Introduction
The neuromuscular junction (NMJ) is a specialized synapse that connects motor neurons and skeletal muscle fibers (see Images. Anatomy of Neurons and Anatomy of the Neuromuscular Junction). This synapse facilitates the transfer of electrical signals from the somatic nervous system to the muscle, initiating the process of contraction. The NMJ plays a central role in voluntary movements and is a key focus in understanding both normal skeletal muscle physiology and neuromuscular disorders.[1]
Knowing the structure and function of the NMJ is critical for understanding conditions that disrupt neuromuscular transmission, such as myasthenia gravis, Lambert-Eaton myasthenic syndrome (LEMS), and botulism. Familiarity with the treatments targeting this junction can help improve the clinical management of disorders affecting neuromuscular function. A thorough understanding of NMJ physiology equips clinicians to diagnose, treat, and optimize outcomes for patients with neuromuscular disorders, improving both functional recovery and quality of life.
Issues of Concern
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Issues of Concern
The NMJ is susceptible to various disruptions that impair its function, including autoimmune conditions, exposure to toxins, genetic mutations, and age-related or degenerative changes. Many drugs act on this junction, modifying its function either as their primary action mechanism or as a side effect. These issues can lead to significant clinical consequences, including muscle weakness, paralysis, and altered muscle control.
Cellular Level
The NMJ involves 2 main cell types: the motor neuron and the skeletal muscle. These cells are separated by a synaptic cleft, also referred to as the "junctional cleft."[2][3][4]
The motor neuron originates in 2 places. The anterior horn of the spinal cord sends branches to the somatic skeletal muscles below the head. The brainstem gives rise to the cranial nerves supplying the cranial muscles responsible for functions such as facial expression, mastication, and eye movement. Upon reaching the muscle, the motor neuron loses its myelin sheath and branches into 100 to 200 terminal ends.
The primary neurotransmitter at the neuromuscular junction, acetylcholine (ACh), facilitates the transmission of electrical signals from the motor neuron to the skeletal muscle fiber, ultimately triggering muscle contraction. ACh is synthesized in the somatic neuron from choline and acetyl coenzyme A (acetyl-CoA) through the catalytic activity of the enzyme choline acetyltransferase. After synthesis, acetylcholine undergoes a series of modifications before being packaged into vesicles. These vesicles, each storing approximately 5,000 to 10,000 molecules of acetylcholine, are docked at specialized regions on the presynaptic membrane called "active zones."
The presynaptic and synaptic vesicle membranes are equipped with soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) family proteins. The presynaptic membrane contains syntaxin and synaptosome-associated protein 25 (SNAP-25), while the vesicle membrane houses synaptobrevin. These SNARE proteins are critical for vesicle docking and fusion.
The muscle fiber contains a specialized membrane region known as the "motor end plate," characterized by thickening and extensive folding into junctional folds. These folds are densely packed with nicotinic acetylcholine receptors (nAChRs), which function as ligand-gated ion channels. These receptors bind ACh released from the motor neuron, leading to muscle membrane depolarization and the subsequent initiation of muscle contraction.
The synaptic cleft, a narrow gap approximately 50 nm wide, separates the nerve terminal from the muscle cell membrane. ACh is released into this space and binds to nAChRs on the motor end plate. The cleft also contains acetylcholinesterase (AChE), an enzyme that rapidly degrades ACh, ensuring that muscle fiber stimulation is brief and precisely regulated.
Mechanism
The mechanism of neurotransmission at the NMJ is a finely tuned process that includes several steps (see Image. Neuromuscular Junction Signaling). This process begins with action potential propagation, culminating in muscle contraction and acetylcholine breakdown, as explained below.
Action Potential Propagation to the Nerve Terminal
An action potential travels down the motor neuron and reaches the nerve terminal. The voltage change causes the voltage-gated calcium channels in the nerve terminal membrane to open.
Calcium Influx and Vesicle Fusion
Calcium enters the nerve terminal and binds to synaptotagmin, causing a conformational change in the latter that allows it to interact with SNARE proteins. This interaction triggers the fusion of synaptic vesicles with the presynaptic membrane, releasing ACh into the synaptic cleft via exocytosis.
Acetylcholine Binding to Receptors
ACh then diffuses across the synaptic cleft and binds to the nAChRs located on the motor end plate of the muscle fiber. ACh binding opens the receptor channels, allowing sodium ions to enter the muscle cell, thus depolarizing the membrane.
Generation of Endplate Potential
The influx of sodium ions depolarizes the postsynaptic membrane, shifting the potential from -90 mV to -40 mV and creating a local depolarization known as the endplate potential (EPP). A single EPP is sufficient to reach the threshold and generate an action potential that propagates along the muscle fiber.
Muscle Contraction
The action potential travels deep into the muscle fiber through the transverse tubules (T-tubules), which are invaginations of the sarcolemma. The dihydropyridine (DHP) receptor, located in the T-tubule membrane, detects the depolarization of the sarcolemma, producing a conformational change in the DHP receptor.
The DHP receptor is mechanically coupled to the ryanodine receptor located on the sarcoplasmic reticulum membrane. The conformational change in the DHP receptor triggers the opening of the ryanodine receptor, allowing calcium ions to be released from the sarcoplasmic reticulum into the muscle cell's cytoplasm. The released calcium binds and causes a conformational change in troponin, a regulatory protein on the actin filament. This change moves tropomyosin away from the myosin-binding sites on actin, exposing the binding sites and enabling the initiation of cross-bridge cycling between actin and myosin. This interaction generates the force required for muscle contraction.
Acetylcholine Breakdown
ACh is rapidly broken down by AChE in the synaptic cleft after binding to nAChRs and triggering muscle depolarization, preventing continuous stimulation of the muscle fiber. The breakdown products are then taken up into the presynaptic terminal for recycling.[5][6]
Related Testing
Evaluating NMJ function involves diagnostic techniques that assess both the site's structural and physiological integrity (see Image. Neuromuscular Transmission Monitor). These tests are essential for identifying disorders that impair synaptic transmission. Below are common diagnostic tests used for this purpose.
Electromyography
Electromyography (EMG) involves inserting a fine-needle electrode into a muscle to monitor its electrical activity at rest and during contraction. A decremental response to repetitive nerve stimulation may signify NMJ dysfunction.
Repetitive Nerve Stimulation
Repetitive nerve stimulation (RNS) assesses the NMJ's response to repeated electrical stimulation of a motor nerve. In certain NMJ disorders, muscle action potential amplitude decreases progressively with repeated stimulation.
Single-Fiber Electromyography
Single-fiber electromyography (SFEMG) is a more sensitive diagnostic tool for NMJ disorders. By measuring jitter, or the time variation between action potentials of 2 muscle fibers innervated by the same motor neuron, SFEMG can detect synaptic transmission abnormalities, such as increased jitter or fiber blocking.[7]
Serological Tests
Blood tests can detect autoantibodies against acetylcholine receptors or muscle-specific kinase (MuSK), a receptor tyrosine kinase that plays a critical role in the formation and maintenance of the NMJ. These tests help confirm autoimmunity as the cause of some cases of NMJ dysfunction.
Genetic Testing
Genetic conditions affecting the NMJ have distinctive clinical presentations, such as hypotonia, fatigable weakness, and delayed motor milestones in children. For congenital syndromes, genetic testing can identify mutations affecting NMJ proteins, such as the acetylcholine receptors, MuSK, or ryanodine receptors.
Muscle Biopsy
A muscle biopsy may show abnormalities in NMJ morphology, such as reduced receptor density or structural disorganization, in cases of suspected congenital or acquired NMJ disorders. These findings are particularly useful when serological or genetic analysis results are inconclusive.
Pathophysiology
Disruptions at the NMJ can arise from various pathological mechanisms that impair neuromuscular transmission, leading to muscle weakness, fatigue, or paralysis. This section explores the pathophysiological mechanisms underlying NMJ disorders.
Deficient Acetylcholine Release
Insufficient ACh release from motor neurons diminishes postsynaptic nAChR activation, resulting in reduced or absent EPPs and subsequent muscle weakness. Reduced ACh release is observed in conditions such as LEMS and botulinum neurotoxin exposure.[8][9]
Autoimmune Disruption
Autoantibodies against critical NMJ components impair neuromuscular function, often leading to fluctuating weakness and fatigue. The most common examples are LEMS, discussed previously, and myasthenia gravis, which is caused by autoantibodies against the nAChRs or MuSK.[10] Myasthenia gravis may thus be classified based on the molecular targets of these autoantibodies.
Acetylcholine receptor antibody-positive myasthenia gravis
The majority of myasthenia gravis cases are caused by autoantibodies against the extracellular domain of the nAChRs at the motor end plate. The antibodies interfere with receptor function in multiple ways:
- Blockade of acetylcholine binding: Some autoantibodies directly block the acetylcholine binding sites on the receptors, preventing efficient receptor activation.
- Receptor internalization and degradation: Antibodies promote accelerated endocytosis of the nAChRs, reducing receptor density on the postsynaptic membrane.
- Complement activation: The binding of autoantibodies activates the complement cascade, resulting in the formation of membrane attack complexes. These complexes damage the postsynaptic membrane, further decreasing receptor availability and impairing endplate structure.
Muscle-specific kinase antibody-positive myasthenia gravis
A subset of myasthenia gravis cases results from the action of autoantibodies targeting MuSK, a protein crucial for nAChR clustering at the motor endplate. Disruption of this process decreases the ability to generate an EPP in response to ACh release.
Congenital Myasthenic Syndromes
Congenital myasthenic syndrome (CMS) often manifests in infancy or childhood with hypotonia, feeding difficulties, and fatigable weakness. CMS represents a heterogeneous group of inherited disorders caused by mutations in genes encoding proteins critical for NMJ function.
These genetic disorders involve mutations in NMJ proteins like collagen Q (COLQ) or docking protein 7 (DOK7). For example, COLQ mutations disrupt AChE anchoring, impairing neurotransmitter degradation. The pathophysiology varies based on the site of the defect, as CMS can involve proteins in the presynaptic terminal, synaptic cleft, or muscle fiber membrane.[11]
Mutations in choline acetyltransferase (CHAT), an enzyme essential for ACh synthesis, lead to inadequate neurotransmitter release. Mutations of the receptor-associated protein of the synapse (RAPSN) disrupt nAChR clustering. Mutations in the COLQ gene, which encodes the collagen-tail subunit of AChE, impair AChE localization, prolonging ACh activity in the cleft, desensitizing nAChRs, and impairing synaptic transmission. Management depends on the specific genetic subtype, with cholinesterase inhibitors being effective in certain types.
Excessive Acetylcholinesterase Activity
AChE hyperactivity results in the rapid degradation of ACh, limiting its ability to bind to and activate nAChRs on the muscle fiber. Excessive activity of these enzymes may result from exposure to specific drugs or genetic abnormalities affecting AChE regulation, such as COLQ mutations discussed above.
Inhibition of Acetylcholinesterase
AChE inhibition prevents ACh degradation, leading to excessive neurotransmitter accumulation in the cleft. This action initially results in nAChR overstimulation but ultimately leads to receptor desensitization and synaptic fatigue.
Clinical Significance
The pathophysiological mechanisms underlying NMJ dysfunction give rise to various clinical conditions, each with distinct presentations and therapeutic challenges. Understanding the mechanisms underlying these diseases is crucial for precise diagnosis and effective management.
Myasthenia Gravis
Myasthenia gravis is an autoimmune disorder characterized by autoantibodies targeting AChRs or MuSK at the NMJ. Clinically, this condition presents as fluctuating muscle weakness that worsens with activity and improves with rest. The initial symptoms typically include extraocular muscle dysfunction, causing ptosis and diplopia. The condition may later affect the bulbar muscles, leading to dysphagia, and the limb muscles, causing generalized weakness.[12]
The detection of AChR or MuSK antibodies supports the diagnosis. Neurophysiological tests, such as RNS and SFEMG, reveal impaired NMJ transmission through a decremental compound muscle action potential (CMAP) response and increased jitter, respectively.
The treatment of myasthenia gravis focuses on symptom management, using AChE inhibitors (eg, pyridostigmine) to increase ACh availability at the NMJ and enhance muscle strength. However, these inhibitors are generally avoided in MuSK-positive myasthenia gravis because they increase the risk of a cholinergic crisis and can worsen symptoms.
Another treatment approach is to use immunosuppressive therapies, including corticosteroids like prednisone, as a first-line treatment to reduce autoantibody production. Other options are steroid-sparing agents, such as azathioprine or mycophenolate mofetil, and the monoclonal antibodies rituximab and eculizumab. Rituximab targets B cells to reduce the production of pathogenic autoantibodies and is particularly effective in MuSK antibody-positive myasthenia gravis. Eculizumab is a complement inhibitor and is used in AChR antibody-positive myasthenia gravis with refractory symptoms.
Acute myasthenic crises can be life-threatening and are generally managed with rapid-acting treatments, including plasmapheresis and intravenous immunoglobulin (IVIG), to reduce circulating autoantibodies quickly. Thymectomy is considered for patients with thymomas or even generalized myasthenia gravis with nondiseased thymus, as it can help reduce the severity of symptoms and improve long-term outcomes.[13]
Myasthenia gravis treatment research is a rapidly developing field, with multiple new drugs approved by the Food and Drug Administration (FDA) in the last couple of years. These novel agents include efgartigimod, rozanolixizumab, ravulizumab, and zilucoplan. Efgartigimod and rozanolixizumab are neonatal Fc receptor (FcRn) antagonists that lower pathogenic antibodies by enhancing their degradation. Rozanolixizumab is approved for both AChR and MuSK antibody-positive myasthenia gravis. Ravulizumab and zilucoplan are complement inhibitors that block complement-mediated damage at the NMJ, specifically for AChR antibody-positive myasthenia gravis.[14][15]
Lambert-Eaton Myasthenic Syndrome
LEMS is an autoimmune disorder characterized by autoantibodies targeting voltage-gated calcium channels (VGCCs) in the terminal ends of motor neurons at the NMJ, impairing ACh release. Clinically, this condition presents with proximal muscle weakness that improves with activity and depressed reflexes. Early symptoms often include weakness in the pelvic and shoulder girdle muscles, which can progress to involve other muscles, including the extraocular and oropharyngeal muscles, leading to ptosis or mild diplopia and swallowing dysfunction. Autonomic symptoms, including dry mouth and constipation, are also common.[16]
Diagnosis may be confirmed through the detection of VGCC antibodies, with neurophysiological tests like RNS showing an incremental response in CMAP, reflecting the improved ACh release with repeated nerve stimulation. When clinical suspicion for LEMS remains high despite the absence of detectable antibodies, an underlying malignancy must also be considered, as malignancy-associated LEMS is often seronegative due to the immune response being driven by tumor-related antigens. In fact, most LEMS cases are linked to small-cell lung cancer (SCLC).[17]
Treatment aims to enhance neuromuscular transmission through medications such as guanidine or 3,4-diaminopyridine, which increase acetylcholine release by reducing potassium efflux in motor neurons, prolonging the action potential, and promoting calcium influx. Addressing the underlying malignancy is crucial in cases associated with small-cell lung carcinoma. Immunosuppressive therapies, including corticosteroids and steroid-sparing agents, may be utilized to reduce autoimmune activity. Plasmapheresis and IVIG are additional treatment options for severe or refractory cases.
Botulism
Botulism is caused by Clostridium botulinum (C. botulinum), an anaerobic gram-positive, spore-forming bacterium. C. botulinum produces several serotypes of botulinum neurotoxin. These toxins target proteins essential for the docking and fusion of synaptic vesicles with the presynaptic membrane, thus inhibiting ACh release at the NMJ. Botulism presents with descending, symmetric flaccid paralysis, starting with cranial nerve involvement, presenting with symptoms like diplopia and dysarthria, and progressing to respiratory failure in severe cases.
Diagnosis is based on clinical presentation and the detection of botulinum toxin or C. botulinum spores in serum, stool, gastrointestinal contents, or wound exudates. Treatment involves the prompt administration of botulinum antitoxin, which neutralizes circulating toxins, and supportive care that may include mechanical ventilation, if necessary. Intravenous human-derived botulism immunoglobulin is recommended for infant botulism.
Organophosphate Toxicity
Organophosphates are a class of chemicals commonly used as insecticides. These substances irreversibly inhibit AChE, leading to the accumulation of ACh and, subsequently, the overstimulation of both muscarinic and nicotinic receptors. Symptoms of organophosphate poisoning include cholinergic crisis, with muscarinic effects such as salivation, lacrimation, and bradycardia, as well as nicotinic effects like muscle fasciculations and weakness.[18] Treatment involves atropine, which counteracts muscarinic effects, and pralidoxime, which can reactivate AChE if administered early.
Pharmacological Implications
Drugs targeting the NMJ have a wide range of clinical applications, from treating neuromuscular disorders to deliberately inducing muscle paralysis. These drugs include AChE inhibitors, neuromuscular blockers (NMBs), and botulinum toxin.
AChE inhibitors increase ACh levels by preventing the hydrolysis of the neurotransmitter by AChE. As mentioned, these drugs are commonly used to increase ACh availability in conditions like myasthenia gravis.
NMBs are used in anesthesiology to induce muscle paralysis and are classified as either depolarizing or nondepolarizing. Succinylcholine is a depolarizing NMB that acts as an AChR agonist, causing sustained depolarization and desensitization of the motor endplate. However, the use of this agent is associated with complications such as malignant hyperthermia in susceptible individuals. Malignant hyperthermia requires prompt treatment with dantrolene, a muscle relaxant that inhibits calcium release from the sarcoplasmic reticulum.
In contrast, nondepolarizing NMBs, such as rocuronium and vecuronium, function as competitive AChR antagonists, blocking ACh binding and preventing muscle contraction. These agents are preferred in patients with conditions like pseudocholinesterase deficiency and ryanodine receptor mutations that make succinylcholine unsafe.[19]
Botulinum toxin, which causes botulism, may also be used therapeutically if administered in safe amounts. Specifically, botulinum toxin type A is widely used in medical and cosmetic procedures due to its ability to induce localized muscle paralysis and relieve sustained muscle contraction. In aesthetics, botulinum toxin is injected in strategic areas, reducing wrinkles by temporarily paralyzing the muscles that cause facial lines. Other indications for administering this neurotoxin include spasticity, dystonia, blepharospasm, and chronic migraines.[20]
Media
(Click Image to Enlarge)
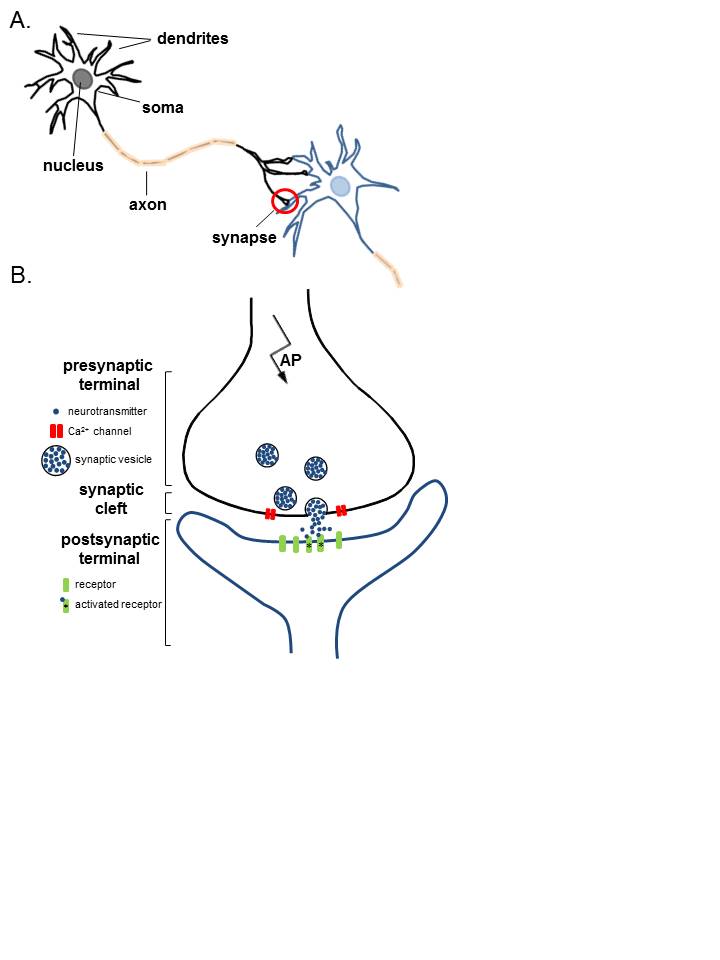
Anatomy of Neurons. A. Two connected neurons. Neurons have a soma that contains a nucleus, an axon, and a dendritic tree. A single synapse (red circle) is formed at the point where an axon's neuron (black) connects to another neuron's dendrite, soma, or axon (blue). B. A magnified view of a single synapse. On the arrival of an action potential at the presynaptic terminal, calcium triggers the release of neurotransmitters from the synaptic vesicles into the synaptic cleft. Neurotransmitters diffuse across the synaptic cleft to activate postsynaptic receptors.
Contributed by K Aubrey, MD
(Click Image to Enlarge)
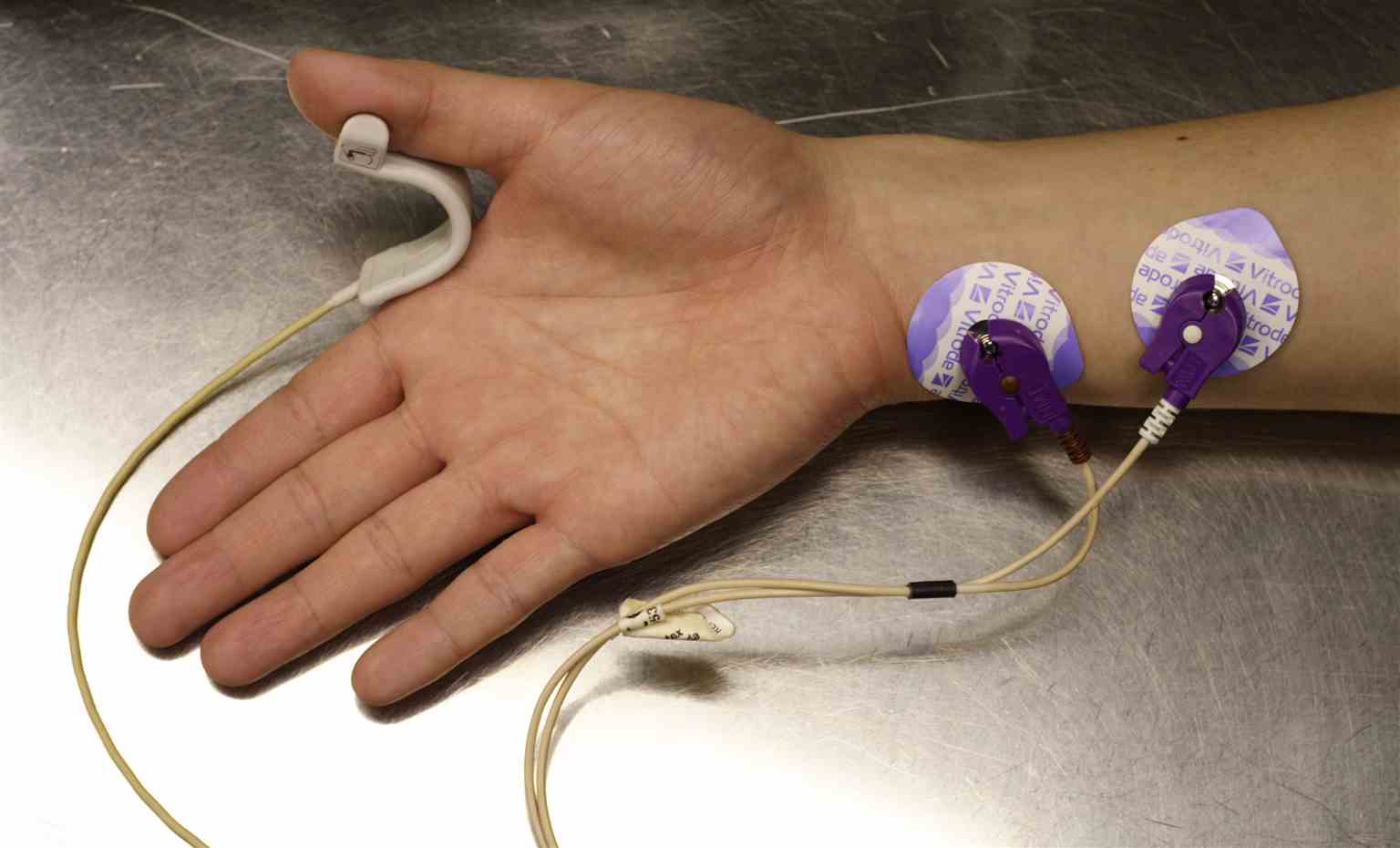
Neuromuscular Transmission Monitor. Probes are placed on the right hand and forearm to measure neuromuscular blockade.
Ignis, Public Domain, via Wikimedia Commons
(Click Image to Enlarge)

Anatomy of the Neuromuscular Junction. This illustration shows the essential parts of the neuromuscular junction, including the nerve terminal, synaptic vesicles, neuronal and sarcolemmal membranes, and the acetylcholine (ACh) receptors.
Paul Hege, Public Domain, via Wikimedia Commons
{kind=link}
(Click Image to Enlarge)

Neuromuscular Junction Signaling. This image shows the processes occurring at the neuromuscular junction that initiate muscle contraction.
Wikiwikiwiki12344321, Public Domain, via Wikimedia Commons
{kind=link}
References
El-Wahsh S, Fraser C, Vucic S, Reddel S. Neuromuscular junction disorders: mimics and chameleons. Practical neurology. 2024 Nov 17:24(6):467-477. doi: 10.1136/pn-2024-004148. Epub 2024 Nov 17 [PubMed PMID: 39174301]
Ratliff WA, Saykally JN, Kane MJ, Citron BA. Neuromuscular Junction Morphology and Gene Dysregulation in the Wobbler Model of Spinal Neurodegeneration. Journal of molecular neuroscience : MN. 2018 Sep:66(1):114-120. doi: 10.1007/s12031-018-1153-8. Epub 2018 Aug 13 [PubMed PMID: 30105628]
Hirsch NP. Neuromuscular junction in health and disease. British journal of anaesthesia. 2007 Jul:99(1):132-8 [PubMed PMID: 17573397]
Slater CR. The Structure of Human Neuromuscular Junctions: Some Unanswered Molecular Questions. International journal of molecular sciences. 2017 Oct 19:18(10):. doi: 10.3390/ijms18102183. Epub 2017 Oct 19 [PubMed PMID: 29048368]
Jimsheleishvili S, Marwaha K, Sherman AL. Physiology, Neuromuscular Transmission. StatPearls. 2025 Jan:(): [PubMed PMID: 31082177]
Fagerlund MJ, Eriksson LI. Current concepts in neuromuscular transmission. British journal of anaesthesia. 2009 Jul:103(1):108-14. doi: 10.1093/bja/aep150. Epub [PubMed PMID: 19546202]
Juel VC. Evaluation of neuromuscular junction disorders in the electromyography laboratory. Neurologic clinics. 2012 May:30(2):621-39. doi: 10.1016/j.ncl.2011.12.012. Epub [PubMed PMID: 22361377]
Meisel A, Sieb JP, Le Masson G, Postila V, Sacconi S. The European Lambert-Eaton Myasthenic Syndrome Registry: Long-Term Outcomes Following Symptomatic Treatment. Neurology and therapy. 2022 Sep:11(3):1071-1083. doi: 10.1007/s40120-022-00354-8. Epub 2022 May 5 [PubMed PMID: 35511347]
Harris RA, Dabritz HA. Infant Botulism: In Search of Clostridium botulinum Spores. Current microbiology. 2024 Aug 13:81(10):306. doi: 10.1007/s00284-024-03828-0. Epub 2024 Aug 13 [PubMed PMID: 39138824]
Loser V, Vicino A, Théaudin M. Autoantibodies in neuromuscular disorders: a review of their utility in clinical practice. Frontiers in neurology. 2024:15():1495205. doi: 10.3389/fneur.2024.1495205. Epub 2024 Nov 1 [PubMed PMID: 39555481]
Ramdas S, Beeson D, Dong YY. Congenital myasthenic syndromes: increasingly complex. Current opinion in neurology. 2024 Oct 1:37(5):493-501. doi: 10.1097/WCO.0000000000001300. Epub 2024 Jul 25 [PubMed PMID: 39051439]
Level 3 (low-level) evidenceAttarian S. New treatment strategies in Myasthenia gravis. Revue neurologique. 2024 Nov:180(9):971-981. doi: 10.1016/j.neurol.2024.09.006. Epub 2024 Oct 7 [PubMed PMID: 39379218]
Alcasid NJ, Vasic I, Brennan PG, Velotta JB. The clinical significance of open vs. minimally invasive surgical approaches in the management of thymic epithelial tumors and myasthenia gravis. Frontiers in surgery. 2024:11():1457029. doi: 10.3389/fsurg.2024.1457029. Epub 2024 Dec 11 [PubMed PMID: 39723340]
Keritam O, Vincent A, Zimprich F, Cetin H. A clinical perspective on muscle specific kinase antibody positive myasthenia gravis. Frontiers in immunology. 2024:15():1502480. doi: 10.3389/fimmu.2024.1502480. Epub 2024 Dec 5 [PubMed PMID: 39703505]
Level 3 (low-level) evidenceDavalos L, Kushlaf H. Advances in Disease-Modifying Therapeutics for Chronic Neuromuscular Disorders. Seminars in respiratory and critical care medicine. 2024 Dec 21:():. doi: 10.1055/a-2463-3385. Epub 2024 Dec 21 [PubMed PMID: 39708835]
Level 3 (low-level) evidenceLipka AF, Verschuuren JJGM. Lambert-Eaton myasthenic syndrome. Handbook of clinical neurology. 2024:200():307-325. doi: 10.1016/B978-0-12-823912-4.00012-8. Epub [PubMed PMID: 38494285]
Jayarangaiah A, Lui F, Theetha Kariyanna P. Lambert-Eaton Myasthenic Syndrome. StatPearls. 2025 Jan:(): [PubMed PMID: 29939668]
Chen Y, Yang Z, Nian B, Yu C, Maimaiti D, Chai M, Yang X, Zang X, Xu D. Mechanisms of Neurotoxicity of Organophosphate Pesticides and Their Relation to Neurological Disorders. Neuropsychiatric disease and treatment. 2024:20():2237-2254. doi: 10.2147/NDT.S479757. Epub 2024 Nov 21 [PubMed PMID: 39588175]
van den Bersselaar LR, Snoeck MMJ, Gubbels M, Riazi S, Kamsteeg EJ, Jungbluth H, Voermans NC. Anaesthesia and neuromuscular disorders: what a neurologist needs to know. Practical neurology. 2020 Oct 27:():. pii: practneurol-2020-002633. doi: 10.1136/practneurol-2020-002633. Epub 2020 Oct 27 [PubMed PMID: 33109742]
Meretsky CR, Umali JP, Schiuma AT. A Systematic Review and Comparative Analysis of Botox Treatment in Aesthetic and Therapeutic Applications: Advantages, Disadvantages, and Patient Outcomes. Cureus. 2024 Aug:16(8):e67961. doi: 10.7759/cureus.67961. Epub 2024 Aug 27 [PubMed PMID: 39328645]
Level 1 (high-level) evidence