Introduction
Neurofibromas are the most prevalent benign peripheral nerve sheath tumor. Often appearing as a soft, skin-colored papule or small subcutaneous nodule, they arise from endoneurium and the connective tissues of peripheral nerve sheaths. Neurofibromas are comprised of Schwann cells, fibroblasts, perineural cells, and mast cells in a variably myxoid background.[1] A mutation in the NF1 gene causes neurofibromas. There are three main types of neurofibromas: localized (most common), diffuse, and plexiform. Although the majority of neurofibromas occur sporadically and have an extremely low risk of malignant transformation, the plexiform type is pathognomonic for neurofibromatosis type 1 (NF 1). It carries an increased risk of malignant transformation.[2] The complete excision of the lesion is curative.
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
Approximately 90% of cases occur sporadically, while the remaining cases are associated with neurofibromatosis type 1 or 2.[3][2]
Epidemiology
Neurofibromas are the most common tumor of the peripheral nerve sheath, affecting men and women equally, without racial or ethnic predilection. Age of onset is highly variable; however, localized lesions most commonly occur in adults aged 20 to 40 years. The diffuse and plexiform types occur more frequently in children, with the plexiform type rarely occurring after age 5.[4]
Pathophysiology
In both sporadic and syndromic cases, neurofibromas are a result of a deletion in the NF1 gene. In sporadic cases, only the lesional cells carry the NF1 mutation. In syndromic cases, neurofibromas are the result of a germline mutation in NF1, encoding the tumor suppressor protein neurofibromin, on chromosome 17q11.2.[5][6]
Histopathology
Macroscopic: Often an unencapsulated, well-circumscribed, grey-tan firm mass. Typically ovoid or fusiform, with a pale gelatinous cut surface. Usually, no areas of degeneration, necrosis, or hemorrhage are grossly identified. There may be transected nerve fibers attached to the mass forming portion of the lesion.[7] The plexiform type is often large, with multiple tortuous nerve fascicles, grossly described as a "bag of worms."
Microscopic Exam Histologic Features [8][7]
- Loose and haphazard spindled cells with poorly defined cell borders
- Background of myxoid to pale pink collagenous matrix
- Coarse collagen bundles are present and often described as “shredded carrots.”
- Low to moderate cellularity
- Mast cells commonly found within the lesion
- Small, hyperchromatic, wavy nuclei, resembling “diving dolphins,” sometimes with nuclear enlargement and smudgy chromatin
- Nuclei may also be described as “buckled” or “comma-shaped.”
- Rarely, may encounter multinucleated giant cells
- Absent to minimal mitoses
- May also occur within the nerve (intraneural localized neurofibroma)
- Well circumscribed lesion in the dermis or subcutaneous tissue
- Dermal lesions typically unencapsulated with a "grenz zone" of uninvolved dermis between lesion and epidermis
- Subcutaneous lesions often have a true capsule
Diffuse Type[9]
- Poorly defined, expansile proliferation around adnexal structures, extending into the subcutaneous tissue and infiltrating adipose
- May entrap nerves or be intraneural (intraneural diffuse neurofibroma)
- Characteristic pseudomeissnerian corpuscles, comprised of fibrillary and whorled Schwann cells
Plexiform Type[10]
- Multiple intertwined hypertrophic nerve fascicles
- Serpentine pattern with multiple nodules
- May have predominantly myxoid or edematous background with thick collagen fibers
- May have atypia (nuclear enlargement, hyperchromasia) related to the degenerative change
Variants Most Commonly Associated With Localized Type And Less Commonly Diffuse Type Neurofibromas[11][12][13][14][15][16][17][18]
- Cellular: Increased cellularity, with or without atypia, no significant increase in mitotic activity
- Pigmented: Histologically and immunohistochemically shows melanin production
- Atypical/Bizarre: Hyperchromatic, pleomorphic, atypical nuclei with degenerative change, arranged in a distinct lamellar pattern
- Epithelioid: Cohesive nests of epithelioid tumor cells
- Granular cell: Contains granular cells, eosinophilic and similar in appearance to those comprising granular cell tumors
- Lipomatous: Diffusely scattered adipocytes, intrinsic to the tumor
- Dendritic cell: Dendritic cell morphology with pseudorosettes
- Hybrid neurofibroma/schwannoma: Nodules resembling schwannoma within a typical neurofibroma
Immunohistochemical Evaluation[8][13][19]
- S100 (+) in Schwann cells (approximately 50% of tumor cells)
- CD34 (+) in spindled fibroblasts with distinct “fingerprint” immunopositivity.
- The “fingerprint” is due to positive staining between whorled collagen bundles, resembling a human fingerprint - this “fingerprint,” if present in greater than 60% of the lesion, is useful in diagnosing neurofibroma and distinguishing neurofibroma from early desmoplastic melanoma
- EMA (+) in occasional perineural cells
- Myelin basic protein (+)
- Neurofilament protein (+) in intratumoral axons
- Acid mucopolysaccharides (+) in mucinous stroma
History and Physical
Patients with neurofibromas are often asymptomatic; however, irritation, mild pruritus, pain, or paresthesia, can occur. The presentation may vary by type of neurofibroma, but the most common chief complaint is cosmetic appearance.[20]
Localized lesions commonly present as a painless skin-colored or violaceous papule, nodule, or subcutaneous mass. Solitary lesions are typically less than 2 cm and have a characteristic “buttonhole sign,” where on palpation, the lesion retracts into the subcutis and reappears on the release of pressure. Localized neurofibromas can occur anywhere on the body, with a predilection for the trunk, head/neck, and extremities.[21] Lesions are often thought to be nevi or acrochordons clinically.
Diffuse neurofibromas most commonly present on the head/neck as ill-defined, indurated plaques with thickened skin. Mild numbness or tingling may occur if the lesion is large.[22]
Plexiform neurofibromas most commonly occur on the head/neck, trunk, and extremities. They surround multiple nerve fascicles and can grow to be large. If superficial, they commonly present as a skin-colored or hyperpigmented nodular swelling. Deeper lesions arising from spinal nerve roots may become highly irregular and tortuous. Deep lesions may present with pain, numbness, paresthesias, mass effect, and sequelae of spinal nerve compression.[21]
Evaluation
Evaluation of solitary superficial lesions is often by physical exam and/or excisional biopsy with microscopic examination. Larger lesions require biopsy and/or additional imaging by CT/MRI to assess the extent of involvement and plan for surgical excision.[23][24]
Treatment / Management
For most cases, complete surgical excision is the preferred treatment, with local recurrence extremely rare. There are currently no alternative therapies for cutaneous neurofibromas.[25](B3)
In rare cases of diffuse or plexiform neurofibromas where complete surgical excision is not possible, lesions are often totally resected for cosmetic or symptomatic relief. These patients require monitoring to watch for rapid growth or recurrence at the discretion of the clinical providers. Interferon-alpha has been studied as an adjunct therapy for plexiform neurofibromas, with variable results.[26][21](B3)
Differential Diagnosis
Malignant peripheral nerve sheath tumor: An aggressive neurogenic neoplasm arising from peripheral nerve or pre-existing nerve sheath tumor, including neurofibromas. Approximately 50% of cases are associated with NF1. A history of rapid growth in a prior stable neurofibroma is suspicious. Histologically there may be areas of admixed neurofibroma. The malignant portion will show increased cellularity, mitoses, and necrosis. Immunohistochemistry: S100 focally positive in approximately 50% of tumors.[7][27] Epithelioid MPNST can be negative for INI-1, about 30% of the time.
Schwannoma: A benign peripheral nerve sheath tumor comprised predominantly of Schwann cells. Associated with somatic and germline mutations in NF2. Histologically often more cellular, circumscribed, with Verocay bodies and Antoni A and B areas. Immunohistochemistry: Diffuse, uniform staining with S100.[28]
Perineuroma: A rare, benign mesenchymal tumor comprised of perineural cells. No definitive association with neurofibromatosis. Immunohistochemistry: EMA(+), Claudin-1(+), GLUT1(+) S100(-), CD34(+/-).[29]
Dermatofibroma: A benign proliferation of fibroblasts and histiocytes within the dermis. Often presents as an indurated papule. Immunohistochemistry: FXIIIA(+), CD163(+), CD68(+), CD34(-).[30]
Dermatofibrosarcoma protuberans: A low-grade fibroblastic sarcoma affecting the dermis and subcutis, with COL1A1-PDGFB gene fusion. This lesion is locally aggressive with a high rate of recurrence (50%) and increased risk of progression and metastasis. Immunohistochemistry: Diffuse CD34(+), S100(-), FXIIIA(-).[30]
Superficial leiomyoma: A benign dermal smooth muscle neoplasm, often arising from arrector pili (pilo-leiomyoma). Immunohistochemistry: SMA(+), MSA(+), Desmin (+).[31]
Neurotized melanocytic nevus: A benign nevus with melanocytic nests and loose neurotized stroma. Immunohistochemistry: Melanocytic markers(+).
Ganglioneuroma: A benign tumor of neural crest origin, comprised of ganglion cells arising from nerves. Most commonly found in the posterior mediastinum and retroperitoneum. Immunohistochemistry: S100 protein stains Schwann cells and synaptophysin stains ganglion cells.
Plexiform fibrohistiocytic tumor: An infiltrative mesenchymal neoplasm, most commonly at the dermal-subcutaneous junction, comprised of fibroblasts and histiocytes. Immunohistochemistry: SMA(+), S100 protein (-).[32]
Desmoplastic melanoma: An invasive melanoma that often resembles a dermal scar. Frequently there is an associated melanoma in-situ. Often large at diagnosis, there can be peripheral lymphoid aggregates and cytologic atypia. Immunohistochemistry: S100 and SOX10 (+).[33][34]
Prognosis
All types of neurofibromas are benign. Local recurrence is extremely rare complete excision of the lesion, and the risk of malignant transformation is exceedingly low; however, malignant transformation affects approximately 10% of patients with NF1.[35][36] NF1 should be considered for any patient presenting with a plexiform neurofibroma. Plexiform types are also the most common precursor to malignant peripheral nerve sheath tumors. Patients with multiple localized neurofibromas should also be candidates for further neurofibromatosis testing.[37][38]
Complications
The complications related to localized neurofibromas are mild and typically related to surgical excision of the lesion: pain, bleeding, scarring, and local infection. In plexiform lesions, complications arise from the inherent risks of surgery, and in rare cases, the inability to surgically remove the entire lesion. In patients with NF1 and persistent lesions, there is an increased risk of malignant transformation to malignant peripheral nerve sheath tumors.[35][36]
Deterrence and Patient Education
There are no definitive risk factors for sporadic neurofibromas that develop in the absence of NF1. In the setting of multiple localized, diffuse, or plexiform neurofibromas, café au lait macules, intertriginous freckling, optic glioma, Lisch nodules, or skeletal dysplasias, neurofibromatosis should also merit diagnostic consideration.[39][38]
Enhancing Healthcare Team Outcomes
Appropriate diagnosis and treatment of neurofibromas depend on the collaborative efforts of an interprofessional healthcare team. It is crucial for the clinician to communicate history and physical exam findings to the pathologist, and for the pathologist to communicate diagnosis and pertinent details to the clinician, primary care provider, and nurse practitioner, and nursing staff. An accurate history and physical, diagnosis, and interprofessional communication ensure the best outcome for the patient.[3] As plexiform neurofibroma is associated with NF1, care should be taken to communicate that fact to clinicians if rendering this diagnosis. Similarly, it is best to not render this diagnosis lightly without an appropriate clinical and gross description of the lesion. Dermatology and plastic surgery nurses assist with surgery, arranging for follow up, and educating patients and their families. [Level 5]
Media
(Click Image to Enlarge)
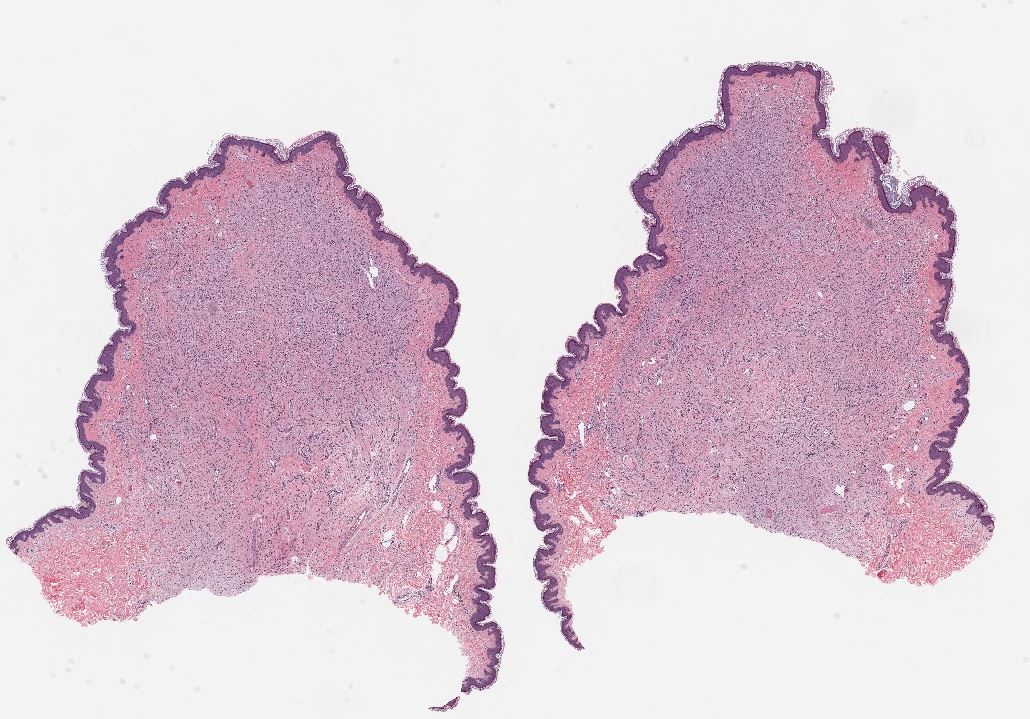
Neurofibroma, H&E stain, 10x magnification. Contributed by Lynn Messersmith, DO
(Click Image to Enlarge)
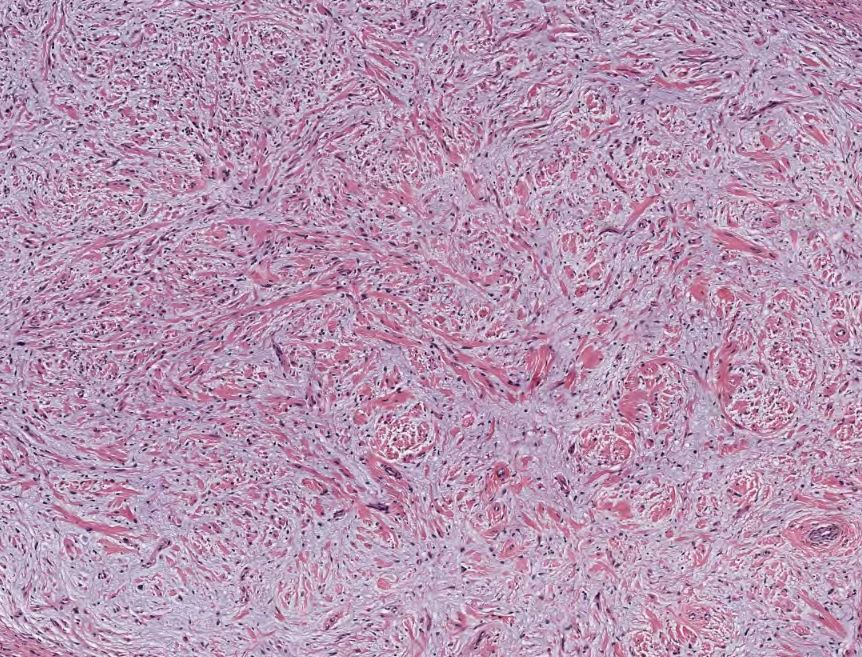
Neurofibroma, H&E stain, 40x magnification. Contributed by Lynn Messersmith, DO
(Click Image to Enlarge)
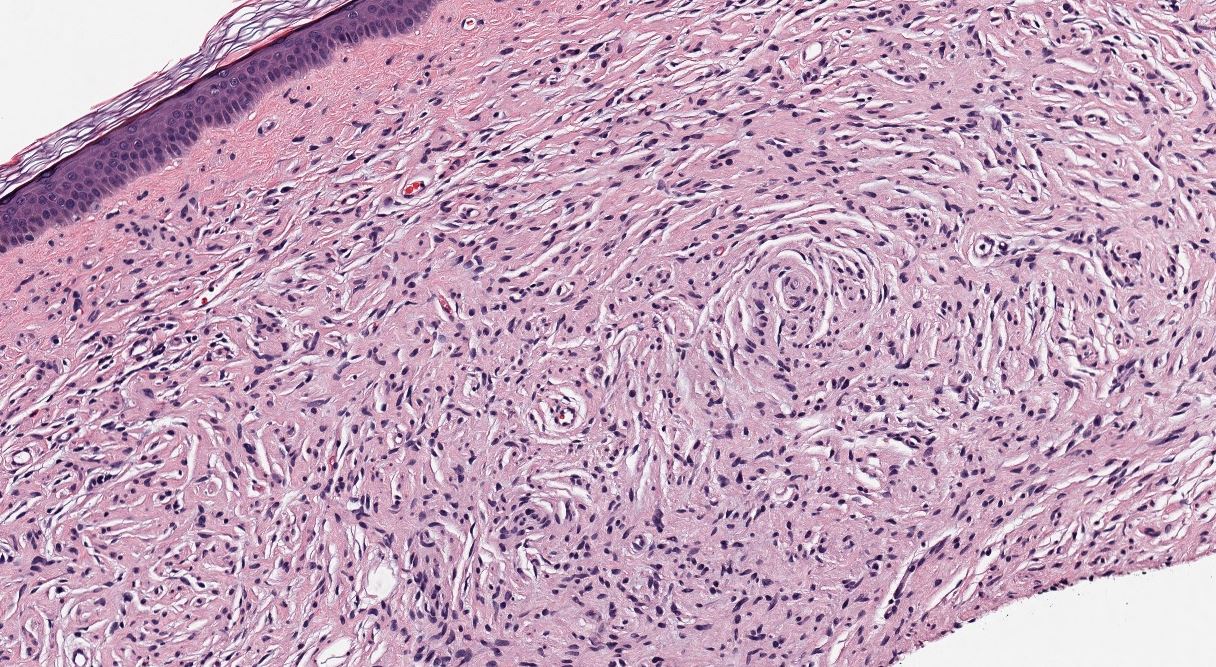
Neurofibroma, H&E stain, 100X magnification. Contributed by Lynn Messersmith, DO
(Click Image to Enlarge)
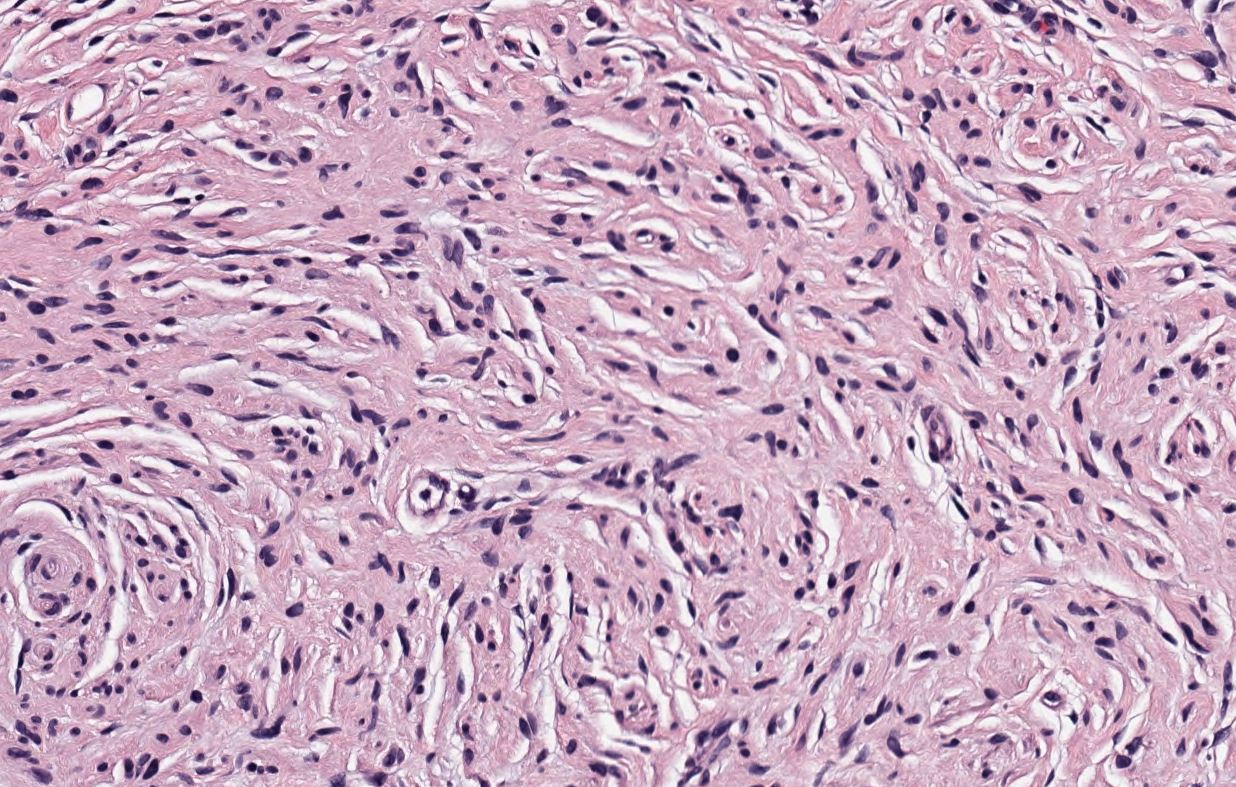
Neurofibroma, H&E stain, 200X magnification. Contributed by Lynn Messersmith, DO
(Click Image to Enlarge)

Neurofibromatosis
Contributed by S Verma, MBBS, DVD, FRCP, FAAD
References
Ferner RE, O'Doherty MJ. Neurofibroma and schwannoma. Current opinion in neurology. 2002 Dec:15(6):679-84 [PubMed PMID: 12447105]
Level 3 (low-level) evidenceLassmann H, Jurecka W, Lassmann G, Gebhart W, Matras H, Watzek G. Different types of benign nerve sheath tumors. Light microscopy, electron microscopy and autoradiography. Virchows Archiv. A, Pathological anatomy and histology. 1977 Sep 28:375(3):197-210 [PubMed PMID: 143775]
Skovronsky DM, Oberholtzer JC. Pathologic classification of peripheral nerve tumors. Neurosurgery clinics of North America. 2004 Apr:15(2):157-66 [PubMed PMID: 15177315]
Hernández-Martín A, Duat-Rodríguez A. An Update on Neurofibromatosis Type 1: Not Just Café-au-Lait Spots, Freckling, and Neurofibromas. An Update. Part I. Dermatological Clinical Criteria Diagnostic of the Disease. Actas dermo-sifiliograficas. 2016 Jul-Aug:107(6):454-64. doi: 10.1016/j.ad.2016.01.004. Epub 2016 Mar 12 [PubMed PMID: 26979265]
Ferner RE, Gutmann DH. Neurofibromatosis type 1 (NF1): diagnosis and management. Handbook of clinical neurology. 2013:115():939-55. doi: 10.1016/B978-0-444-52902-2.00053-9. Epub [PubMed PMID: 23931823]
Level 3 (low-level) evidenceTucker T, Riccardi VM, Brown C, Fee J, Sutcliffe M, Vielkind J, Wechsler J, Wolkenstein P, Friedman JM. S100B and neurofibromin immunostaining and X-inactivation patterns of laser-microdissected cells indicate a multicellular origin of some NF1-associated neurofibromas. Journal of neuroscience research. 2011 Sep:89(9):1451-60. doi: 10.1002/jnr.22654. Epub 2011 Jun 14 [PubMed PMID: 21674567]
Woodruff JM. Pathology of tumors of the peripheral nerve sheath in type 1 neurofibromatosis. American journal of medical genetics. 1999 Mar 26:89(1):23-30 [PubMed PMID: 10469433]
Magro G, Amico P, Vecchio GM, Caltabiano R, Castaing M, Kacerovska D, Kazakov DV, Michal M. Multinucleated floret-like giant cells in sporadic and NF1-associated neurofibromas: a clinicopathologic study of 94 cases. Virchows Archiv : an international journal of pathology. 2010 Jan:456(1):71-6. doi: 10.1007/s00428-009-0859-y. Epub 2009 Nov 25 [PubMed PMID: 19937344]
Level 2 (mid-level) evidenceShiurba RA, Eng LF, Urich H. The structure of pseudomeissnerian corpuscles. An immunohistochemical study. Acta neuropathologica. 1984:63(2):174-6 [PubMed PMID: 6428156]
Tchernev G, Chokoeva AA, Patterson JW, Bakardzhiev I, Wollina U, Tana C. Plexiform Neurofibroma: A Case Report. Medicine. 2016 Feb:95(6):e2663. doi: 10.1097/MD.0000000000002663. Epub [PubMed PMID: 26871793]
Level 3 (low-level) evidenceLin BT, Weiss LM, Medeiros LJ. Neurofibroma and cellular neurofibroma with atypia: a report of 14 tumors. The American journal of surgical pathology. 1997 Dec:21(12):1443-9 [PubMed PMID: 9414187]
Level 2 (mid-level) evidenceMotoi T, Ishida T, Kawato A, Motoi N, Fukayama M. Pigmented neurofibroma: review of Japanese patients with an analysis of melanogenesis demonstrating coexpression of c-met protooncogene and microphthalmia-associated transcription factor. Human pathology. 2005 Aug:36(8):871-7 [PubMed PMID: 16112003]
Jokinen CH, Argenyi ZB. Atypical neurofibroma of the skin and subcutaneous tissue: clinicopathologic analysis of 11 cases. Journal of cutaneous pathology. 2010 Jan:37(1):35-42. doi: 10.1111/j.1600-0560.2009.01293.x. Epub 2009 Mar 25 [PubMed PMID: 19469864]
Level 3 (low-level) evidenceLaskin WB, Fetsch JF, Lasota J, Miettinen M. Benign epithelioid peripheral nerve sheath tumors of the soft tissues: clinicopathologic spectrum of 33 cases. The American journal of surgical pathology. 2005 Jan:29(1):39-51 [PubMed PMID: 15613855]
Level 3 (low-level) evidenceFinkel G, Lane B. Granular cell variant of neurofibromatosis: ultrastructure of benign and malignant tumors. Human pathology. 1982 Oct:13(10):959-63 [PubMed PMID: 6290371]
Level 3 (low-level) evidenceVal-Bernal JF, González-Vela MC. Cutaneous lipomatous neurofibroma: characterization and frequency. Journal of cutaneous pathology. 2005 Apr:32(4):274-9 [PubMed PMID: 15769276]
Michal M, Fanburg-Smith JC, Mentzel T, Kutzner H, Requena L, Zamecnik M, Miettinen M. Dendritic cell neurofibroma with pseudorosettes: a report of 18 cases of a distinct and hitherto unrecognized neurofibroma variant. The American journal of surgical pathology. 2001 May:25(5):587-94 [PubMed PMID: 11342769]
Level 3 (low-level) evidenceHarder A, Wesemann M, Hagel C, Schittenhelm J, Fischer S, Tatagiba M, Nagel C, Jeibmann A, Bohring A, Mautner VF, Paulus W. Hybrid neurofibroma/schwannoma is overrepresented among schwannomatosis and neurofibromatosis patients. The American journal of surgical pathology. 2012 May:36(5):702-9. doi: 10.1097/PAS.0b013e31824d3155. Epub [PubMed PMID: 22446939]
Level 2 (mid-level) evidenceYeh I, McCalmont TH. Distinguishing neurofibroma from desmoplastic melanoma: the value of the CD34 fingerprint. Journal of cutaneous pathology. 2011 Aug:38(8):625-30. doi: 10.1111/j.1600-0560.2011.01700.x. Epub 2011 Apr 4 [PubMed PMID: 21457155]
Wolkenstein P, Zeller J, Revuz J, Ecosse E, Leplège A. Quality-of-life impairment in neurofibromatosis type 1: a cross-sectional study of 128 cases. Archives of dermatology. 2001 Nov:137(11):1421-5 [PubMed PMID: 11708944]
Level 2 (mid-level) evidenceStaser K, Yang FC, Clapp DW. Pathogenesis of plexiform neurofibroma: tumor-stromal/hematopoietic interactions in tumor progression. Annual review of pathology. 2012:7():469-95. doi: 10.1146/annurev-pathol-011811-132441. Epub 2011 Nov 7 [PubMed PMID: 22077553]
Level 3 (low-level) evidenceKumar BS, Gopal M, Talwar A, Ramesh M. Diffuse neurofibroma of the scalp presenting as circumscribed alopecic patch. International journal of trichology. 2010 Jan:2(1):60-2. doi: 10.4103/0974-7753.66919. Epub [PubMed PMID: 21188030]
Level 3 (low-level) evidenceRaffensperger J, Cohen R. Plexiform neurofibromas in childhood. Journal of pediatric surgery. 1972 Apr:7(2):144-51 [PubMed PMID: 4623404]
Serletis D, Parkin P, Bouffet E, Shroff M, Drake JM, Rutka JT. Massive plexiform neurofibromas in childhood: natural history and management issues. Journal of neurosurgery. 2007 May:106(5 Suppl):363-7 [PubMed PMID: 17566202]
Level 3 (low-level) evidenceAllaway RJ, Gosline SJC, La Rosa S, Knight P, Bakker A, Guinney J, Le LQ. Cutaneous neurofibromas in the genomics era: current understanding and open questions. British journal of cancer. 2018 Jun:118(12):1539-1548. doi: 10.1038/s41416-018-0073-2. Epub 2018 Apr 26 [PubMed PMID: 29695767]
Level 3 (low-level) evidenceKebudi R, Cakir FB, Gorgun O. Interferon-α for unresectable progressive and symptomatic plexiform neurofibromas. Journal of pediatric hematology/oncology. 2013 Apr:35(3):e115-7. doi: 10.1097/MPH.0b013e318270cd24. Epub [PubMed PMID: 23042022]
Level 3 (low-level) evidenceHornick JL. Limited biopsies of soft tissue tumors: the contemporary role of immunohistochemistry and molecular diagnostics. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc. 2019 Jan:32(Suppl 1):27-37. doi: 10.1038/s41379-018-0139-y. Epub 2019 Jan 2 [PubMed PMID: 30600320]
Jo VY, Fletcher CDM. SMARCB1/INI1 Loss in Epithelioid Schwannoma: A Clinicopathologic and Immunohistochemical Study of 65 Cases. The American journal of surgical pathology. 2017 Aug:41(8):1013-1022. doi: 10.1097/PAS.0000000000000849. Epub [PubMed PMID: 28368924]
Level 3 (low-level) evidenceZelger B, Weinlich G, Zelger B. Perineuroma. A frequently unrecognized entity with emphasis on a plexiform variant. Advances in clinical pathology : the official journal of Adriatic Society of Pathology. 2000 Jan:4(1):25-33 [PubMed PMID: 10936896]
Level 3 (low-level) evidenceSadullahoğlu C, Dere Y, Atasever TR, Öztop MT, Karaaslan Ö. The Role of CD34 and D2-40 in the Differentiation of Dermatofibroma and Dermatofibrosarcoma Protuberans. Turk patoloji dergisi. 2017:1(1):223-227. doi: 10.5146/tjpath.2017.01402. Epub [PubMed PMID: 28832078]
Magro G. Differential Diagnosis of Benign Spindle Cell Lesions. Surgical pathology clinics. 2018 Mar:11(1):91-121. doi: 10.1016/j.path.2017.09.005. Epub 2017 Dec 9 [PubMed PMID: 29413661]
Remstein ED, Arndt CA, Nascimento AG. Plexiform fibrohistiocytic tumor: clinicopathologic analysis of 22 cases. The American journal of surgical pathology. 1999 Jun:23(6):662-70 [PubMed PMID: 10366148]
Level 3 (low-level) evidenceAbrams JS, Tait N, Silva H, Eisenberger M, Van Echo DA, Olver IN, Aisner J. Phase II trial of N-methylformamide in advanced renal cancer. American journal of clinical oncology. 1989 Feb:12(1):41-2 [PubMed PMID: 2912020]
Ochoa CE, Joseph RW. Desmoplastic melanoma: a brief review and the efficacy of immunotherapy. Expert review of anticancer therapy. 2019 Mar:19(3):205-207. doi: 10.1080/14737140.2019.1574573. Epub 2019 Feb 4 [PubMed PMID: 30686076]
Gregorian C, Nakashima J, Dry SM, Nghiemphu PL, Smith KB, Ao Y, Dang J, Lawson G, Mellinghoff IK, Mischel PS, Phelps M, Parada LF, Liu X, Sofroniew MV, Eilber FC, Wu H. PTEN dosage is essential for neurofibroma development and malignant transformation. Proceedings of the National Academy of Sciences of the United States of America. 2009 Nov 17:106(46):19479-84. doi: 10.1073/pnas.0910398106. Epub 2009 Oct 21 [PubMed PMID: 19846776]
Level 3 (low-level) evidenceSpurlock G, Knight SJ, Thomas N, Kiehl TR, Guha A, Upadhyaya M. Molecular evolution of a neurofibroma to malignant peripheral nerve sheath tumor (MPNST) in an NF1 patient: correlation between histopathological, clinical and molecular findings. Journal of cancer research and clinical oncology. 2010 Dec:136(12):1869-80. doi: 10.1007/s00432-010-0846-3. Epub 2010 Mar 15 [PubMed PMID: 20229272]
Level 3 (low-level) evidenceGesundheit B, Parkin P, Greenberg M, Baruchel S, Senger C, Kapelushnik J, Smith C, Klement GL. The role of angiogenesis in the transformation of plexiform neurofibroma into malignant peripheral nerve sheath tumors in children with neurofibromatosis type 1. Journal of pediatric hematology/oncology. 2010 Oct:32(7):548-53. doi: 10.1097/MPH.0b013e3181e887c7. Epub [PubMed PMID: 20686424]
Schaefer IM, Fletcher CD. Malignant peripheral nerve sheath tumor (MPNST) arising in diffuse-type neurofibroma: clinicopathologic characterization in a series of 9 cases. The American journal of surgical pathology. 2015 Sep:39(9):1234-41. doi: 10.1097/PAS.0000000000000447. Epub [PubMed PMID: 25929351]
Level 3 (low-level) evidenceGarozzo D. Peripheral nerve tumors in neurofibromatosis 1: An overview on management and indications for surgical treatment in our experience. Neurology India. 2019 Jan-Feb:67(Supplement):S38-S44. doi: 10.4103/0028-3886.250697. Epub [PubMed PMID: 30688231]
Level 3 (low-level) evidence