Introduction
Motor neuron disease (MND) is said to be a progressive neurological disorder that presents with both lower motor neurons (anterior horn cells that project from the brainstem and the spinal cord to the muscle) and upper motor neuron signs (neurons that project to the brainstem and spinal cord from higher cortical centers).[1]
While the anterior horn cell and the corticospinal tract have been shown to be the primary site of involvement, the involvement of other parts of the nervous system (cortical, autonomic, cerebellar, and extrapyramidal system) has also been documented.[1] The four main phenotypes of motor neuron disease, based upon the site of origin and the severity of neurological involvement, are as follows: Amyotrophic lateral sclerosis, progressive bulbar palsy, progressive muscular atrophy, and primary lateral sclerosis.[2]
Other systems used for the classification of this disease have used the site of onset (spinal or bulbar onset), degree of adherence to the El-Escorial (and Airlie house) criteria, and pattern of heritability (sporadic versus familial) as criteria for characterizing this neurodegenerative illness with a complex genetic basis.[3] MND has been shown to be a disease of middle age with a mean age of 58 to 63 years at the time of onset for sporadic Amyotrophic lateral sclerosis (ALS), and 40-60 years of age for familial ALS.[4]
There has been a growing ethical debate along with pleas to formulate policies pertaining to the provision of Euthanasia and physician-assisted suicide by the families of patients and their advocates, which has brought the discussion surrounding this disease into the public domain.[5]
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
Existing research points towards an underlying genetic basis. Four genes are associated with up to 70 percent cases of Familial ALS, namely the C9ORF72, TARDBP, SOD1, and FUS. Over 25 genes and loci have been identified in relation to the predisposition of the disease, amongst which the C9ORF72 (Chromosome 9 open reading frame), has been associated with 40 percent cases of familial and 10 percent cases of sporadic MND. This mutation, which leads to a hexanucleotide repeat expansion, has also been found in cases presenting which Fronto-temporal dementia which has lead to a hypothesis that these neurological disorders exist as part of a dynamic spectrum of neurological syndromes.[6] This observation also provides evidence for the involvement of extra motor structures in MND spectrum disorders.
While this particular locus has less than 30 repeats in healthy individuals, patients with C9 related ALS, have a c9orf72 expansion mutation that has hundreds to thousands of repeats.[7] The presence of this expansion leads to ALS in two different ways. For example, TDP-43 accumulation and repeat-associated non-ATG translation of nuclear RNA foci leading to the formation of repeat derived RNA and dipeptide repeat proteins, which have been postulated to cause neurotoxicity.[8]
TARDBP gene encodes the TAR DNA-binding protein 43, SOD-1 gene encodes for superoxide dismutase. In contrast, the FUS gene codes for the similarly named (Fused in sarcoma) RNA binding protein.[8]
A study of population-based registries of ALS has led a degree of credibility to the hypothesis that ALS follows a complex genetic inheritance, where a variety of environmental factors interact with genetic mutations (present in “at-risk” variants), through a multistep process that decides the pattern of the disease manifestation. The occurrence of rare variants makes extensive genome-wide studies less suited for the study of this disease.[9]
Dysregulation of micro RNA and variations in ion channels that predispose to cellular excitotoxicity has also been postulated as the etiology underlying motor neuron disease.[10]
Familial forms of ALS are also characterized by penetrance of less than 50 percent and genetic pleiotropy (where a single gene can lead to the manifestation of multiple phenotypical traits). In contrast, sporadic forms have been associated with oligogenic (determination of a phenotypic trait by more than one gene) and polygenic inheritance.[11]
Among environmental and lifestyle factors that have been associated with the development of ALS, cyanotoxins, and related compounds deserve mention. The association between the ALS and pre-morbid physical prowess is also an area where some research has been done. An increased incidence in athletes, who have been exposed to repetitive head trauma was proposed, and discarded subsequently, due to the absence of evidence.[12] Amongst other environmental factors, cigarette smoking, and past history of military service have been consistently shown to be associated with an increased propensity for developing ALS.[4]
Epidemiology
The incidence of motor neuron disease has been shown to approach 2 to 3 per 100,000 population, while a lower frequency (less than 1 per 100,000) of ALS has been demonstrated in the South and East Asian community.[13] Ancestral origin has been shown to have a significant impact on the disease risk in ALS, with higher survival in racially heterogeneous or admixed populations (as compared to the White or Black race community).[14] A shorter survival has also been reported in European ALS patients (2 years) as compared to the Asian population (4 years).[14]
Approximately 10 to 15 percent of individuals with ALS have familial disease.[15] The estimated lifetime risk of sporadic ALS is one in 400.[4] While bulbar onset ALS has been shown to be more common in females, spinal onset illness has been shown to be common in males.[16] Progressive muscular atrophy (PMA) represents 2.5 to 11 percent of cases with MND. With an incidence of 0.02 per 100,000 population, it is a much rarer form of the disease. It is predominantly seen in males, with a male to female ratio ranging from 3 to 7.5 to 1. The median age of onset is of 68 years, which is older than that of ALS patients.[17]
Pathophysiology
Motor neuron injury arising as a result of derangement of multiple interlinked intracellular processes that have been attributed to genetic mutations leading to impaired protein homeostasis (SIGMAR 1M, CHMP2B,c9orf72), aberrant RNA metabolism (SETS, FUS, ANG), mitochondrial dysfunction (SOD1, CHCHD10, TARDBP), dysregulated vesicle transport (SOD1, ALS2, FIG4), impaired DNA repair (NEK1, C21ORF2, SPG11), oxidative stress (SOD 1, ALS2, TARDBP2 mutation), which have all been implicated in the development of neurotoxicity.[18]
Out of the processes mentioned above, the two which have been postulated as being central to the etiopathogenesis of the neuronal injury include defects in protein homeostasis and RNA metabolism.[19] Neuronal hyperexcitability and axonal dysfunction have also been implicated in the etiopathogenesis.[20]
These processes lead to the failure of the motor neuron axon in maintaining its projections leading to retraction of the axon and ultimately resulting in denervation of the target. This leads to denervation of the neuron when the lower motor neuron and loss of supraspinal control of the upper motor neuron, hypertonia, and spastic weakness when the upper motor neuron is involved.[21]
Histopathology
The neuropathological hallmark of this disease is the presence of TDP-43 (transactive response DNA binding protein 43) containing ubiquitinylated proteinaceous inclusions in motor neurons.[22] Nuclear depletion of TDP-43 along with cytoplasmic deposition of atypical cytoskeletal aggregates has been observed in residual motor neurons. Neurofilaments hyaline aggregate inclusions, deposition of misfolded SOD 1 proteins, p62, or sequestosome 1 coded by SQSTM1 have also been identified as the molecular constituents of ubiquitinated aggregates observed in afferent motor neurons.[23] It has also been hypothesized that the high molecular weight complexes, which precede the development of these aggregates are responsible for the neurotoxicity.[24]
Skeletal muscle and cortical motor atrophy, corticospinal, and corticobulbar tract hyalinization and sclerosis, hypoglossal nerve, and ventral root thinning have been observed on gross examination in pathology specimens.[25] Microscopically, diseased skeletal muscle fibers demonstrate changes of denervation and reinnervation with the grouping of atrophic and angular fibers.[26]
The high expression of Ephrin type a receptor 4 and matrix metalloproteinase 9 along with lower expression osteopontin and Insulin-like growth factor 2 along with the large size of the cell and the need to maintain long axonal projections could make the motor neuron more vulnerable to the effect of these derangements.[27] It has been seen that motor neurons that comprise the fast fatigable motor units are damaged earlier in the disease process. The importance of this, and relation to other etiological environmental factors, is yet to be elucidated.[27]
History and Physical
The diagnosis of MND hinges upon critical clinical observation.[28] The characterization of weakness should address the following questions - asymmetrical versus symmetrical, proximal versus distal, upper versus lower limb predominance, presence, or absence of bulbar symptoms.[29]
Amyotrophic lateral sclerosis (ALS):
While a majority of ALS patients present with limb onset disease (60-80 percent), about one third to a fifth present with bulbar involvement in the form of dysarthria and dysphagia (along with emotional lability). A small minority of patients present with signs of respiratory muscle weakness. Patients may present with predominantly upper motor neuron (UMN) symptoms (hyperreflexia and spastic weakness) with the lower motor neuron (LMN) signs becoming commoner with the progression of the disease.[16] Presentation with lower motor neuron signs of flaccid weakness, fasciculations, and muscle wasting may also be seen.
The initial symptom usually involves the distal muscles asymmetrically manifestation, such as difficulty in turning doorknobs, writing, turning keys in locks, picking up objects from overhead shelves along with impairment of gait, imbalance, and frequent falls which might manifest as functional disability and impairment of activities of daily living.[30] Progressive dysphagia may lead to a reduction in oral intake and ensuing weight loss.[31] Amyotrophic lateral sclerosis has also been related to a hypercatabolic state, the etiopathogenesis of which has not been elucidated.[32] The ability to communicate effectively might be hindered by the development of dysarthria.[33]
Respiratory insufficiency may present with breathlessness along with symptoms of carbon dioxide retention (headache) and hypoxia (confusion). Complications arising as a result of respiratory impairment (Respiratory failure and pneumonia) are the leading cause of death.[34] Impairment of executive function manifesting as inattention, working memory, organization, and planning capabilities are seen in a third of patients.[35] A tenth of newly diagnosed patients may present with symptoms of Frontotemporal dementia.[36]
Behavioral traits such as apathy, changes in personality and mood, disinhibition, and obsessive behavior are also being recognized increasingly in this subset of patients.[37] The symptoms present in nearly forty percent of patients at presentation can be clustered into five different groups, which have been known to map to known neuroanatomical networks.[38]
A degree of severe autonomic impairment with the potential to impact the quality of life is usually not seen in patients with ALS. Upper motor neuron involvement has been associated with more severe autonomic deficits.[39]
Progressive muscular atrophy (PMA):
Also known as the LMN phenotype, and represents 5 percent of the MND cases. It presents with progressively worsening LMN signs. The upper motor neuron signs are known to develop in 20-30 percent of the cases, usually within 5 to 10 years of the onset of the disease. Patients present with typical LMN symptoms of progressive flaccid paralysis, muscle atrophy, hyporeflexia/areflexia, and fasciculations.
Asymmetric weakness and atrophy in the distal limbs are seen at presentation. It is associated with a relatively better prognosis and characterized by a slower rate of progression. The asymmetrical proximal muscle weakness pattern is seen in a fifth of the patients. Bulbar involvement portends a poorer prognosis.[40] Axial or respiratory muscle involvement is uncommon at the onset but may predict a poor prognosis when present.[41] Cognitive impairment in MND is linked with UMN involvement and is rarely seen in PMA.[42]
Flail arm and flail leg variants are other lower motor neuron variants of MND.[40] While, the flail arm variant is characterized by progressive, predominant upper limb involvement, proximal to distal progression, the flail leg variant is known to present with progressive, asymmetric weakness of the lower limbs.
Family history of a similar neurological illness, personal account of smoking, exposure to toxins (cyanotoxins) assumes importance, occupational history of military service and past history of repetitive head trauma (as in pugilists) also needs to be ruled out in patients suspected to have a working diagnosis of ALS.[43] Past medical history of HIV also needs to be ruled out as it has been shown to be associated with an ALS like syndrome, which is shown to improve with antiretroviral therapy.[44]
Physical examination:
An examination may reveal a constellation of upper motor neuron and lower motor neuron signs. Upper motor neuron signs range from hyperreflexia, spastic weakness, and exaggerated jaw jerk to clonus. In contrast, lower motor neuron involvement is characterized by areflexia or hyporeflexia, flaccid weakness, wasting, and fasciculations.[45]
Bulbar involvement is characterized by dysarthria and dyspnoea, which might also be accompanied by emotional liability.[16] A neuropsychiatric assessment may present with signs of executive dysfunction and behavioral abnormalities.[46] Extraocular muscle and sphincter involvement are rare in the typical variant of the disease.[47]
Identification of specific patterns of limb weakness may provide an aid to the physical diagnosis of ALS. In this split hand pattern, there is a preferential involvement of the first dorsal interossei and the thenar muscles with sparing of hypothenar muscles. In contrast, the split leg pattern is characterized by preferential ankle plantar flexion involvement with the sparing of the dorsiflexors.[48]
Physical signs in ALS:
- Fasciculations, if generalized (can be easily missed over the shoulders)[49]
- Bilateral wasting of the tongues – lateral borders, particularly when there are also fasciculations with a brisk jaw jerk and orbicularis oculi reflexes
- The “split hand”- preferential wasting of the lateral border of the hand, first dorsal interosseous and abductor policies Brevis, though to reflect the cortical organization
- Head drop due to weakness of neck extensors, may consider an alternative diagnosis of myasthenia gravis. Care should be exercised to not attribute this sign to degenerative cervical sign[29]
- Emotionality – exaggerated response to emotional stimuli, usually crying, bulbar weakness may be present. An abnormal response to the glabellar tap may be seen[50]
- Cognitive or behavioral disorders – Frontotemporal dementia overlap features[36]
Evaluation
Essential investigations advised in the evaluation of a patient are complete blood count, erythrocyte sedimentation rate, C–reactive protein, full biochemical count (including thyroid function tests and calcium), creatine kinase (more than 1000iu/l are very unusual in MND patients), immunoglobulins and serum electrophoresis. The electromyography, nerve conduction studies, MRI of the brain, and spine are also advised.[51]
Nerve conduction studies can be used in assessing the type of pattern of involvement, the existence of demyelination or focal motor conduction block, and the existence of subclinical sensory abnormalities.[40] Optional investigations include vitamin B12, folate, anti-acetylcholine receptor antibodies, HIV, Lyme’s serology, lumbar puncture, and muscle biopsy, which might be useful in ruling out other differential diagnoses.
The diagnosis of ALS is based upon recognition of a specific pattern of weakness, ruling out other etiologies that may present with similar symptoms and ascertainment of disease progression. Various diagnostic criteria include the El Escorial, Airlie house, Awaji, and ALS diagnostic index.[2] These criteria require the demonstration of the three features. Those are the demonstration of clinical, electrophysiological or neuropathological lower motor neuron degeneration, evidence of clinical involvement of upper motor neurons, and progression of the disease from one region to the other by neurological or electrophysiological testing. For the purpose of diagnosis, specific patterns of involvement confined to one particular area have been identified, namely the bulbar, cervical, thoracic, and lumbar region.[2]
While the categories of diagnostic certainty, ranged from suspected ALS to definitive ALS in the El Escorial criteria, the Airlie house criteria demarcate these clinical features and laboratory supported findings into categories ranging from definitive ALS to possible ALS.[2] The Awaji criteria allow the inclusion of electrophysiological parameters into the diagnostic parameters, while the ALS diagnostic index includes transcranial magnetic stimulation defined standards that can be used to determine the functional integrity of the upper motor neuron. Threshold tracking TMS (transcranial magnetic stimulation) has demonstrated evidence of cortical hyperexcitability in MND and may provide objective evidence of clinically inapparent UMN dysfunction.[3]
Biomarkers:
Various biomarkers such as neurofilament light peptide and phosphorylated neurofilament heavy neuropeptides in cerebrospinal fluid (CSF) have been used to differentiate ALS from its mimics, namely, cervical myelopathy, multifocal motor neuropathy, and inclusion body myositis.[52] Levels of these CSF biomarkers have been found to be specific and sensitive to diagnose patients with ALS and have also been shown to correlate with disease progression.
Though abnormalities on FDG PET (F-fluorodeoxyglucose positron emission tomography) imaging and spectral electroencephalography have identified selective network disruptions in structural and functional connectomes (corticospinal, orbitofrontal, frontostriatal and orbitotemporal circuits), they have not been found to be clinically useful in establishing a diagnosis of motor neuron disease with certainty.[53]
Treatment / Management
Disease-modifying Therapies
Riluzole, an NMDA (N-methyl-D-aspartate) receptor antagonist, reduces glutaminergic transmission by acting on the presynaptic voltage-gated sodium channels. Relative glutamate excess has been shown to lead to upper and lower motor neuron excitotoxicity contributing to neuronal cell death.[54] It has been shown to be associated with a statistically significant improvement in tracheostomy free survival.[55] Though sustained use for 18 months was not associated with improved muscle strength, it has been shown to improve survival by three months (equitable to a 9 percent increase in one-year survival).[56] It was found to be very useful in those with moderate functional impairment in those with advanced disease when used on a compassionate basis.[57](A1)
The standard dose of Riluzole in the management of ALS is 50 mg two times daily.[58] The most common adverse effects include nausea, asthenia, and derangement in liver enzymes.[57] Pancreatitis and fulminant hepatic failure have also been reported as rare adverse effects.[59] Regular blood testing every month for the first three months, followed by three monthly for nine months and yearly, is recommended.[60] An increase of over three times the upper limit of normal in hepatic enzymes can be managed by dose reduction, followed by withdrawal followed by the re-introduction of the drug[60]. Riluzole is contraindicated in hepatic and renal impairments and in those who are breastfeeding and lactating. Further trials have been advised to test the efficacy of different dosing regimens.[61](A1)
Edaravone has also been shown to impede disease progression in those patients with early-onset disease and rapid disease progression.[62] Its role in the management of all patients with ALS remains to be addressed.[63] The drug acts as an antioxidant, which has been shown to prevent the nitration of tyrosine residues in experimental animal models.[64](B3)
Symptom Directed Treatment
Spasticity:
Spasticity may be present in most of the patients with primary lateral sclerosis, though it may only be present with a lesser frequency in patients with amyotrophic lateral sclerosis.[65] Tools to measure spasticity have included the modified Ashworth scale, numerical rating scale spasticity responses, and the Rasch based scale for patient-reported spasticity, among others.[66] Gabapentin, baclofen, tizanidine, benzodiazepines, and levetiracetam have been advised for the management of spasticity. The practice of these drugs is limited by the occurrence of fatigue and weakness.[67] Baclofen may sometime induce hyperpolarisation of some of the afferent terminals and also inhibits monosynaptic and polysynaptic reflexes usually at the spinal level.[68] Intrathecal baclofen, wherein the drug is infused into the cerebrospinal fluid through a surgically placed infusion pump, has also been used in the management of spasticity in primary progressive aphasia.[69] Stages of the disease.[16]
Siallorhoea is caused by the inability to swallow saliva, which may be the result of tongue spasticity, weakness of the facial, mouth and pharyngeal muscles, loss of pharyngeal coordination, and function.[70] Anticholinergic drugs such as atropine, hyoscine, glycopyrrolate, and amitryptiline have been found to be beneficial in the management of sialorrhea. Salivary gland irradiation and botulinum toxin injections have also been proposed as treatment modalities.[16] Anticholinergic drugs have been associated with urinary retention and constipation. They are contraindicated in patients with heart block and benign prostatic hypertrophy.[1] Optimum advice on diet, swallowing, positioning, posture, hydration, and oral care should be provided. Nebulizers, humidifiers, and mucolytics should be considered.[71](A1)
Pain:
15% to 85% of patients, more commonly nociceptive than neuropathic in nature. Later stages of the ailment may be characterized by musculoskeletal pain, which arises as a result of the loss of protective sheath comprising of muscles that protect the bones and joints. Muscle contractures and bone stiffness may also be a source of pain. Decubitus ulcers seen in 16 percent of patients may also be associated with pain. Increased intensity of pain in the later stages of the disease has also been associated with assisted suicide.[72] While, NSAIDs, opioids, and cannabinoids have been used for nociceptive pain, gabapentinoids, and tricyclic antidepressants have been used in neuropathic pain. The dearth of randomized data on this topic is a matter of some concern. Intra-articular injections of lidocaine, along with physical therapy and assistive range of motion exercises, have also been helpful.[72](A1)
Muscle cramps:
Cramps are caused by the instability of motor units.[1] Quinine sulfate, mexiletine, and levetiracetam have been used in ameliorating these symptoms. Caution is advised with the use of quinine sulfate as it has been associated with QT prolongation, arrhythmias, and bradycardia.
Dysphagia:
Strategies to mitigate this particularly distressing symptom include dietary changes. That consists of the change in consistency of the diet, high calories, and high protein diets, fluid thickeners along with exercises to facilitate swallowing. Insertion of a percutaneous gastrostomy tube for enteral nutrition has also been advised in those with a weight loss in excess of five percent and in whom swallowing can be associated with the occurrence of dangerous complications. In those in whom these procedures are contraindicated or those with respiratory insufficiency, feeding through a central venous catheter should be tried.[1]
Dysarthria:
Up to 30 percent of the patients with ALS develop dysarthria. This might extend to 80 percent of patients during the course of the disease.[1] Speech therapy, use of customized software for augmentative and alternative software have been shown to enhance the quality of life in ALS.[73]
Coagulation abnormalities (Deep venous thrombosis):
The annual incidence of coagulation abnormalities has been in the range of 3 and 11 percent. The use of prophylaxis, compression stockings, and standard doses of anticoagulation as per existing guidelines have been advised.[74]
Neuropsychiatric disturbances:
Neuropsychiatric disturbances such as pseudobulbar affect and depression have been reported.[75] The pseudobulbar effect has been postulated to be caused due to the disruption of the corticopontocerebellar pathways. Impaired volitional control and cerebellar demodulation have been considered central to the neuropathogenesis of this symptom.[76] It has also been described as dysmetria caused due to cerebellar dysmodulation. Depression may be an early manifestation of frontal lobe dysfunction and may signify widespread cortical involvement.[77] Tricyclic depressants and selective serotonin reuptake inhibitors (SSRIs) have been used in the management of depression. Pseudobulbar effects have been managed with the use of these antidepressants.[78] Dextromethorphan and quinidine have also been shown to be useful in the management of this symptom.(B2)
Though no specific agents have been found to be useful in the treatment of cognitive symptoms, SSRIs have been found to be helpful in controlling symptoms such as control of loss of inhibition, overeating, and compulsive behaviors. In contrast, antipsychotics have been found helpful in the treatment of restlessness.
Respiratory symptoms:
The weakness of respiratory muscles may present in the form of the loss of ability to cough and respiratory insufficiency.[79] The monitoring of physiological parameters of respiratory function has been advised as respiratory symptoms have been found to poorly correlate with respiratory muscle function.[80] Supine Forced vital capacity (FVC) may be used as a marker of diaphragmatic weakness.[81] Methods to assist the patient while coughing (cough assist devices such as physiotherapy maneuvers like tussive squeeze, mechanical insufflator – exsufflator devices) have been advised.[82] The usage of non-invasive ventilation (NIV) has been associated with improvement in parameters of respiratory function, which is also associated with an improvement in the quality of life, along with a substantial increase in survival.(A1)
The presence of dyspnoea, orthopnoea, and daytime fatigue, vital capacity less than 80 percent of the normal, partial pressure of CO2 more than 45 mm of Hg, nocturnal hypoventilation (when the oxygen saturation is less than 88-90 percent for more than 5 percent of the time is asleep), maximal inspiratory pressure less than 60 percent predicted, sitting fixed vital capacity less than 50 percent, sniff nasal inspiratory pressure less than 40 percent have been listed as various indications for non-invasive ventilation. NIV is poorly tolerated in 30 percent of patients due to anxiety, emotional lability from pseudobulbar damage, hypersalivation, claustrophobia, and soreness of the nasal bridge.[83] The presence of bulbar dysfunction at the onset and cognitive impairment has been shown to correlate with poor compliance.[84]
Persistent respiratory insufficiency, despite the use of NIV and severe bulbar dysfunction, has been reported as indications for tracheostomy.[85](B2)
The use of diaphragmatic muscle pacing, where four electrodes are placed on the motor roots of the phrenic nerve on the abdominal surface of the diaphragm has also been studied.[86] The procedure involves the implantation of intramuscular electrodes on the abdominal surface of the hemidiaphragm at motor points. Intramuscular diaphragmatic pacing aims to make the diaphragm contract, strengthen the force of contraction, and lead to a decrease in the patient’s dependence upon mechanical ventilation. After implantation, the patient undergoes a conditioning program that involves gradual weaning from the ventilator. Current evidence suggests long term safety concerns related to the occurrence of adverse events such as capnothorax or pneumothorax, acute respiratory failure requiring mechanical ventilation, venous thromboembolism, and gastrostomy tube placement. Suture granuloma, infection at the stimulation cable entry point, and superficial wound infection has also been reported.[86]
Fatigue:
Fatigue has been reported as a frequent and debilitating symptom characterized by reversible weakness. It is characterized by a feeling of tiredness, which is not relieved by rest. Both pharmacological measures (Modafinil) and non-pharmacological measures (resistance exercise, respiratory exercise, and repetitive transcranial magnetic stimulation) have been used in the management of fatigue. The quality of evidence for the effectiveness of these interventions has been reported to be very low, and more research is needed on this topic.[87](A1)
Palliative Medicine Interventions
Palliative medicine interventions include adequate symptom control, prognostication about goals of care, discussions surrounding degree of aggressiveness of treatment, advanced care planning, provision of quality home-based care, possible liaison with organizations providing community palliative services, anticipatory prescribing, provision of support to family members (addressing caregiver burden), and bereavement support.
Formal care at the end of life should incorporate measures to improve the quality of life (such as a system to enable effective communication) and focus upon the management of commonly encountered symptoms. The use of opioids and benzodiazepines for breathlessness, glycopyrrolate for increased secretions, antipsychotics for delirium, and other non-pharmacological treatment options may be indicated in patients approaching their end of life.[88]
Reduction in ICU utilization at end of life, the number of deaths inside the hospital setting (transition in the place of care), and costs of treatment are essential indicators of the efficacy of provision of high-quality care.[89] For patients being treated in the intensive care setting, discussions centering upon withdrawal of life-sustaining measures and DNCPR (Do not perform cardiopulmonary resuscitation) decisions may be indicated.[90]
Physiotherapy
Exercise programs such as structured exercise programs may improve stiffness, prevent contractures, reduce discomfort, and optimize the quality of life. Provision of the orthosis, if necessary, should be facilitated without delay.[91]
Communication
Communication should be assessed by a speech and language therapist. Constant review during interdisciplinary meets is advised. The provision of augmentative and alternative communication strategies that cause the least interference with the patient’s quality of life is recommended.[92]
Future therapeutic options
The use of antisense oligonucleotides in SOD 1 related and C9orf related ALS are subjects of research in future clinical trials.[93](B3)
Use of stem cell therapy
The proposed mechanism of the utility of stem cell therapy includes the differentiation of stem cells into spinal neurons that synapse with existing motor neurons to establish or maintain neurocircuitry and provide neurotrophic support. Differentiation of stem cells into non-neuronal cells can also impact disease progression by providing neurotrophic support, preventing oligodendrocyte dysfunction and toxicity. Peripheral stem cell transplantation can maintain the neuromuscular junction. The immunomodulatory effect of mesenchymal stem cells mobilized from the bone marrow can also attenuate the inflammatory response via the production of anti-inflammatory mediators. The commonly used stem cell therapies include olfactory ensheathing stem cells, endogenous mesenchymal stem cells, autologous mesenchymal stem cells, and neural progenitor cells. The most significant number of trials of stem cell therapy involves the use of mesenchymal stem cells.[94](B3)
Differential Diagnosis
The most common disease that mimics the amyotrophic lateral sclerosis is none other than degenerative spondylotic myeloradiculopathy in the cervical and/or lumbosacral spine, which may present with radicular pain. This may be characterized by progressive symptoms that plateau later with the progression of the disease. Although imaging has been advised to differentiate between the two entities, there may be the presence of co-existent degenerative illness in those with ALS.[51]
Presentation with symmetrical findings, prominent extensor plantar responses (cervical myelopathy), presence of sensory deficit, presence of substantial weakness in the absence of wasting (multifocal motor neuropathy and myasthenia gravis) and disproportionate involvement of quadriceps (inclusion body myositis) should prompt further investigation for an alternative diagnosis.
An acute presentation of lower motor neuron syndrome should prompt consideration of immune, toxic, metabolic, and infective etiology. The major differentials include MND, multifocal motor neuropathy, and monomelic amyotrophy.[40] The diagnosis of multifocal motor neuropathy should be considered in patients with early involvement of finger and wrist extensors, focal motor conduction block, presence of positive symptoms such as cramps, spasms, and presence of anti-IgM GM -1 antibody.[95]
Motor neuron disease mimics:
a. Presenting with progressive, painless weakness[51]
i) Genetic disorders – Spinal muscular atrophy, Kennedy’s disease, hexosaminidase deficiency, mitochondrial disease, triple-A (Allgrove syndrome), polyglucosan body disease, hereditary spastic paraparesis, and adrenoleukodystrophy.
ii) Toxicity or metabolic disorders – Radiation myelopathy, thyrotoxicosis, hyperparathyroidism, lead poisoning, mercury poisoning, and copper deficiency.
iii) Infectious diseases – Post-polio syndrome, Lyme disease, and HIV.
iv) Immunological or inflammatory syndromes – Paraproteinemic neuropathy, conduction block neuropathy, Sjogren’s syndrome, Inclusion body myositis, Gluten sensitivity, myasthenia gravis, and polymyositis.
v) Structural diseases – Spondylotic myelopathy, the base of skull lesions, foramen magnum lesions, intrinsic or extrinsic cord tumors, and lumbosacral radiculopathies.
vi) Miscellaneous condition – Cramp fasciculation syndromes, paraneoplastic disease, lymphoproliferative disease, and paraneoplastic neuromuscular disease.
b. Presenting with a generalized or focal single (monomelic) limb onset lower motor neuron syndromes[51]
i) Brachial neuritis / Parsonage-Turner syndrome[96]
Sudden onset typically idiopathic inflammatory condition affecting the brachial plexus, characterized by severe pain followed by weakness and wasting in the absence of trauma. Five percent of cases may be painless. History of preceding viral illness or vaccination may be present. In 10 percent of the cases, there may be a bilateral pathology. A history of bilateral phrenic nerve paralysis, producing diaphragmatic palsy leading to orthopnoea, may also be present.
ii) Hirayama’s disease[97]
Benign juvenile-onset monomelic amyotrophy of the upper limb or focal segmental spinal muscular atrophy may be progressive for 1 to 3 years, and then remain stable, may not show progression or recovery. Sub-clinical involvement of another limb may be apparent on NCS/EMG. May present with cold paresis or oblique amyotrophy of the forearm, commonly seen in Japan and the Indian subcontinent.
iii) Kennedy’s syndrome[98]
Spinobulbar muscular atrophy, trinucleotide repeat mutation in the first exon of the androgen receptor gene, develop symptoms in the 4th and 5th decade, LMN syndrome, slowly progressive, bulbar weakness, tongue wasting, disproportionate bulbar involvement, lower facial/chin (mentalis) fasciculation characteristic, sensory as well as motor neuropathy may be seen. Gynecomastia, partial feminization, small testes, may cause respiratory insufficiency, associated with average life expectancy.
iv) Inclusion body myositis[99]
Slowly evolving painless symmetrical weakness, distal weakness, weakness distal or characteristic involvement of the quadriceps and long finger flexors (medial forearm), dysphagia, creatine kinase modestly elevated to within 2-10 times the normal range (important diagnostic clue). Usually aged over 50 years, 3 to 1 female to male predilection, EMG may appear neurogenic with fibrillation potentials. Muscle biopsy may show rimmed vacuoles, with inflammatory changes, relentless progression over the years, significant disability, and impairment of quality of life. Rare hereditary forms of inclusion body myositis may be associated with MND and frontotemporal dementia, in which mutations in the gene encoding for valosin containing protein and muscle biopsies stain for the protein TDP-43. This is the characteristic protein that accumulates in the central nervous system in such patients.
c. Upper motor neuron signs[51]
i) Hereditary spastic paraparesis[29]
Younger onset, slowly progressive paraplegia, dorsal column involvement, sphincter dysfunction, may include symptoms of ataxia or dementia, especially with a positive family history (autosomal dominant). Forty percent have mutations in the SPAST gene; minimal upper limb involvement does not include severe corticobulbar involvement.
ii) Primary progressive multiple sclerosis[29]
Slowly progressive, positive CSF findings (oligoclonal bands) and demyelination.
iii) Metabolic myelopathies[29]
Vitamin B12 and copper associated myelopathies, slowly progressive myelopathy, typically associated with sensory neuropathy, and adult-onset slowly progressive myelopathy,
iv) X linked adrenoleucodystrophy[29]
A peroxisomal disorder involving the ABCD1 transporter, long-chain fatty acid transporter, presents typically in the third to a fourth decade, confined to lower limbs, may present with sensory ataxia and bladder symptoms. Secondary Wallerian degeneration may result in hyperintensities in the associated cerebral corticospinal tracts.
Prognosis
Negative prognostic indicators – These include the bulbar onset or respiratory onset disease, presence of executive impairment, frontotemporal dementia, and weight loss, worsening respiratory function indicated by a reduction in serially assessed forced vital capacity, and also sniff nasal inspiratory pressure.
Symptoms such as speech and swallowing problems, weight loss, reduced respiratory function, older age, lower amyotrophic lateral sclerosis functional rating scale, shorter time from developing first symptoms to time of diagnosis have been identified as factors that might indicate a poor prognosis.[100]
King’s criteria that include the number of body regions affected and the presence of respiratory/nutritional failure and Milano-Torino system (records changes in domains of bulbar, fine motor, gross motor, and respiratory function), are used in clinical staging. While the King’s system has been shown to be sensitive to changes early in the trajectory of the disease, the MITOS system has been shown to be more sensitive to detect changes in the late stages of the disease).[101]
These staging systems have come under criticism for not including the domains of cognitive and affective function within their purview. However, scales such as the Edinburgh cognitive and behavioral functional (ALS) stream (ECAS), the detection of affective and cognitive dysfunction is vital as the presence of executive impairment has been associated with a more progressive downward disease trajectory while being shown to be related to specific needs that have the impact on increasing caregiver burden.[102]
Complications
Most patients with ALS die of respiratory failure within three years of the onset of the disease.[34] Progressive weakness and wasting of the limb and respiratory muscles have been attributed to be the underlying precipitants of respiratory failure. Severe dysphagia may lead to weight loss, choking, and aspiration.[103] Prolonged effortless mealtimes and coughing on attempting to swallow are other factors that have the ability to impact the patient's quality of care.[104]
Progressive deterioration in the activities of daily living, inability to ambulate, issues related to prolonged immobilization such as superficial skin infections, decubitus ulcers, and deep venous thrombosis are also seen in these patients.[105][106]
Enhancing Healthcare Team Outcomes
A multidisciplinary team consisting of neurologists, respiratory physicians, physical medicine, and rehabilitation specialists (respiratory physiotherapists), gastroenterologists, dieticians may have an active role in the management of the patient. Palliative medicine input may be required for the management of pain, difficult to control symptoms (such as resistant breathlessness at the end of life), and psychosocial issues.[88] The inclusion of social care practitioners and staff who have been trained to manage patients, in the setting of their homes, in the multidisciplinary team has also been advised.[107] Identification of the unique needs of family and provision of coordinated care in the home setting may become necessary as the patient approaches the terminal stage of decline.[108] Good end of life care may be provided by specialist palliative teams.[109]
Media
(Click Video to Play)
Clinical Examination of Ankle Clonus. Clinical examination of ankle clonus is typically demonstrated in upper motor neuron lesions or serotonin syndrome.
Contributed by RS Menon, MD
(Click Video to Play)
(Click Image to Enlarge)
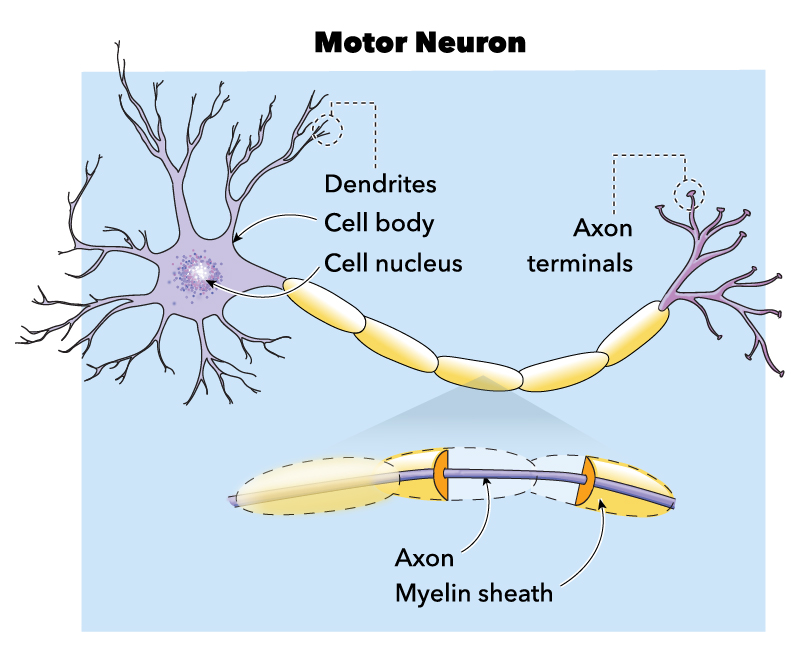
Figure showing Labeled Motor Neuron.
Contributed by Katherine Humphreys
References
Hardiman O, Al-Chalabi A, Chio A, Corr EM, Logroscino G, Robberecht W, Shaw PJ, Simmons Z, van den Berg LH. Amyotrophic lateral sclerosis. Nature reviews. Disease primers. 2017 Oct 5:3():17071. doi: 10.1038/nrdp.2017.71. Epub 2017 Oct 5 [PubMed PMID: 28980624]
Statland JM, Barohn RJ, McVey AL, Katz JS, Dimachkie MM. Patterns of Weakness, Classification of Motor Neuron Disease, and Clinical Diagnosis of Sporadic Amyotrophic Lateral Sclerosis. Neurologic clinics. 2015 Nov:33(4):735-48. doi: 10.1016/j.ncl.2015.07.006. Epub 2015 Sep 8 [PubMed PMID: 26515618]
Traynor BJ, Codd MB, Corr B, Forde C, Frost E, Hardiman OM. Clinical features of amyotrophic lateral sclerosis according to the El Escorial and Airlie House diagnostic criteria: A population-based study. Archives of neurology. 2000 Aug:57(8):1171-6 [PubMed PMID: 10927797]
Ingre C, Roos PM, Piehl F, Kamel F, Fang F. Risk factors for amyotrophic lateral sclerosis. Clinical epidemiology. 2015:7():181-93. doi: 10.2147/CLEP.S37505. Epub 2015 Feb 12 [PubMed PMID: 25709501]
Maessen M, Veldink JH, Onwuteaka-Philipsen BD, Hendricks HT, Schelhaas HJ, Grupstra HF, van der Wal G, van den Berg LH. Euthanasia and physician-assisted suicide in amyotrophic lateral sclerosis: a prospective study. Journal of neurology. 2014 Oct:261(10):1894-901. doi: 10.1007/s00415-014-7424-6. Epub 2014 Jul 15 [PubMed PMID: 25022937]
Renton AE, Chiò A, Traynor BJ. State of play in amyotrophic lateral sclerosis genetics. Nature neuroscience. 2014 Jan:17(1):17-23. doi: 10.1038/nn.3584. Epub 2013 Dec 26 [PubMed PMID: 24369373]
van Blitterswijk M, DeJesus-Hernandez M, Rademakers R. How do C9ORF72 repeat expansions cause amyotrophic lateral sclerosis and frontotemporal dementia: can we learn from other noncoding repeat expansion disorders? Current opinion in neurology. 2012 Dec:25(6):689-700. doi: 10.1097/WCO.0b013e32835a3efb. Epub [PubMed PMID: 23160421]
Level 3 (low-level) evidenceChen S, Sayana P, Zhang X, Le W. Genetics of amyotrophic lateral sclerosis: an update. Molecular neurodegeneration. 2013 Aug 13:8():28. doi: 10.1186/1750-1326-8-28. Epub 2013 Aug 13 [PubMed PMID: 23941283]
Level 3 (low-level) evidenceChiò A, Mazzini L, D'Alfonso S, Corrado L, Canosa A, Moglia C, Manera U, Bersano E, Brunetti M, Barberis M, Veldink JH, van den Berg LH, Pearce N, Sproviero W, McLaughlin R, Vajda A, Hardiman O, Rooney J, Mora G, Calvo A, Al-Chalabi A. The multistep hypothesis of ALS revisited: The role of genetic mutations. Neurology. 2018 Aug 14:91(7):e635-e642. doi: 10.1212/WNL.0000000000005996. Epub 2018 Jul 25 [PubMed PMID: 30045958]
LoRusso E, Hickman JJ, Guo X. Ion channel dysfunction and altered motoneuron excitability in ALS. Neurological disorders & epilepsy journal. 2019:3(2):. pii: 124. Epub 2019 Jul 30 [PubMed PMID: 32313901]
Volk AE, Weishaupt JH, Andersen PM, Ludolph AC, Kubisch C. Current knowledge and recent insights into the genetic basis of amyotrophic lateral sclerosis. Medizinische Genetik : Mitteilungsblatt des Berufsverbandes Medizinische Genetik e.V. 2018:30(2):252-258. doi: 10.1007/s11825-018-0185-3. Epub 2018 Jul 13 [PubMed PMID: 30220791]
Bozzoni V, Pansarasa O, Diamanti L, Nosari G, Cereda C, Ceroni M. Amyotrophic lateral sclerosis and environmental factors. Functional neurology. 2016 Jan-Mar:31(1):7-19 [PubMed PMID: 27027889]
Alonso A, Logroscino G, Jick SS, Hernán MA. Incidence and lifetime risk of motor neuron disease in the United Kingdom: a population-based study. European journal of neurology. 2009 Jun:16(6):745-51 [PubMed PMID: 19475756]
Level 2 (mid-level) evidenceGundogdu B, Al-Lahham T, Kadlubar F, Spencer H, Rudnicki SA. Racial differences in motor neuron disease. Amyotrophic lateral sclerosis & frontotemporal degeneration. 2014 Mar:15(1-2):114-8. doi: 10.3109/21678421.2013.837930. Epub 2013 Sep 25 [PubMed PMID: 24067242]
Level 2 (mid-level) evidenceBoylan K. Familial Amyotrophic Lateral Sclerosis. Neurologic clinics. 2015 Nov:33(4):807-30. doi: 10.1016/j.ncl.2015.07.001. Epub 2015 Sep 8 [PubMed PMID: 26515623]
Wijesekera LC, Leigh PN. Amyotrophic lateral sclerosis. Orphanet journal of rare diseases. 2009 Feb 3:4():3. doi: 10.1186/1750-1172-4-3. Epub 2009 Feb 3 [PubMed PMID: 19192301]
Liewluck T, Saperstein DS. Progressive Muscular Atrophy. Neurologic clinics. 2015 Nov:33(4):761-73. doi: 10.1016/j.ncl.2015.07.005. Epub [PubMed PMID: 26515620]
Donnelly CJ, Grima JC, Sattler R. Aberrant RNA homeostasis in amyotrophic lateral sclerosis: potential for new therapeutic targets? Neurodegenerative disease management. 2014:4(6):417-37. doi: 10.2217/nmt.14.36. Epub [PubMed PMID: 25531686]
Level 3 (low-level) evidenceLiu YJ, Tsai PY, Chern Y. Energy Homeostasis and Abnormal RNA Metabolism in Amyotrophic Lateral Sclerosis. Frontiers in cellular neuroscience. 2017:11():126. doi: 10.3389/fncel.2017.00126. Epub 2017 May 4 [PubMed PMID: 28522961]
van den Bos MAJ, Geevasinga N, Higashihara M, Menon P, Vucic S. Pathophysiology and Diagnosis of ALS: Insights from Advances in Neurophysiological Techniques. International journal of molecular sciences. 2019 Jun 10:20(11):. doi: 10.3390/ijms20112818. Epub 2019 Jun 10 [PubMed PMID: 31185581]
Level 3 (low-level) evidenceMukherjee A, Chakravarty A. Spasticity mechanisms - for the clinician. Frontiers in neurology. 2010:1():149. doi: 10.3389/fneur.2010.00149. Epub 2010 Dec 17 [PubMed PMID: 21206767]
Prasad A, Bharathi V, Sivalingam V, Girdhar A, Patel BK. Molecular Mechanisms of TDP-43 Misfolding and Pathology in Amyotrophic Lateral Sclerosis. Frontiers in molecular neuroscience. 2019:12():25. doi: 10.3389/fnmol.2019.00025. Epub 2019 Feb 14 [PubMed PMID: 30837838]
Blokhuis AM, Groen EJ, Koppers M, van den Berg LH, Pasterkamp RJ. Protein aggregation in amyotrophic lateral sclerosis. Acta neuropathologica. 2013 Jun:125(6):777-94. doi: 10.1007/s00401-013-1125-6. Epub 2013 May 15 [PubMed PMID: 23673820]
McAlary L, Plotkin SS, Yerbury JJ, Cashman NR. Prion-Like Propagation of Protein Misfolding and Aggregation in Amyotrophic Lateral Sclerosis. Frontiers in molecular neuroscience. 2019:12():262. doi: 10.3389/fnmol.2019.00262. Epub 2019 Nov 1 [PubMed PMID: 31736708]
Saberi S, Stauffer JE, Schulte DJ, Ravits J. Neuropathology of Amyotrophic Lateral Sclerosis and Its Variants. Neurologic clinics. 2015 Nov:33(4):855-76. doi: 10.1016/j.ncl.2015.07.012. Epub [PubMed PMID: 26515626]
Jensen L, Jørgensen LH, Bech RD, Frandsen U, Schrøder HD. Skeletal Muscle Remodelling as a Function of Disease Progression in Amyotrophic Lateral Sclerosis. BioMed research international. 2016:2016():5930621. doi: 10.1155/2016/5930621. Epub 2016 Apr 18 [PubMed PMID: 27195289]
Nijssen J, Comley LH, Hedlund E. Motor neuron vulnerability and resistance in amyotrophic lateral sclerosis. Acta neuropathologica. 2017 Jun:133(6):863-885. doi: 10.1007/s00401-017-1708-8. Epub 2017 Apr 13 [PubMed PMID: 28409282]
McDermott CJ, Shaw PJ. Diagnosis and management of motor neurone disease. BMJ (Clinical research ed.). 2008 Mar 22:336(7645):658-62. doi: 10.1136/bmj.39493.511759.BE. Epub [PubMed PMID: 18356234]
Turner MR, Talbot K. Mimics and chameleons in motor neurone disease. Practical neurology. 2013 Jun:13(3):153-64. doi: 10.1136/practneurol-2013-000557. Epub 2013 Apr 24 [PubMed PMID: 23616620]
Barohn RJ, Dimachkie MM, Jackson CE. A pattern recognition approach to patients with a suspected myopathy. Neurologic clinics. 2014 Aug:32(3):569-93, vii. doi: 10.1016/j.ncl.2014.04.008. Epub [PubMed PMID: 25037080]
Ngo ST, Mi JD, Henderson RD, McCombe PA, Steyn FJ. Exploring targets and therapies for amyotrophic lateral sclerosis: current insights into dietary interventions. Degenerative neurological and neuromuscular disease. 2017:7():95-108. doi: 10.2147/DNND.S120607. Epub 2017 Jul 25 [PubMed PMID: 30050381]
Ferri A, Coccurello R. What is "Hyper" in the ALS Hypermetabolism? Mediators of inflammation. 2017:2017():7821672. doi: 10.1155/2017/7821672. Epub 2017 Sep 7 [PubMed PMID: 29081604]
Tomik B, Guiloff RJ. Dysarthria in amyotrophic lateral sclerosis: A review. Amyotrophic lateral sclerosis : official publication of the World Federation of Neurology Research Group on Motor Neuron Diseases. 2010:11(1-2):4-15. doi: 10.3109/17482960802379004. Epub [PubMed PMID: 20184513]
Niedermeyer S, Murn M, Choi PJ. Respiratory Failure in Amyotrophic Lateral Sclerosis. Chest. 2019 Feb:155(2):401-408. doi: 10.1016/j.chest.2018.06.035. Epub 2018 Jul 7 [PubMed PMID: 29990478]
Garcia-Willingham NE, Roach AR, Kasarskis EJ, Segerstrom SC. Self-Regulation and Executive Functioning as Related to Survival in Motor Neuron Disease: Preliminary Findings. Psychosomatic medicine. 2018 Sep:80(7):665-672. doi: 10.1097/PSY.0000000000000602. Epub [PubMed PMID: 29771729]
Ferrari R, Kapogiannis D, Huey ED, Momeni P. FTD and ALS: a tale of two diseases. Current Alzheimer research. 2011 May:8(3):273-94 [PubMed PMID: 21222600]
Caga J, Hsieh S, Lillo P, Dudley K, Mioshi E. The Impact of Cognitive and Behavioral Symptoms on ALS Patients and Their Caregivers. Frontiers in neurology. 2019:10():192. doi: 10.3389/fneur.2019.00192. Epub 2019 Mar 11 [PubMed PMID: 30915018]
Burke T, Pinto-Grau M, Lonergan K, Bede P, O'Sullivan M, Heverin M, Vajda A, McLaughlin RL, Pender N, Hardiman O. A Cross-sectional population-based investigation into behavioral change in amyotrophic lateral sclerosis: subphenotypes, staging, cognitive predictors, and survival. Annals of clinical and translational neurology. 2017 May:4(5):305-317. doi: 10.1002/acn3.407. Epub 2017 Apr 11 [PubMed PMID: 28491898]
Level 2 (mid-level) evidencePiccione EA, Sletten DM, Staff NP, Low PA. Autonomic system and amyotrophic lateral sclerosis. Muscle & nerve. 2015 May:51(5):676-9. doi: 10.1002/mus.24457. Epub 2015 Mar 31 [PubMed PMID: 25211238]
Level 2 (mid-level) evidenceGarg N, Park SB, Vucic S, Yiannikas C, Spies J, Howells J, Huynh W, Matamala JM, Krishnan AV, Pollard JD, Cornblath DR, Reilly MM, Kiernan MC. Differentiating lower motor neuron syndromes. Journal of neurology, neurosurgery, and psychiatry. 2017 Jun:88(6):474-483. doi: 10.1136/jnnp-2016-313526. Epub 2016 Dec 21 [PubMed PMID: 28003344]
Kim WK, Liu X, Sandner J, Pasmantier M, Andrews J, Rowland LP, Mitsumoto H. Study of 962 patients indicates progressive muscular atrophy is a form of ALS. Neurology. 2009 Nov 17:73(20):1686-92. doi: 10.1212/WNL.0b013e3181c1dea3. Epub [PubMed PMID: 19917992]
Level 2 (mid-level) evidencede Vries BS, Rustemeijer LMM, Bakker LA, Schröder CD, Veldink JH, van den Berg LH, Nijboer TCW, van Es MA. Cognitive and behavioural changes in PLS and PMA:challenging the concept of restricted phenotypes. Journal of neurology, neurosurgery, and psychiatry. 2019 Feb:90(2):141-147. doi: 10.1136/jnnp-2018-318788. Epub 2018 Aug 3 [PubMed PMID: 30076267]
Chen H, Richard M, Sandler DP, Umbach DM, Kamel F. Head injury and amyotrophic lateral sclerosis. American journal of epidemiology. 2007 Oct 1:166(7):810-6 [PubMed PMID: 17641152]
Level 2 (mid-level) evidenceBowen LN, Tyagi R, Li W, Alfahad T, Smith B, Wright M, Singer EJ, Nath A. HIV-associated motor neuron disease: HERV-K activation and response to antiretroviral therapy. Neurology. 2016 Oct 25:87(17):1756-1762 [PubMed PMID: 27664983]
Tao QQ, Wu ZY. Amyotrophic Lateral Sclerosis: Precise Diagnosis and Individualized Treatment. Chinese medical journal. 2017 Oct 5:130(19):2269-2272. doi: 10.4103/0366-6999.215323. Epub [PubMed PMID: 28937029]
Benbrika S, Desgranges B, Eustache F, Viader F. Cognitive, Emotional and Psychological Manifestations in Amyotrophic Lateral Sclerosis at Baseline and Overtime: A Review. Frontiers in neuroscience. 2019:13():951. doi: 10.3389/fnins.2019.00951. Epub 2019 Sep 10 [PubMed PMID: 31551700]
Bali T, Miller TM. Management of amyotrophic lateral sclerosis. Missouri medicine. 2013 Sep-Oct:110(5):417-21 [PubMed PMID: 24279194]
Benny R, Shetty K. The split hand sign. Annals of Indian Academy of Neurology. 2012 Jul:15(3):175-6. doi: 10.4103/0972-2327.99700. Epub [PubMed PMID: 22919187]
Singh N, Ray S, Srivastava A. Clinical Mimickers of Amyotrophic Lateral Sclerosis-Conditions We Cannot Afford to Miss. Annals of Indian Academy of Neurology. 2018 Jul-Sep:21(3):173-178. doi: 10.4103/aian.AIAN_491_17. Epub [PubMed PMID: 30258257]
Finegan E, Chipika RH, Li Hi Shing S, Hardiman O, Bede P. Pathological Crying and Laughing in Motor Neuron Disease: Pathobiology, Screening, Intervention. Frontiers in neurology. 2019:10():260. doi: 10.3389/fneur.2019.00260. Epub 2019 Mar 21 [PubMed PMID: 30949121]
Williams TL. Motor neurone disease: diagnostic pitfalls. Clinical medicine (London, England). 2013 Feb:13(1):97-100 [PubMed PMID: 23472505]
Lee Y, Lee BH, Yip W, Chou P, Yip BS. Neurofilament Proteins as Prognostic Biomarkers in Neurological Disorders. Current pharmaceutical design. 2020:25(43):4560-4569. doi: 10.2174/1381612825666191210154535. Epub [PubMed PMID: 31820696]
Verber NS, Shepheard SR, Sassani M, McDonough HE, Moore SA, Alix JJP, Wilkinson ID, Jenkins TM, Shaw PJ. Biomarkers in Motor Neuron Disease: A State of the Art Review. Frontiers in neurology. 2019:10():291. doi: 10.3389/fneur.2019.00291. Epub 2019 Apr 3 [PubMed PMID: 31001186]
Rammes G, Zieglgänsberger W, Parsons CG. The fraction of activated N-methyl-D-aspartate receptors during synaptic transmission remains constant in the presence of the glutamate release inhibitor riluzole. Journal of neural transmission (Vienna, Austria : 1996). 2008 Aug:115(8):1119-26. doi: 10.1007/s00702-008-0059-y. Epub 2008 May 21 [PubMed PMID: 18493706]
Level 3 (low-level) evidenceHinchcliffe M, Smith A. Riluzole: real-world evidence supports significant extension of median survival times in patients with amyotrophic lateral sclerosis. Degenerative neurological and neuromuscular disease. 2017:7():61-70. doi: 10.2147/DNND.S135748. Epub 2017 May 29 [PubMed PMID: 30050378]
Dharmadasa T, Kiernan MC. Riluzole, disease stage and survival in ALS. The Lancet. Neurology. 2018 May:17(5):385-386. doi: 10.1016/S1474-4422(18)30091-7. Epub 2018 Mar 7 [PubMed PMID: 29525493]
Miller RG, Mitchell JD, Lyon M, Moore DH. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). The Cochrane database of systematic reviews. 2002:(2):CD001447 [PubMed PMID: 12076411]
Level 1 (high-level) evidenceMiller RG, Mitchell JD, Moore DH. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). The Cochrane database of systematic reviews. 2012 Mar 14:2012(3):CD001447. doi: 10.1002/14651858.CD001447.pub3. Epub 2012 Mar 14 [PubMed PMID: 22419278]
Level 1 (high-level) evidenceFalcão de Campos C, de Carvalho M. Riluzole-induced recurrent pancreatitis. Journal of clinical neuroscience : official journal of the Neurosurgical Society of Australasia. 2017 Nov:45():153-154. doi: 10.1016/j.jocn.2017.08.032. Epub 2017 Sep 1 [PubMed PMID: 28867362]
Siniscalchi A. [Tolerability of riluzole: a review of the literature]. La Clinica terapeutica. 2004 Jan:155(1):25-8 [PubMed PMID: 15147078]
Bensimon G, Doble A. The tolerability of riluzole in the treatment of patients with amyotrophic lateral sclerosis. Expert opinion on drug safety. 2004 Nov:3(6):525-34 [PubMed PMID: 15500412]
Level 3 (low-level) evidenceTakei K, Watanabe K, Yuki S, Akimoto M, Sakata T, Palumbo J. Edaravone and its clinical development for amyotrophic lateral sclerosis. Amyotrophic lateral sclerosis & frontotemporal degeneration. 2017 Oct:18(sup1):5-10. doi: 10.1080/21678421.2017.1353101. Epub [PubMed PMID: 28872907]
Turnbull J. Is edaravone harmful? (A placebo is not a control). Amyotrophic lateral sclerosis & frontotemporal degeneration. 2018 Nov:19(7-8):477-482. doi: 10.1080/21678421.2018.1517179. Epub 2018 Oct 29 [PubMed PMID: 30373406]
Sawada H. Clinical efficacy of edaravone for the treatment of amyotrophic lateral sclerosis. Expert opinion on pharmacotherapy. 2017 May:18(7):735-738. doi: 10.1080/14656566.2017.1319937. Epub [PubMed PMID: 28406335]
Level 3 (low-level) evidenceStatland JM, Barohn RJ, Dimachkie MM, Floeter MK, Mitsumoto H. Primary Lateral Sclerosis. Neurologic clinics. 2015 Nov:33(4):749-60. doi: 10.1016/j.ncl.2015.07.007. Epub 2015 Sep 8 [PubMed PMID: 26515619]
Balci BP. Spasticity Measurement. Noro psikiyatri arsivi. 2018:55(Suppl 1):S49-S53. doi: 10.29399/npa.23339. Epub [PubMed PMID: 30692856]
Hardiman O, van den Berg LH, Kiernan MC. Clinical diagnosis and management of amyotrophic lateral sclerosis. Nature reviews. Neurology. 2011 Oct 11:7(11):639-49. doi: 10.1038/nrneurol.2011.153. Epub 2011 Oct 11 [PubMed PMID: 21989247]
Malak M, Barzegar M. Baclofen Induced Encephalopathy in a 6-Year-Old Boy with Advanced Renal Failure. Iranian journal of child neurology. 2015 Spring:9(2):61-3 [PubMed PMID: 26221166]
Medical Advisory Secretariat. Intrathecal baclofen pump for spasticity: an evidence-based analysis. Ontario health technology assessment series. 2005:5(7):1-93 [PubMed PMID: 23074476]
Young CA, Ellis C, Johnson J, Sathasivam S, Pih N. Treatment for sialorrhea (excessive saliva) in people with motor neuron disease/amyotrophic lateral sclerosis. The Cochrane database of systematic reviews. 2011 May 11:(5):CD006981. doi: 10.1002/14651858.CD006981.pub2. Epub 2011 May 11 [PubMed PMID: 21563158]
Level 1 (high-level) evidenceDorst J, Ludolph AC, Huebers A. Disease-modifying and symptomatic treatment of amyotrophic lateral sclerosis. Therapeutic advances in neurological disorders. 2018:11():1756285617734734. doi: 10.1177/1756285617734734. Epub 2017 Oct 9 [PubMed PMID: 29399045]
Level 3 (low-level) evidenceBrettschneider J, Kurent J, Ludolph A. Drug therapy for pain in amyotrophic lateral sclerosis or motor neuron disease. The Cochrane database of systematic reviews. 2013 Jun 5:2013(6):CD005226. doi: 10.1002/14651858.CD005226.pub3. Epub 2013 Jun 5 [PubMed PMID: 23740607]
Level 1 (high-level) evidenceKiousi V, Arnaoutoglou M, Printza A. Speech and language intervention for language impairment in patients in the FTD-ALS spectrum. Hellenic journal of nuclear medicine. 2019 Jan-Apr:22 Suppl():133-146 [PubMed PMID: 30877731]
De Luca C, Virtuoso A, Maggio N, Papa M. Neuro-Coagulopathy: Blood Coagulation Factors in Central Nervous System Diseases. International journal of molecular sciences. 2017 Oct 12:18(10):. doi: 10.3390/ijms18102128. Epub 2017 Oct 12 [PubMed PMID: 29023416]
Mioshi E, Caga J, Lillo P, Hsieh S, Ramsey E, Devenney E, Hornberger M, Hodges JR, Kiernan MC. Neuropsychiatric changes precede classic motor symptoms in ALS and do not affect survival. Neurology. 2014 Jan 14:82(2):149-55. doi: 10.1212/WNL.0000000000000023. Epub 2013 Dec 11 [PubMed PMID: 24336140]
Floeter MK, Katipally R, Kim MP, Schanz O, Stephen M, Danielian L, Wu T, Huey ED, Meoded A. Impaired corticopontocerebellar tracts underlie pseudobulbar affect in motor neuron disorders. Neurology. 2014 Aug 12:83(7):620-7. doi: 10.1212/WNL.0000000000000693. Epub 2014 Jul 9 [PubMed PMID: 25008395]
Level 2 (mid-level) evidenceRoos E, Mariosa D, Ingre C, Lundholm C, Wirdefeldt K, Roos PM, Fang F. Depression in amyotrophic lateral sclerosis. Neurology. 2016 Jun 14:86(24):2271-7. doi: 10.1212/WNL.0000000000002671. Epub 2016 Apr 22 [PubMed PMID: 27164661]
Ahmed A, Simmons Z. Pseudobulbar affect: prevalence and management. Therapeutics and clinical risk management. 2013:9():483-9. doi: 10.2147/TCRM.S53906. Epub 2013 Nov 29 [PubMed PMID: 24348042]
Nichols NL, Van Dyke J, Nashold L, Satriotomo I, Suzuki M, Mitchell GS. Ventilatory control in ALS. Respiratory physiology & neurobiology. 2013 Nov 1:189(2):429-37. doi: 10.1016/j.resp.2013.05.016. Epub 2013 May 18 [PubMed PMID: 23692930]
Level 3 (low-level) evidencede Carvalho M, Swash M, Pinto S. Diaphragmatic Neurophysiology and Respiratory Markers in ALS. Frontiers in neurology. 2019:10():143. doi: 10.3389/fneur.2019.00143. Epub 2019 Feb 21 [PubMed PMID: 30846968]
Lechtzin N, Wiener CM, Shade DM, Clawson L, Diette GB. Spirometry in the supine position improves the detection of diaphragmatic weakness in patients with amyotrophic lateral sclerosis. Chest. 2002 Feb:121(2):436-42 [PubMed PMID: 11834654]
Lacombe M, Del Amo Castrillo L, Boré A, Chapeau D, Horvat E, Vaugier I, Lejaille M, Orlikowski D, Prigent H, Lofaso F. Comparison of three cough-augmentation techniques in neuromuscular patients: mechanical insufflation combined with manually assisted cough, insufflation-exsufflation alone and insufflation-exsufflation combined with manually assisted cough. Respiration; international review of thoracic diseases. 2014:88(3):215-22. doi: 10.1159/000364911. Epub 2014 Aug 21 [PubMed PMID: 25171575]
Level 1 (high-level) evidenceCheng HWB, Chan KY, Chung YKJ, Choi CW, Chan CH, Cheng SC, Chan WH, Fung KS, Wong KY, Chan OMI, Man CW. Supportive & palliative interventions in motor neurone disease: what we know from current literature? Annals of palliative medicine. 2018 Jul:7(3):320-331. doi: 10.21037/apm.2017.10.01. Epub 2017 Oct 31 [PubMed PMID: 29156920]
Sancho J, Martínez D, Bures E, Díaz JL, Ponz A, Servera E. Bulbar impairment score and survival of stable amyotrophic lateral sclerosis patients after noninvasive ventilation initiation. ERJ open research. 2018 Apr:4(2):. pii: 00159-2017. doi: 10.1183/23120541.00159-2017. Epub 2018 Apr 16 [PubMed PMID: 29670892]
Bach JR, Bianchi C, Aufiero E. Oximetry and indications for tracheotomy for amyotrophic lateral sclerosis. Chest. 2004 Nov:126(5):1502-7 [PubMed PMID: 15539719]
Level 2 (mid-level) evidenceLe Pimpec-Barthes F, Legras A, Arame A, Pricopi C, Boucherie JC, Badia A, Panzini CM. Diaphragm pacing: the state of the art. Journal of thoracic disease. 2016 Apr:8(Suppl 4):S376-86. doi: 10.21037/jtd.2016.03.97. Epub [PubMed PMID: 27195135]
Gibbons C, Pagnini F, Friede T, Young CA. Treatment of fatigue in amyotrophic lateral sclerosis/motor neuron disease. The Cochrane database of systematic reviews. 2018 Jan 2:1(1):CD011005. doi: 10.1002/14651858.CD011005.pub2. Epub 2018 Jan 2 [PubMed PMID: 29293261]
Level 1 (high-level) evidenceKaram CY, Paganoni S, Joyce N, Carter GT, Bedlack R. Palliative Care Issues in Amyotrophic Lateral Sclerosis: An Evidenced-Based Review. The American journal of hospice & palliative care. 2016 Feb:33(1):84-92. doi: 10.1177/1049909114548719. Epub 2014 Sep 8 [PubMed PMID: 25202033]
Zwicker J, Qureshi D, Talarico R, Bourque P, Scott M, Chin-Yee N, Tanuseputro P. Dying of amyotrophic lateral sclerosis: Health care use and cost in the last year of life. Neurology. 2019 Dec 3:93(23):e2083-e2093. doi: 10.1212/WNL.0000000000008582. Epub 2019 Oct 31 [PubMed PMID: 31672715]
Mazutti SR, Nascimento AF, Fumis RR. Limitation to Advanced Life Support in patients admitted to intensive care unit with integrated palliative care. Revista Brasileira de terapia intensiva. 2016 Sep:28(3):294-300 [PubMed PMID: 27626949]
Bello-Haas VD. Physical therapy for individuals with amyotrophic lateral sclerosis: current insights. Degenerative neurological and neuromuscular disease. 2018:8():45-54. doi: 10.2147/DNND.S146949. Epub 2018 Jul 16 [PubMed PMID: 30890895]
Beukelman D, Fager S, Nordness A. Communication Support for People with ALS. Neurology research international. 2011:2011():714693. doi: 10.1155/2011/714693. Epub 2011 Apr 14 [PubMed PMID: 21603029]
Cappella M, Ciotti C, Cohen-Tannoudji M, Biferi MG. Gene Therapy for ALS-A Perspective. International journal of molecular sciences. 2019 Sep 6:20(18):. doi: 10.3390/ijms20184388. Epub 2019 Sep 6 [PubMed PMID: 31500113]
Level 3 (low-level) evidenceLunn JS, Sakowski SA, Feldman EL. Concise review: Stem cell therapies for amyotrophic lateral sclerosis: recent advances and prospects for the future. Stem cells (Dayton, Ohio). 2014 May:32(5):1099-109. doi: 10.1002/stem.1628. Epub [PubMed PMID: 24448926]
Level 3 (low-level) evidenceLawson VH, Arnold WD. Multifocal motor neuropathy: a review of pathogenesis, diagnosis, and treatment. Neuropsychiatric disease and treatment. 2014:10():567-76. doi: 10.2147/NDT.S39592. Epub 2014 Apr 5 [PubMed PMID: 24741315]
Feinberg JH, Radecki J. Parsonage-turner syndrome. HSS journal : the musculoskeletal journal of Hospital for Special Surgery. 2010 Sep:6(2):199-205. doi: 10.1007/s11420-010-9176-x. Epub 2010 Jul 30 [PubMed PMID: 21886536]
Aundhakar SC, Mahajan SK, Chhapra DA. Hirayama's Disease: A Rare Clinical Variant of Amyotrophic Lateral Sclerosis. Advanced biomedical research. 2017:6():95. doi: 10.4103/2277-9175.211797. Epub 2017 Jul 28 [PubMed PMID: 28828346]
Breza M, Koutsis G. Kennedy's disease (spinal and bulbar muscular atrophy): a clinically oriented review of a rare disease. Journal of neurology. 2019 Mar:266(3):565-573. doi: 10.1007/s00415-018-8968-7. Epub 2018 Jul 13 [PubMed PMID: 30006721]
Dimachkie MM, Barohn RJ. Inclusion body myositis. Neurologic clinics. 2014 Aug:32(3):629-46, vii. doi: 10.1016/j.ncl.2014.04.001. Epub 2014 Jun 6 [PubMed PMID: 25037082]
Chiò A, Logroscino G, Hardiman O, Swingler R, Mitchell D, Beghi E, Traynor BG, Eurals Consortium. Prognostic factors in ALS: A critical review. Amyotrophic lateral sclerosis : official publication of the World Federation of Neurology Research Group on Motor Neuron Diseases. 2009 Oct-Dec:10(5-6):310-23. doi: 10.3109/17482960802566824. Epub [PubMed PMID: 19922118]
Level 2 (mid-level) evidenceChiò A, Hammond ER, Mora G, Bonito V, Filippini G. Development and evaluation of a clinical staging system for amyotrophic lateral sclerosis. Journal of neurology, neurosurgery, and psychiatry. 2015 Jan:86(1):38-44. doi: 10.1136/jnnp-2013-306589. Epub 2013 Dec 13 [PubMed PMID: 24336810]
Crockford C, Newton J, Lonergan K, Chiwera T, Booth T, Chandran S, Colville S, Heverin M, Mays I, Pal S, Pender N, Pinto-Grau M, Radakovic R, Shaw CE, Stephenson L, Swingler R, Vajda A, Al-Chalabi A, Hardiman O, Abrahams S. ALS-specific cognitive and behavior changes associated with advancing disease stage in ALS. Neurology. 2018 Oct 9:91(15):e1370-e1380. doi: 10.1212/WNL.0000000000006317. Epub 2018 Sep 12 [PubMed PMID: 30209236]
Jackson CE, McVey AL, Rudnicki S, Dimachkie MM, Barohn RJ. Symptom Management and End-of-Life Care in Amyotrophic Lateral Sclerosis. Neurologic clinics. 2015 Nov:33(4):889-908. doi: 10.1016/j.ncl.2015.07.010. Epub [PubMed PMID: 26515628]
Cichero JAY. Age-Related Changes to Eating and Swallowing Impact Frailty: Aspiration, Choking Risk, Modified Food Texture and Autonomy of Choice. Geriatrics (Basel, Switzerland). 2018 Oct 12:3(4):. doi: 10.3390/geriatrics3040069. Epub 2018 Oct 12 [PubMed PMID: 31011104]
Chen JH, Wu SC, Chen HJ, Kao CH, Tseng CH, Tsai CH. Risk of developing pressure sore in amyotrophic lateral sclerosis patients - a nationwide cohort study. Journal of the European Academy of Dermatology and Venereology : JEADV. 2018 Sep:32(9):1589-1596. doi: 10.1111/jdv.14911. Epub 2018 May 1 [PubMed PMID: 29512203]
Gladman M, Dehaan M, Pinto H, Geerts W, Zinman L. Venous thromboembolism in amyotrophic lateral sclerosis: a prospective study. Neurology. 2014 May 13:82(19):1674-7. doi: 10.1212/WNL.0000000000000405. Epub 2014 Apr 11 [PubMed PMID: 24727309]
Smithson WH. Integrating the algorithm into community practice. Neurology. 1999:53(8 Suppl 5):S63-6; discussion S67-71 [PubMed PMID: 10560642]
Hogden A, Foley G, Henderson RD, James N, Aoun SM. Amyotrophic lateral sclerosis: improving care with a multidisciplinary approach. Journal of multidisciplinary healthcare. 2017:10():205-215. doi: 10.2147/JMDH.S134992. Epub 2017 May 19 [PubMed PMID: 28579792]
Oliver DJ. Palliative care in motor neurone disease: where are we now? Palliative care. 2019:12():1178224218813914. doi: 10.1177/1178224218813914. Epub 2019 Jan 21 [PubMed PMID: 30718958]