44] Neutrophil mobility can also be assessed in the medium alone and in the presence of a chemoattractant.
Autoantibodies
Autoantibodies are common in neutropenia and are more commonly detected indirectly. Several limitations to direct antibody testing exist, including low neutrophil count and passive absorption of labeled antibodies to the neutrophil surface, creating false-positive results.[6] A positive ANA is highly suggestive of autoimmune disease, and any manifestations suggesting connective tissue disorders should be investigated with appropriate antibody testing. Patients with thyroid disease are also more likely to have autoimmune neutropenia than the general population. Neonates with neutropenia should be tested for maternal IgG antibodies to fetal neutrophils. Infants and children younger than 3 years in whom autoimmune neutropenia of infancy is suspected should be tested for antineutrophil antibodies (although negative antibodies do not rule this condition out).[3]
Complement System Evaluation
Autoimmune neutropenia often involves the activation of the complement system. The complement factors C3 and C4 can be measured by immunoprecipitation tests, ELISA, or Western blotting. CH50 (total complement activity, also called CH100) is a screening assay for the overall activation of the classical complement pathway.[45]
Complement system functional studies
- Classical pathway assay (using IgM on a microtiter plate)
- Alternative pathway assay (using LPS on a microtiter plate)
- Mannose pathway assay (using mannose on a microtiter plate)
Microbiological studies
Cultures can be drawn from the blood, urine, stool, sputum, and cerebrospinal fluid.
Other laboratory investigations
- Complete blood cell count (to determine the absolute neutrophil count)
- Blood chemistry
- Tumoral markers (although not highly sensitive for each tumor), including CEA, CA 19-9, CA-125, and PSA
- Levels of cytokines (granulocyte-colony stimulating factor)
- Nutritional deficiencies should be evaluated, including iron, copper, B12, and folate levels
- Infectious causes, especially human immunodeficiency virus and hepatitis B/C should be evaluated
A peripheral blood smear is often performed to assess for pseudo neutropenia, which can occur due to in vitro aggregation related to cold agglutins. It may also reveal other hematologic abnormalities.
Bone marrow biopsy is performed at the discretion of the hematologist and is especially helpful in distinguishing between low neutrophil reserve and normal reserve bone marrow etiologies. Autoimmune neutropenia will often show a normal bone marrow. Conditions like myelofibrosis, myelodysplastic syndrome, and WHIM syndrome will exhibit characteristic pathology respective to each disease state in addition to variably decreased granulocytic precursors, owing to a shift toward the production of other marrow precursors relevant to the disease.
Genetic Testing
- Fluorescent in situ hybridization (FISH)
- Deoxyribonucleic acid testing (for most congenital disorders)
Radiologic Imaging Studies
- Chest x-rays can evaluate for suspected lung infections. However, early chest x-rays may have a lower sensitivity for pneumonia compared to computed tompgraphy (CT) scans.[46]
- Diagnostic ultrasonography can evaluate for hepatosplenomegaly.
- A CT scan can evaluate suspected abdominal pathologies, including inflammatory bowel syndrome and other suspected malabsorptive states.
Treatment / Management
Administration of granulocyte-colony stimulating factor (G-CSF) is the treatment mainstay for neutropenia.[47][48] G-CSF is primarily effective in defective myelopoiesis.[3] The lowest possible G-CSF dose should be given to increase the absolute neutrophil count (ANC) to the desired level, usually greater than 1000 cells/μL.[49] A recommended starting dose for adults is 5 μg/kg/day, increasing to doses of 10 μg/kg/day if needed for ANC below 1000 cells/uL, but decisions on precise dosing should be tailored to individual patients. The dose for cyclic neutropenia is approximately 1 to 3 µg/kg/d given every 1 to 3 days.[3] (B2)
Treatment for autoimmune neutropenia with G-CSF is initiated based on the occurrence of infections or stomatitis. Usually, low doses are required as bone marrow reserves should be preserved; a dosage of 0.5 to 3 µg/kg/d of G-CSF can be given every 1 to 3 days to increase the ANC to levels greater than 1000/μL. Immunosuppressive agents are sometimes used in adults for primary autoimmune or chronic idiopathic neutropenia, primarily glucocorticoids, cyclosporine, cyclophosphamide, or methotrexate. However, relapses are common, and the decision for immunosuppressive therapy is patient-specific.[3][6] Treatment for autoimmune neutropenia secondary to rheumatologic disorders usually involves immunosuppressants for the primary disease. G-CSF has actually been noted to aggravate underlying immune disorders, such as Sweet syndrome.[6]
Prophylactic use of antibiotics and antifungals are routinely utilized in patients with impaired neutrophil function, such as in chronic granulomatous disease.[39][50] Interferon-gamma has been used in the past, particularly for chronic granulomatous disease, as it theoretically increases the oxidative burst activity of neutrophils, increasing their effectiveness. However, its use is limited by frequent adverse effects such as fevers, myalgia, and malaise.[51][52] Trimethoprim/sulfamethoxazole is commonly used in chronic granulomatous disease as it covers 5 of the most common infecting pathogens.[39] The utilization of antimicrobials is compulsory with recurrent infections. Please see StatPearls' companion review, "Febrile Neutropenia," for further information on this subject. (B3)
Hematopoietic stem cell transplant (HSCT) is considered in certain neutropenia cases, such as those unresponsive to G-CSF, those with recurrent severe infections, or those who have transformed into a myeloid malignancy. Some clinicians consider high dosing requirements of G-CSF (>15 µg/kg/d) and certain ELANE variants to also be indications.[30] Neutropenic patients are especially at risk of post-transplant infections, and aggressive post-HSCT prophylaxis is crucial. Graft rejection is also prevalent for all causes of neutropenia.[3] Transplantation at a high-volume center is recommended.
A newer class of medication, CXCR4 antagonists, have been studied for the treatment of certain genetic disorders that impair the migration/trafficking of neutrophils to the periphery. Gain-of-function mutations in CXCR4, such as WHIM syndrome, cause neutrophil retention or myelokathesis in the bone marrow. In April 2024, the CXCR4 antagonist mavorixafor was approved for the treatment of WHIM syndrome.[53] Another CXCR4 antagonist, plerixafor, has been used for stem cell mobilization in autologous stem cell transplants.[54] However, plerixafor has not been shown superior to C-GSF in a small trial.[55] Gene therapy remains investigational for certain causes of neutropenia, but enrollment in clinical trials at high-volume centers is advised for patients with certain neutropenias, including those with chronic granulomatous disease, who are refractory to primary therapies or who are not candidates for HSCT.(A1)
Differential Diagnosis
Neutropenia can differentiate from antibody deficiency disorders, where a class or different classes of immunoglobulins are below the normal range or absence. These disorders may present clinically by recurrent infections with bacteria and fungi; some are opportunistic pathogens, so the use of antimicrobials to treat infectious diseases is standard.[56] A patient with neutropenia may have an intact acquired immune response but a low number or impaired function of neutrophils. Some complement system deficiencies, eg, C3 deficiency, can manifest with multiple extracellular bacterial infections and neutropenia, but can be ruled out by quantification and functional assessment of the complement system.[2][57]
Prognosis
The prognosis of neutropenia disorders depends on the cause and organs involved. Neutropenia due to chemotherapy or drugs generally resolves following growth factor support or cessation of therapy. Cyclic neutropenia, Duffy antigen null neutropenia, and drug-related neutropenia have good prognoses and are rarely fatal. Serious infections occur in a significant number of patients with neutropenia, and many require repeated admissions. Without treatment, neutropenia is associated with a high risk of death from opportunistic infections.
Complications
The primary complications of neutropenia are recurrent and fatal bacterial, viral, and fungal infections.[39] Another major concern is the development of myelodysplastic syndrome or acute myeloid leukemia, the risk of which is especially high in congenital neutropenias secondary to mutations in ELANE, HAX1, WAS, GATA2, G6PC3, and SBDS. These syndromes are a common cause of mortality in patients with congenital neutropenia. Required monitoring includes complete blood counts with differentials every 3 to 4 months and bone marrow biopsies yearly to monitor for concerning changes.[3][30]
Osteoporosis and osteopenia are common findings in patients with congenital neutropenias, although the exact pathophysiology is unknown. Fortunately, pathological fractures are not common, but patients should still be monitored closely for such.[22] G-CSF, which is commonly used in severe combined neutropenia, can cause bone pain, which can significantly affect patient compliance.[55] Second-generation antihistamines, such as loratidine, may be used to alleviate this discomfort.
Deterrence and Patient Education
Patients with any condition causing neutropenia should be extensively educated regarding infection control practices, such as rigorous handwashing and using masks when appropriate. Vaccines should be up to date, and oral hygiene is especially important. All patients with a family history or diagnosis of congenital neutropenia should be offered genetic counseling and prenatal testing where appropriate. Relatives of these patients can also be tested for disorders and for possible stem cell transplant donation.
Enhancing Healthcare Team Outcomes
Neutropenia is a serious life-threatening disorder with high morbidity and mortality. Thus, the condition is best managed by an interprofessional team that includes primary care providers, hematologists/oncologists, infectious disease specialists, radiologists, oncology nurses, pharmacists, and other health care providers who are crucial to effective care. Nurses caring for these patients must monitor vital signs, and administer medications and blood products. They should know what signs and symptoms to monitor for and when to call the specialist. They can also educate the patient about precautions and warning signs. Pharmacists review the prescription for dose, contraindications, and interactions. They may also help identify any possible drug contributors to neutropenia.
A strategic approach is equally crucial, involving evidence-based strategies to optimize treatment plans and minimize adverse effects. Ethical considerations must guide decision-making, ensuring informed consent and respecting patient autonomy in treatment choices. Each healthcare professional must be aware of their responsibilities and contribute their unique expertise to the patient's care plan, fostering a multidisciplinary approach. Effective interprofessional communication is paramount, allowing seamless information exchange and collaborative decision-making among the team members. Care coordination plays a pivotal role in ensuring that the patient's journey from diagnosis to treatment and follow-up is well-managed, minimizing errors and enhancing patient safety. By embracing these principles of skill, strategy, ethics, responsibilities, interprofessional communication, and care coordination, healthcare professionals can deliver patient-centered care, ultimately improving patient outcomes and enhancing team performance in the management of neutropenia.
Media
(Click Image to Enlarge)
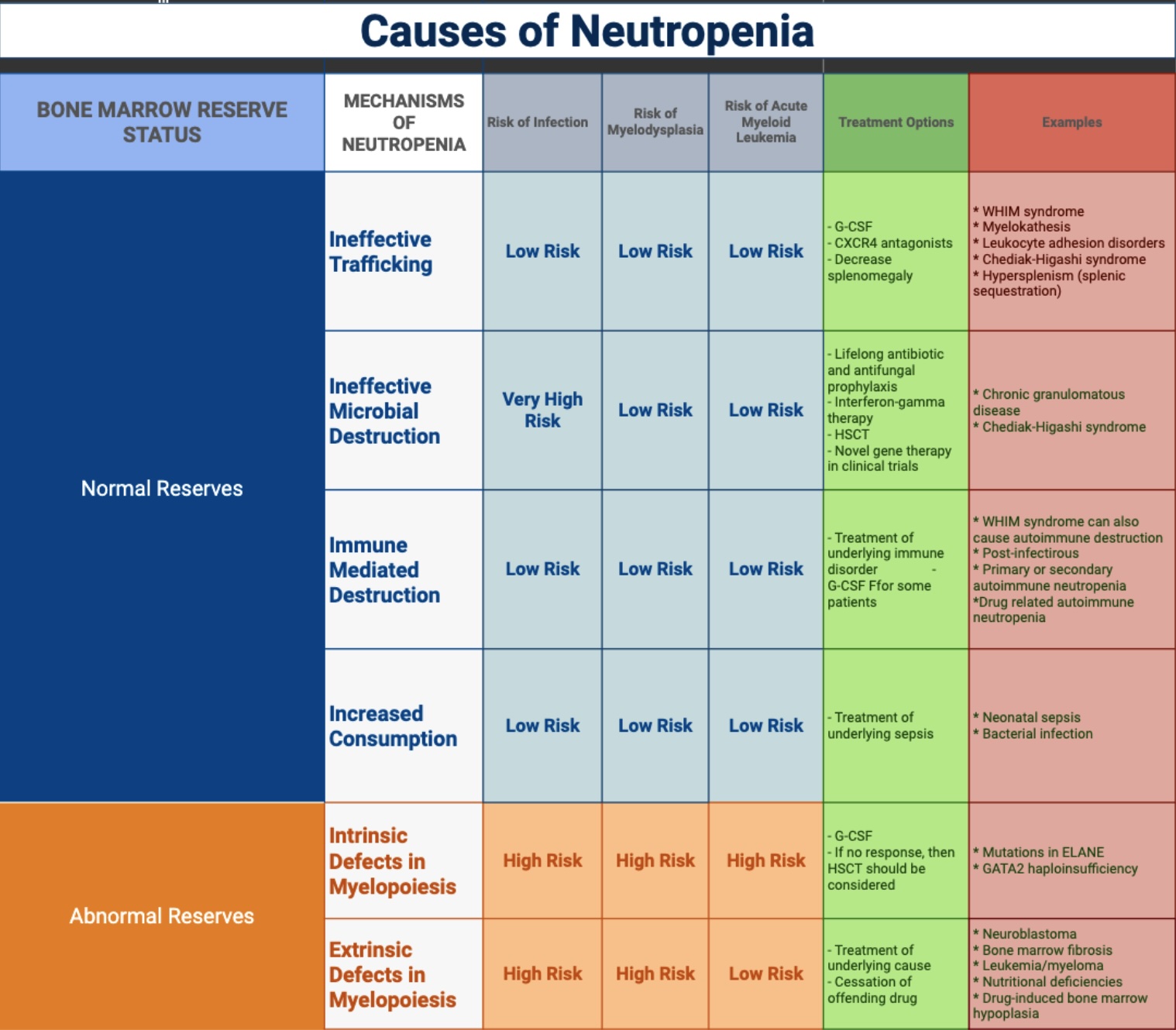
Causes of Neutropenia. Neutropenia can be caused by many different conditions, including infections, cancer, autoimmune diseases, medications, and nutritional deficiencies.
Adapted from: Connelly JA, Walkovich K. Diagnosis and therapeutic decision-making for the neutropenic patient. Hematology Am Soc Hematol Edu Program. 2021(1):492-503.
Arnold DE, Heimall JR. A review of chronic granulomatous disease. Adv Ther. 2017;34(12):2543-2557.
Mart G, Malkan UY, Buyukasik Y. Determination of etiology in patients admitted due to isolated leukopenia. Medicine (Baltimore). 2022;101(33):e30116.
References
Filippi MD. Neutrophil transendothelial migration: updates and new perspectives. Blood. 2019 May 16:133(20):2149-2158. doi: 10.1182/blood-2018-12-844605. Epub 2019 Mar 21 [PubMed PMID: 30898863]
Level 3 (low-level) evidenceJustiz Vaillant AA, Qurie A. Immunodeficiency. StatPearls. 2025 Jan:(): [PubMed PMID: 29763203]
Connelly JA, Walkovich K. Diagnosis and therapeutic decision-making for the neutropenic patient. Hematology. American Society of Hematology. Education Program. 2021 Dec 10:2021(1):492-503. doi: 10.1182/hematology.2021000284. Epub [PubMed PMID: 34889413]
Rezaei N, Moazzami K, Aghamohammadi A, Klein C. Neutropenia and primary immunodeficiency diseases. International reviews of immunology. 2009:28(5):335-66. doi: 10.1080/08830180902995645. Epub [PubMed PMID: 19811314]
Bohn G, Welte K, Klein C. Severe congenital neutropenia: new genes explain an old disease. Current opinion in rheumatology. 2007 Nov:19(6):644-50 [PubMed PMID: 17917547]
Level 3 (low-level) evidenceFioredda F, Dufour C, Höglund P, Papadaki HA, Palmblad J. Autoimmune Neutropenias: Update on Clinical and Biological Features in Children and Adults. HemaSphere. 2023 Jan:7(1):e814. doi: 10.1097/HS9.0000000000000814. Epub 2022 Dec 19 [PubMed PMID: 36570693]
Nargund AR, Madhumathi DS, Premalatha CS, Rao CR, Appaji L, Lakshmidevi V. Accelerated phase of chediak higashi syndrome mimicking lymphoma--a case report. Journal of pediatric hematology/oncology. 2010 Aug:32(6):e223-6. doi: 10.1097/MPH.0b013e3181e62663. Epub [PubMed PMID: 20661157]
Level 3 (low-level) evidenceMart G, Malkan UY, Buyukasik Y. Determination of etiology in patients admitted due to isolated leukopenia. Medicine. 2022 Aug 19:101(33):e30116. doi: 10.1097/MD.0000000000030116. Epub [PubMed PMID: 35984149]
Solomon N, Hayes J. Levamisole: A High Performance Cutting Agent. Academic forensic pathology. 2017 Sep:7(3):469-476. doi: 10.23907/2017.039. Epub 2017 Sep 1 [PubMed PMID: 31239995]
Moore DC. Drug-Induced Neutropenia: A Focus on Rituximab-Induced Late-Onset Neutropenia. P & T : a peer-reviewed journal for formulary management. 2016 Dec:41(12):765-768 [PubMed PMID: 27990078]
Islam LN, Nabi AH, Rahman MM, Khan MA, Kazi AI. Association of clinical complications with nutritional status and the prevalence of leukopenia among arsenic patients in Bangladesh. International journal of environmental research and public health. 2004 Sep:1(2):74-82 [PubMed PMID: 16696181]
Johannsen CF, Petersen TS, Nielsen J, Jørgensen A, Jimenez-Solem E, Fink-Jensen A. Clozapine- and non-clozapine-associated neutropenia in patients with schizophrenia: a retrospective cohort study. Therapeutic advances in psychopharmacology. 2022:12():20451253211072341. doi: 10.1177/20451253211072341. Epub 2022 Mar 5 [PubMed PMID: 35273789]
Level 2 (mid-level) evidenceOloyede E, Blackman G, Whiskey E, Bachmann C, Dzahini O, Shergill S, Taylor D, McGuire P, MacCabe J. Clozapine haematological monitoring for neutropenia: a global perspective. Epidemiology and psychiatric sciences. 2022 Nov 25:31():e83. doi: 10.1017/S204579602200066X. Epub 2022 Nov 25 [PubMed PMID: 36426600]
Level 3 (low-level) evidenceSilva E, Higgins M, Hammer B, Stephenson P. Clozapine rechallenge and initiation despite neutropenia- a practical, step-by-step guide. BMC psychiatry. 2020 Jun 5:20(1):279. doi: 10.1186/s12888-020-02592-2. Epub 2020 Jun 5 [PubMed PMID: 32503471]
Mijovic A, MacCabe JH. Clozapine-induced agranulocytosis. Annals of hematology. 2020 Nov:99(11):2477-2482. doi: 10.1007/s00277-020-04215-y. Epub 2020 Aug 20 [PubMed PMID: 32815018]
Legge SE, Walters JT. Genetics of clozapine-associated neutropenia: recent advances, challenges and future perspective. Pharmacogenomics. 2019 Mar:20(4):279-290. doi: 10.2217/pgs-2018-0188. Epub 2019 Feb 15 [PubMed PMID: 30767710]
Level 3 (low-level) evidenceSella R, Flomenblit L, Goldstein I, Kaplinsky C. Detection of anti-neutrophil antibodies in autoimmune neutropenia of infancy: a multicenter study. The Israel Medical Association journal : IMAJ. 2010 Feb:12(2):91-6 [PubMed PMID: 20550032]
Level 2 (mid-level) evidenceHsieh MM, Everhart JE, Byrd-Holt DD, Tisdale JF, Rodgers GP. Prevalence of neutropenia in the U.S. population: age, sex, smoking status, and ethnic differences. Annals of internal medicine. 2007 Apr 3:146(7):486-92 [PubMed PMID: 17404350]
Level 2 (mid-level) evidenceWeycker D, Barron R, Kartashov A, Legg J, Lyman GH. Incidence, treatment, and consequences of chemotherapy-induced febrile neutropenia in the inpatient and outpatient settings. Journal of oncology pharmacy practice : official publication of the International Society of Oncology Pharmacy Practitioners. 2014 Jun:20(3):190-8. doi: 10.1177/1078155213492450. Epub 2013 Jul 2 [PubMed PMID: 23824496]
Level 2 (mid-level) evidenceRider NL, Jameson MB, Creech CB. Chronic Granulomatous Disease: Epidemiology, Pathophysiology, and Genetic Basis of Disease. Journal of the Pediatric Infectious Diseases Society. 2018 May 9:7(suppl_1):S2-S5. doi: 10.1093/jpids/piy008. Epub [PubMed PMID: 29746675]
Dale DC, Bolyard AA, Shannon JA, Connelly JA, Link DC, Bonilla MA, Newburger PE. Outcomes for patients with severe chronic neutropenia treated with granulocyte colony-stimulating factor. Blood advances. 2022 Jul 12:6(13):3861-3869. doi: 10.1182/bloodadvances.2021005684. Epub [PubMed PMID: 35476051]
Level 3 (low-level) evidenceSkokowa J, Dale DC, Touw IP, Zeidler C, Welte K. Severe congenital neutropenias. Nature reviews. Disease primers. 2017 Jun 8:3():17032. doi: 10.1038/nrdp.2017.32. Epub 2017 Jun 8 [PubMed PMID: 28593997]
Dale DC. How I manage children with neutropenia. British journal of haematology. 2017 Aug:178(3):351-363. doi: 10.1111/bjh.14677. Epub 2017 Apr 17 [PubMed PMID: 28419427]
Hanna S, Etzioni A. Leukocyte adhesion deficiencies. Annals of the New York Academy of Sciences. 2012 Feb:1250():50-5. doi: 10.1111/j.1749-6632.2011.06389.x. Epub 2012 Jan 25 [PubMed PMID: 22276660]
Level 3 (low-level) evidenceMöttönen M, Lanning M, Baumann P, Saarinen-Pihkala UM. Chediak-Higashi syndrome: four cases from Northern Finland. Acta paediatrica (Oslo, Norway : 1992). 2003 Sep:92(9):1047-51 [PubMed PMID: 14599068]
Level 3 (low-level) evidenceSánchez-Guiu I, Antón AI, García-Barberá N, Navarro-Fernández J, Martínez C, Fuster JL, Couselo JM, Ortuño FJ, Vicente V, Rivera J, Lozano ML. Chediak-Higashi syndrome: description of two novel homozygous missense mutations causing divergent clinical phenotype. European journal of haematology. 2014 Jan:92(1):49-58. doi: 10.1111/ejh.12203. Epub 2013 Oct 24 [PubMed PMID: 24112114]
Level 3 (low-level) evidenceSharma P, Nicoli ER, Serra-Vinardell J, Morimoto M, Toro C, Malicdan MCV, Introne WJ. Chediak-Higashi syndrome: a review of the past, present, and future. Drug discovery today. Disease models. 2020 Summer:31():31-36. doi: 10.1016/j.ddmod.2019.10.008. Epub 2019 Dec 9 [PubMed PMID: 33424983]
Rivers E, Worth A, Thrasher AJ, Burns SO. How I manage patients with Wiskott Aldrich syndrome. British journal of haematology. 2019 May:185(4):647-655. doi: 10.1111/bjh.15831. Epub 2019 Mar 12 [PubMed PMID: 30864154]
Keszei M, Record J, Kritikou JS, Wurzer H, Geyer C, Thiemann M, Drescher P, Brauner H, Köcher L, James J, He M, Baptista MA, Dahlberg CI, Biswas A, Lane DP, Song W, Pütsep K, Vandenberghe P, Snapper SB, Westerberg LS. Constitutive activation of WASp in X-linked neutropenia renders neutrophils hyperactive. The Journal of clinical investigation. 2018 Aug 31:128(9):4115-4131. doi: 10.1172/JCI64772. Epub 2018 Aug 20 [PubMed PMID: 30124469]
Warren JT, Link DC. Impaired myelopoiesis in congenital neutropenia: insights into clonal and malignant hematopoiesis. Hematology. American Society of Hematology. Education Program. 2021 Dec 10:2021(1):514-520. doi: 10.1182/hematology.2021000286. Epub [PubMed PMID: 34889405]
Burroughs L, Woolfrey A, Shimamura A. Shwachman-Diamond syndrome: a review of the clinical presentation, molecular pathogenesis, diagnosis, and treatment. Hematology/oncology clinics of North America. 2009 Apr:23(2):233-48. doi: 10.1016/j.hoc.2009.01.007. Epub [PubMed PMID: 19327581]
Heusinkveld LE, Majumdar S, Gao JL, McDermott DH, Murphy PM. WHIM Syndrome: from Pathogenesis Towards Personalized Medicine and Cure. Journal of clinical immunology. 2019 Aug:39(6):532-556. doi: 10.1007/s10875-019-00665-w. Epub 2019 Jul 16 [PubMed PMID: 31313072]
Atallah-Yunes SA, Ready A, Newburger PE. Benign ethnic neutropenia. Blood reviews. 2019 Sep:37():100586. doi: 10.1016/j.blre.2019.06.003. Epub 2019 Jun 21 [PubMed PMID: 31255364]
Roy A, Kar R, Basu D, Srivani S, Badhe BA. Clinico-hematological profile of Chediak-Higashi syndrome: experience from a tertiary care center in south India. Indian journal of pathology & microbiology. 2011 Jul-Sep:54(3):547-51. doi: 10.4103/0377-4929.85090. Epub [PubMed PMID: 21934218]
Khoo AL, Zhao YJ, Teng M, Ying D, Jin J, Chee YL, Poon LM, Lim SE, Koh LP, Chng WJ, Lim BP, Hsu LY, Chai LYA. Evaluation of a risk-guided strategy for empirical carbapenem use in febrile neutropenia. International journal of antimicrobial agents. 2018 Sep:52(3):350-357. doi: 10.1016/j.ijantimicag.2018.04.017. Epub 2018 May 9 [PubMed PMID: 29751120]
Dotta L, Parolini S, Prandini A, Tabellini G, Antolini M, Kingsmore SF, Badolato R. Clinical, laboratory and molecular signs of immunodeficiency in patients with partial oculo-cutaneous albinism. Orphanet journal of rare diseases. 2013 Oct 17:8():168. doi: 10.1186/1750-1172-8-168. Epub 2013 Oct 17 [PubMed PMID: 24134793]
Cabral-Marques O, Schimke LF, de Oliveira EB Jr, El Khawanky N, Ramos RN, Al-Ramadi BK, Segundo GRS, Ochs HD, Condino-Neto A. Flow Cytometry Contributions for the Diagnosis and Immunopathological Characterization of Primary Immunodeficiency Diseases With Immune Dysregulation. Frontiers in immunology. 2019:10():2742. doi: 10.3389/fimmu.2019.02742. Epub 2019 Nov 26 [PubMed PMID: 31849949]
Wan C, Yu HH, Lu MY, Lee JH, Wang LC, Lin YT, Yang YH, Chiang BL. Clinical manifestations and outcomes of pediatric chronic neutropenia. Journal of the Formosan Medical Association = Taiwan yi zhi. 2012 Apr:111(4):220-7. doi: 10.1016/j.jfma.2010.12.003. Epub 2012 Mar 16 [PubMed PMID: 22526211]
Slack MA, Thomsen IP. Prevention of Infectious Complications in Patients With Chronic Granulomatous Disease. Journal of the Pediatric Infectious Diseases Society. 2018 May 9:7(suppl_1):S25-S30. doi: 10.1093/jpids/piy016. Epub [PubMed PMID: 29746681]
Al-Herz W, Nanda A. Skin manifestations in primary immunodeficient children. Pediatric dermatology. 2011 Sep-Oct:28(5):494-501. doi: 10.1111/j.1525-1470.2011.01409.x. Epub 2011 Mar 31 [PubMed PMID: 21453308]
Sameshima T, Iwatani S, Fukushima S, Taniguchi-Ikeda M, Hashimoto M, Yurugi K, Iijima K, Morioka I. Periomphalitis with Delayed Umbilical Cord Separation due to Alloimmune Neonatal Neutropenia. Clinical laboratory. 2016 Nov 1:62(11):2249-2252. doi: 10.7754/Clin.Lab.2016.160422. Epub [PubMed PMID: 28164680]
Zermatten MG, Koenig C, von Allmen A, Agyeman P, Ammann RA. Episodes of fever in neutropenia in pediatric patients with cancer in Bern, Switzerland, 1993-2012. Scientific data. 2019 Jan 15:6():180304. doi: 10.1038/sdata.2018.304. Epub 2019 Jan 15 [PubMed PMID: 30644854]
Wuyts L, Wojciechowski M, Maes P, Matthieu L, Lambert J, Aerts O. Juvenile ecthyma gangrenosum caused by Pseudomonas aeruginosa revealing an underlying neutropenia: case report and review of the literature. Journal of the European Academy of Dermatology and Venereology : JEADV. 2019 Apr:33(4):781-785. doi: 10.1111/jdv.15420. Epub 2019 Mar 5 [PubMed PMID: 30633375]
Level 3 (low-level) evidenceAyatollahi M, Tabei Z, Ramzi M, Kashef S, Haghshenas M. A fast and easy nitroblue tetrazolium method for carrier screening and prenatal detection of chronic granulomatous disease. Archives of Iranian medicine. 2006 Oct:9(4):335-8 [PubMed PMID: 17061605]
Costabile M. Measuring the 50% haemolytic complement (CH50) activity of serum. Journal of visualized experiments : JoVE. 2010 Mar 29:(37):. pii: 1923. doi: 10.3791/1923. Epub 2010 Mar 29 [PubMed PMID: 20351687]
Level 3 (low-level) evidenceBurivong W, Sricharoen T, Thachang A, Soodchuen S, Maroongroge P, Leelasithorn V. Early Radiologic Diagnosis of Pulmonary Infection in Febrile Neutropenic Patients: A Comparison of Serial Chest Radiography and Single CT Chest. Radiology research and practice. 2021:2021():8691363. doi: 10.1155/2021/8691363. Epub 2021 Feb 18 [PubMed PMID: 33680511]
Pilatova K, Bencsikova B, Demlova R, Valik D, Zdrazilova-Dubska L. Myeloid-derived suppressor cells (MDSCs) in patients with solid tumors: considerations for granulocyte colony-stimulating factor treatment. Cancer immunology, immunotherapy : CII. 2018 Dec:67(12):1919-1929. doi: 10.1007/s00262-018-2166-4. Epub 2018 May 10 [PubMed PMID: 29748897]
Jolis L, Carabantes F, Pernas S, Cantos B, López A, Torres P, Funes C, Caballero D, Benedit P, Salar A, PRAXIS Study Group. Incidence of chemotherapy-induced neutropenia and current practice of prophylaxis with granulocyte colony-stimulating factors in cancer patients in Spain: a prospective, observational study. European journal of cancer care. 2013 Jul:22(4):513-21. doi: 10.1111/ecc.12057. Epub 2013 Jun 3 [PubMed PMID: 23730920]
Level 2 (mid-level) evidenceAvalos BR, Lazaryan A, Copelan EA. Can G-CSF cause leukemia in hematopoietic stem cell donors? Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation. 2011 Dec:17(12):1739-46. doi: 10.1016/j.bbmt.2011.07.003. Epub 2011 Jul 13 [PubMed PMID: 21745453]
Perparim K, Hashimoto A, Nasu M. Efficacy of recombinant human granulocyte-colony stimulating factor alone and in combination with antifungal agents against disseminated trichosporonosis in neutropenic mice. Journal of infection and chemotherapy : official journal of the Japan Society of Chemotherapy. 1996:2(4):232-239. doi: 10.1007/BF02355120. Epub 2014 Apr 5 [PubMed PMID: 29681373]
Kang EM, Marciano BE, DeRavin S, Zarember KA, Holland SM, Malech HL. Chronic granulomatous disease: overview and hematopoietic stem cell transplantation. The Journal of allergy and clinical immunology. 2011 Jun:127(6):1319-26; quiz 1327-8. doi: 10.1016/j.jaci.2011.03.028. Epub 2011 Apr 17 [PubMed PMID: 21497887]
Level 3 (low-level) evidenceFiliz S, Uygun DF, Köksoy S, Şahin E, Yeğin O. In vitro interferon γ improves the oxidative burst activity of neutrophils in patients with chronic granulomatous disease with a subtype of gp91phox deficiency. Central-European journal of immunology. 2015:40(1):54-60. doi: 10.5114/ceji.2015.50833. Epub 2015 Apr 22 [PubMed PMID: 26155184]
Badolato R, Alsina L, Azar A, Bertrand Y, Bolyard AA, Dale D, Deyà-Martínez À, Dickerson KE, Ezra N, Hasle H, Kang HJ, Kiani-Alikhan S, Kuijpers TW, Kulagin A, Langguth D, Levin C, Neth O, Olbrich P, Peake J, Rodina Y, Rutten CE, Shcherbina A, Tarrant TK, Vossen MG, Wysocki CA, Belschner A, Bridger GJ, Chen K, Dubuc S, Hu Y, Jiang H, Li S, MacLeod R, Stewart M, Taveras AG, Yan T, Donadieu J. A phase 3 randomized trial of mavorixafor, a CXCR4 antagonist, for WHIM syndrome. Blood. 2024 Jul 4:144(1):35-45. doi: 10.1182/blood.2023022658. Epub [PubMed PMID: 38643510]
Level 1 (high-level) evidenceMerati N, Sivachandran S, Jfri A, Ben-Shoshan M, Vinh DC, Popradi G, Litvinov IV. Plerixafor on a WHIM - Promise or Fantasy of a New CXCR4 Inhibitor for This Rare, but Important Syndrome? Skin therapy letter. 2022 Mar:27(2):1-5 [PubMed PMID: 35385630]
Level 3 (low-level) evidenceMcDermott DH, Velez D, Cho E, Cowen EW, DiGiovanna JJ, Pastrana DV, Buck CB, Calvo KR, Gardner PJ, Rosenzweig SD, Stratton P, Merideth MA, Kim HJ, Brewer C, Katz JD, Kuhns DB, Malech HL, Follmann D, Fay MP, Murphy PM. A phase III randomized crossover trial of plerixafor versus G-CSF for treatment of WHIM syndrome. The Journal of clinical investigation. 2023 Oct 2:133(19):. doi: 10.1172/JCI164918. Epub 2023 Oct 2 [PubMed PMID: 37561579]
Level 1 (high-level) evidenceJustiz Vaillant AA, Ramphul K. Antibody Deficiency Disorder (Archived). StatPearls. 2025 Jan:(): [PubMed PMID: 29939682]
Afzali P, Isaeian A, Sadeghi P, Moazzami B, Parvaneh N, Robatjazi M, Ziaee V. Complement deficiency in pediatric-onset systemic lupus erythematosus. Journal of laboratory physicians. 2018 Apr-Jun:10(2):232-236. doi: 10.4103/JLP.JLP_171_17. Epub [PubMed PMID: 29692593]