Introduction
Microscopic polyangiitis (MPA) is a small vessel necrotizing vasculitis that falls within a larger spectrum of disorders known as antineutrophil cytoplasmic antibody (ANCA)–associated vasculitides. This group also includes granulomatosis with polyangiitis (GPA), MPA, eosinophilic granulomatosis with polyangiitis (EGPA or Churg-Strauss disease), and renal limited vasculitis. This classification of vasculitides is based on the type of vessels involved and the underlying etiology, as established by the International Chapel Hill Consensus Conference on the Nomenclature of Vasculitides (CHCC 2012).[1]
The term "microscopic polyangiitis" was first described as "microscopic polyarteritis" and introduced in the literature by Davson in 1948 to describe the pattern of glomerulonephritis observed in patients with polyarteritis nodosa. This condition was later described as a pattern of necrotizing vasculitis without immune complex deposition, primarily affecting small vessels such as the capillaries, venules, and arterioles. The disease commonly involves glomerulonephritis, pulmonary capillaritis, and other systemic capillary beds, with considerable overlap with GPA.
The absence of granulomatous inflammation in the upper respiratory tract and the presence of pulmonary capillaritis help differentiate MPA from GPA. MPA is more likely to have renal involvement than GPA. MPA is part of a group of disorders known as pulmonary-renal syndrome, which also includes GPA, Goodpasture disease, and systemic lupus erythematosus.[2][3] While ANCAs have a central role in the pathogenesis of ANCA-associated vasculitis, they are neither necessary nor sufficient to cause the disease patterns of ANCA-associated vasculitides. Clinical symptoms and biopsy results are equally important for diagnosis. The role of ANCAs and the distinctions between the various ANCA-associated vasculitides are also discussed in this activity.
Please refer to the following StatPearls' companion resources for further information:
- Granulomatosis With Polyangiitis
- Eosinophilic Granulomatosis With Polyangiitis
- ANCA-Associated Vasculitis
- Rapidly Progressive Glomerulonephritis
- Antineutrophil Cytoplasmic Antibodies ANCA Test
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
The etiopathogenesis of MPA and related vasculitides has largely been attributed to ANCAs, which are host-derived autoantibodies targeting normally shielded neutrophilic antigens. These antibodies react against primary granules in neutrophils and monocytes. The formation of these antibodies has been hypothesized to be a 2-step process.[4] In the first step, neutrophils are exposed to inflammatory cytokines, leading to the surface expression of antigens such as myeloperoxidase (MPO). Next, predisposing genetic, environmental, and other factors lead to ANCA production. In the second step, these ANCAs damage the host vasculature by triggering inflammation and cytokine production.[5]
Although anti-MPO is the primary ANCA associated with MPA, anti-proteinase 3 (PR3) can also be present in some cases. Estimates suggest that approximately 70% of patients with MPA are anti-MPO–positive, 20% are anti-PR3–positive, and 10% are ANCA-negative.[6] Less specific ANCA antigens include cathepsin G, lactoferrin, elastase, defensin, α-enolase, moesin, leukocidin, and bactericidal permeability increasing protein, although their pathogenicity is considered to be low.[6]
Lysosomal-associated membrane protein 2 (LAMP-2) antibodies are of particular interest in ANCA-associated vasculitis because they are present in patients with both anti-MPO and anti-PR3 antibodies and may contribute to the disease pathology. Moreover, LAMP-2 antibodies have been detected in patients with active ANCA-associated vasculitis who are negative for MPO and PR3 antibodies. Some evidence suggests that LAMP-2 antibodies may correlate better with disease activity than anti-MPO or anti-PR3 antibodies.[7][8]
About 90% of patients with active MPA test positive for ANCA; however, not all cases of ANCA positivity correlate with active disease. This suggests that other factors may also contribute to the disease's etiopathogenesis. These factors include (but are not limited to):
- Infectious causes: The considerable overlap between the clinical presentations of various infectious processes and MPA leads to this implication. Chronic nasal carriage of Staphylococcus aureus has been proposed as a potential contributing factor.
- Genetic factors: Recent genome-wide studies conducted in Europe have identified genes such as HLA-DP, HLA-DR3, and alpha-1 antitrypsin as potentially involved in the pathogenesis of ANCA-associated vasculitis.[9][10]
- Environmental factors: UV light, air pollution, and silica exposure have also been suggested as possible triggers for ANCA-associated vasculitis.[8]
- Drugs: Certain medications, including hydralazine, thionamides, sulfasalazine, and minocycline, have been observed to be associated with the incidence of ANCA-associated vasculitis.[10][11] This form of vasculitis is often anti-MPO positive and is covered more extensively in the StatPearls' companion resource, "ANCA-Associated Vasculitis."
Epidemiology
Due to the recent differentiation of MPA from other ANCA-associated vasculitides, comprehensive demographic data for its prevalence in the US population are not widely available. A study conducted in Rochester, Minnesota, over a 20-year period estimated the annual incidence of ANCA-associated vasculitis at 3.3 per 100,000, with a prevalence of 42.1 per 100,000. The incidence of GPA and MPA was approximately 1.5 per 100,000.[12] An increase in the prevalence of MPA over the past 2 decades is likely due to improved recognition and the availability of ANCA testing. The global incidence of MPA is estimated to be 20 per million.[6]
Evidence suggests that MPA is more common in China and Japan, whereas GPA is more prevalent among populations of European descent.[6] The median age of onset for MPA is between 50 and 60. The disease appears to have about equal prevalence between males and females.[8]
Pathophysiology
While anti-MPO is the primary ANCA associated with MPA, anti-PR3 is also present in some cases. Notably, it is estimated that in MPA, approximately 70% of patients are anti-MPO–positive, 20% are anti-PR3–positive, and 10% are ANCA-negative.[6]
The clinical manifestations of MPA result from the activation of primed neutrophils and MPO-ANCA, which interact with receptors on the endothelial surface.[13] This process leads to a range of symptoms affecting the renal, pulmonary, and other capillary beds. Patients may present with an insidious onset of systemic signs such as fever, malaise, or weight loss; however, the onset is more commonly acute, with complaints of arthralgia and flu-like symptoms. MPA is a small vessel vasculitis characterized by pauci-immune, necrotizing inflammation without clinical or pathological evidence of granulomatous involvement, often leading to necrosis and bleeding.[6]
Renal manifestations are extremely common in MPA, with 80% to 90% of individuals experiencing some form of glomerulonephritis at onset or as the disease progresses. The most common renal manifestation is a "pauci-immune" form of rapidly progressive glomerulonephritis. Clinical presentations can range from asymptomatic hematuria, sub-nephrotic proteinuria, and elevated serum creatinine to overt renal failure. Pulmonary manifestations often include alveolar hemorrhage, which may sometimes be the first presenting symptom of the disease.[14]
Histopathology
Histopathological evidence of vasculitis is the gold standard for confirming the diagnosis of MPA and other ANCA-associated vasculitides. The most commonly sampled tissues are from the kidneys, skin, and lungs. Diffuse capillaritis is a typical pulmonary finding in patients with MPA, distinguishing it from GPA, which is characterized by the presence of granulomatous lesions. Skin biopsy yields acute or chronic leukocytoclastic vasculitis with neutrophilic infiltrate in the narrow caliber vessels of the superficial dermis. Renal biopsy findings in MPA typically range from mild focal or segmental to diffuse necrotizing and sclerosing glomerulonephritis, often presenting as crescentic glomerulonephritis. These biopsies usually show minimal to no immune complex deposits on light and immunofluorescent microscopy, a characteristic described as "pauci-immune" (see Image. Histopathological Changes in Microscopic Polyangiitis Showing Fibrosis).[15]
The importance of renal biopsy is underscored by the fact that the severity of renal involvement observed on histopathological evaluation correlates closely with clinical disease activity.[16] This makes biopsy a crucial tool for guiding the management of the condition and decisions regarding tapering immunosuppressive therapy. In some cases, the presence of immune complex deposits in the glomeruli may be associated with more severe systemic signs and symptoms. Please see StatPearls' companion resource, "Rapidly Progressive Glomerulonephritis," for more information.
History and Physical
Patients with MPA may present with constitutional symptoms, including insidious onset fever, arthralgia, myalgia, and weight loss. Other manifestations include urinary abnormalities, cough with or without hemoptysis, skin examination findings consistent with palpable purpura, mononeuritis multiplex, seizures, other nonspecific neurological complaints, abdominal pain, gastrointestinal bleeding, sinusitis, and chest, testicular, and ocular pain. Some patients may present with acute onset of fulminant disease with frank hemoptysis, hematuria, or even renal failure.
Renal Manifestations
The primary renal feature of MPA is rapidly progressive glomerulonephritis.[17] Previous studies reported that 80% to 90% of patients with MPA experience renal manifestations, which can range from asymptomatic urinary sediment to end-stage renal failure requiring renal replacement therapy. Common clinical features of renal involvement due to glomerulonephritis include microscopic hematuria, proteinuria, and the presence of granular or red blood cell (RBC) casts in the urine.[18]
Pulmonary Manifestations
Pulmonary involvement is thought to be observed in 25% to 55% of cases. However, pulmonary symptoms may be more common than previously thought. A single-center study found that 80% of MPA patients were observed to have symptoms of pulmonary involvement, and 92% were found to have radiographic changes.[19] Although the most common pulmonary feature is diffuse alveolar hemorrhage, some patients may develop chronic interstitial fibrosis, causing respiratory failure.[20] Common manifestations of alveolar hemorrhage include hemoptysis, cough, dyspnea, and pleuritic chest pain.[21] Unlike GPA and EGPA, upper respiratory tract signs are rare.
Skin Manifestations
Skin lesions are observed in 30% to 60% of patients with MPA, and these lesions are the initial presenting feature in 15% to 30% of cases.[22][23] These skin manifestations include palpable purpura, livedo reticularis, urticaria, nodules, and ulcers with necrosis. Additionally, skin manifestations have been associated with joint pain in patients with MPA (see Image. Typical Skin Manifestations Observed in Microscopic Polyangiitis).[24]
Gastrointestinal Manifestations
Abdominal pain is the most common gastrointestinal symptom in MPA. Although gastrointestinal bleeding can occur, massive hemorrhage is rare.[25]
Neurological Manifestations
Neurological involvement is common in MPA, with peripheral neuropathy occurring more frequently than central nervous system (CNS) involvement. Predominant peripheral nervous system features include distal symmetrical polyneuropathy and mononeuritis multiplex. Sural nerve biopsies often reveal necrotizing vasculitis in up to 80% of cases, and nerve conduction studies may show acute axonopathy.[26] Rarely, patients may present with posterior reversible encephalopathy syndrome.[27] CNS manifestations can vary and include cerebral hemorrhage, nonhemorrhagic cerebral infarctions, and pachymeningitis.[28]
Physical Examination
Findings from the physical examination of a patient with MPA may reveal systemic signs such as general malaise, fever, or weight loss. Skin examination findings may include leukocytoclastic angiitis, palpable purpura, livedo reticularis, skin ulcerations, necrosis, gangrene, necrotizing nodules, digital ischemia, and urticaria. Pulmonary examination may reveal rales or bronchial breath sounds, particularly in cases of pulmonary capillaritis. Neurological examination might demonstrate motor or sensory deficits localized to a particular dermatome. Cardiovascular findings may include hypertension, signs of heart failure, myocardial infarction, and pericarditis. Findings from the gastrointestinal examination include bleeding, bowel ischemia, and perforation. The ocular examination may reveal retinal hemorrhage, scleritis, and uveitis. Other physical findings will vary depending on the involved vascular bed.
Evaluation
Evaluating a patient with suspected MPA requires a comprehensive clinical, radiological, histopathological, and laboratory assessment. The initial step is a detailed clinical evaluation to determine the site and extent of involvement across various organ systems.
Laboratory Studies
As noted above, although anti-MPO is the primary ANCA associated with MPA, anti-PR3 can also be present in some cases. ANCA levels are most closely correlated with renal activity compared to other organ systems.[8] Other useful laboratory evaluations include:
- The complete blood cell count (CBC) shows leukocytosis and anemia.
- The erythrocyte sedimentation rate (ESR) is usually elevated.
- Blood urea nitrogen (BUN) and serum creatinine levels are elevated in cases of renal failure.
- Urine examination shows abnormal urine sediment, proteinuria, hematuria, leukocyturia, and RBC casts.
- Blood cultures may be performed to rule out bacterial endocarditis.
- C3 and C4 levels are usually normal on complement testing.
Imaging Studies
A chest radiography and computed tomography (CT) scan of the chest should be obtained to assess for pulmonary lesions in patients presenting with hemoptysis or pulmonary fibrosis.[29] These imaging studies help differentiate between GPA and MPA, as GPA often shows cavitary and nodular lesions on radiological evaluation. A CT scan of the abdomen may also be performed to evaluate for pancreatitis or mesenteric angiography to distinguish MPA from polyarteritis nodosa.[30]
Other Studies
- Electrocardiography (ECG) is used to evaluate for myocardial infarction, pericarditis, or heart failure.
- Gastrointestinal endoscopy is performed in cases of gastrointestinal bleeding.
- Electromyography (EMG) and nerve conduction studies are utilized in cases of clinical evidence of neuropathy.[31]
Histopathological Evaluation
A histopathological specimen (skin, renal, or lung biopsy) should be obtained whenever possible to identify evidence of vasculitis and immune deposits. The extent of inflammation observed on renal biopsy can be used to assess disease activity and guide treatment decisions.
Treatment / Management
Currently, the American College of Rheumatology and the European Alliance of Associations for Rheumatology (ACR/EULAR) are developing criteria for the treatment of ANCA-associated vasculitis. Based on new clinical trial data, recent recommendations suggest classifying disease manifestations into organ/life-threatening or non-organ/life-threatening categories rather than "severe" or "not severe." Examples of organ/life-threatening manifestations include glomerulonephritis, pulmonary hemorrhage, cardiac involvement, and retroorbital disease. Non-organ/life-threatening manifestations include skin involvement, myositis, and non-cavitating pulmonary nodules.[32]
The treatment of MPA involves using immunosuppressive agents in various combinations, divided into two phases: induction and maintenance. Commonly used agents include cyclophosphamide, glucocorticoids, rituximab, azathioprine, methotrexate, and, when indicated, plasmapheresis.
The 2022 EULAR recommendations for the management of ANCA-associated vasculitis apply to both GPA and MPA.[32] Treating EGPA follows a different set of guidelines. A key distinction between GPA and MPA is that patients with the clinical syndrome of GPA and PR3-ANCA positivity have a higher relapse risk compared to those with MPA or anti-MPO positivity. Additionally, persistent ANCA positivity, despite clinical remission, is associated with an increased risk of relapse.[32] Although extending immunosuppressive therapy beyond 24 months may reduce relapse risk, the heightened risk of infection may not warrant this prolonged treatment course.
The 2022 EULAR recommendations include:
- For new-onset organ/life-threatening MPA and GPA, remission should be induced with glucocorticoids combined with either rituximab or cyclophosphamide. Rituximab is preferred in cases of relapsing disease. No significant differences are observed in outcomes between using 2-dose or 4-dose infusions of rituximab.[33] (A1)
- For new-onset non-organ/life-threatening MPA, remission should be induced with glucocorticoids and rituximab. In some cases, methotrexate or mycophenolate mofetil can be substituted for rituximab. Cyclophosphamide is generally not preferred due to a higher incidence of adverse effects despite its reduced efficacy. Additionally, a lower prednisolone dose of 0.5 mg/kg/d may also be considered.
- The induction regimen should include oral glucocorticoids at a weight-dependent dose of 50 to 75 mg daily, with prednisolone tapered to 5 mg daily over 4 to 5 months.
- Avacopan is an oral C5a-receptor antagonist that helps block neutrophil attraction and activation. The ADVOCATE trial demonstrated that avacopan can induce remission when used alongside rituximab or cyclophosphamide, reducing the need for glucocorticoid exposure. Avacopan was approved by the US Food and Drug Administration (FDA) in 2021 to treat severe ANCA-associated vasculitis.[34]
- Plasma exchange can induce remission in patients with glomerulonephritis and a serum creatinine level greater than 3.39 mg/dL (300 μmol/L). Routine use of plasmapheresis for alveolar hemorrhage in MPA and GPA is not recommended. However, it is often used as "salvage" therapy, particularly for patients with a serum creatinine level exceeding 5.7 mg/dL.[35] A meta-analysis suggests that plasmapheresis reduces the risk of end-stage renal disease but is associated with an increased risk of infections within the first year.[36] (A1)
- Rituximab is recommended for maintaining remission. Azathioprine or methotrexate may also be considered; however, their use should be reserved for patients with an estimated glomerular filtration rate greater than 60 mL/min/1.73m2.[35]
- For new-onset disease, maintenance therapy should continue for 24 to 48 months after remission begins. A longer treatment duration should be considered for patients with relapsing disease.
- Treatment decisions should be guided by clinical assessment rather than ANCA levels or CD19+ B lymphocyte counts.
- If rituximab is administered, serum immunoglobulin levels should be measured before each dose.
- For patients receiving rituximab, cyclophosphamide, or high-dose glucocorticoids, prophylaxis with trimethoprim-sulfamethoxazole should be provided.
Differential Diagnosis
Many conditions may mimic ANCA-associated vasculitis/MPA and must be excluded before establishing a diagnosis. Some of these processes include:
Infectious Etiologies
- Infective endocarditis
- Rocky Mountain spotted fever and other tick-borne vasculitides
- Disseminated fungal infections
Malignancies
- Atrial myxomas
- Lymphomas
- Carcinomatosis
Drug Toxicities
- Cocaine
- Amphetamines
- Ergot alkaloids
- Levamisole
Other Autoimmune Conditions
- Amyloidosis
- Goodpasture disease [37]
- Sarcoidosis
- Polyarteritis nodosa
- Leukocytoclastic vasculitis
- Granulomatosis with polyangiitis
- Eosinophilic granulomatosis with polyangiitis
- Cryoglobulinemia
Toxicity and Adverse Effect Management
The immunosuppressive agents used in managing ANCA-associated vasculitis have severe adverse effects, which may be even more debilitating than the manifestations of the disease.
Glucocorticoids
- Osteoporosis [38]
- Cataract
- Glaucoma
- Diabetes mellitus
- Electrolyte abnormalities
- Avascular necrosis of bone
Cyclophosphamide
- Bone marrow suppression
- Hemorrhagic cystitis [39]
- Bladder carcinoma
- Myelodysplasia
Methotrexate
- Hepatotoxicity
- Pneumonitis
- Bone marrow suppression
Azathioprine
- Hepatotoxicity
- Bone marrow suppression
Rituximab
- Progressive multifocal leukoencephalopathy
- Opportunistic infections
Prognosis
The 5-year estimated survival rate for patients with MPA ranges from 45% to 76%, which is significantly worse than that for GPA or EGPA, largely due to renal disease.[6][40]
Long-term morbidity in a cohort of 296 patients with MPA or GPA was associated with the severity of the primary disease, the number of relapses, older age, and the duration of glucocorticoid treatment. In a 7-year follow-up post-diagnosis, the mean duration of glucocorticoid therapy was 40.4 months.[41]
Complications
If left untreated, MPA can lead to permanent organ damage, with kidney failure being the most common complication. Other complications depend on the specific organ systems involved. Research has shown that older age, diastolic hypertension, and positive PR3-ANCA status correlate with an increased risk of cardiovascular events.[42]
The medications used to treat MPA can also lead to significant adverse effects. For instance, cyclophosphamide is strongly associated with an increased risk of bladder cancer. Additionally, steroids are known to cause bone loss, hyperglycemia, muscle weakness, and skin issues.
Deterrence and Patient Education
Adherence to medication and therapy is crucial, along with regular follow-ups. Patients should be closely monitored during the prolonged course of immunosuppressive treatment, which typically extends for over a year, with disease activity assessed through ANCA levels. It's essential to inform patients about the potential adverse effects of their medications and emphasize the importance of reporting them promptly. Additionally, patients should be made aware that relapse is common, so they understand the necessity of ongoing follow-up even after achieving remission, as they are not "cured."
Enhancing Healthcare Team Outcomes
MPA is a complex condition best managed by a multidisciplinary team due to its varied clinical presentation and the need for patient-specific treatment. Effective communication among an interprofessional medical team—including clinicians, nurses, radiologists, pathologists, and pharmacists—is essential for optimal care. Nurses are critical in monitoring and documenting vital signs, especially urine output, which is crucial for guiding treatment decisions. Radiologists contribute by performing image-guided tissue biopsies, which are then analyzed by pathologists to confirm the presence of disease. Proper dosing and dispensing of immunosuppressive agents by pharmacists are crucial for the successful induction and maintenance of remission. Failure in this area can result in severe systemic adverse effects. Additionally, pharmacists must perform thorough medication reconciliation to prevent potential drug-drug interactions.
To determine the appropriate treatment, researchers at Johns Hopkins University have developed the revised Birmingham score, which can be used to clinically classify the patient's disease state.[43] All interprofessional healthcare team members must maintain accurate, updated patient records, ensuring all professionals involved in patient care have access to the same patient information. Open communication channels between all healthcare team members are crucial to successful interprofessional care.
In addition, care coordination is pivotal in ensuring seamless and efficient patient care. Physicians, advanced practitioners, nurses, pharmacists, social workers, and other healthcare professionals must collaborate closely to streamline the patient's journey, from diagnosis through treatment and follow-up. This coordination minimizes errors, reduces delays, and enhances patient safety, ultimately leading to improved outcomes and patient-centered care that prioritizes the well-being and satisfaction of those affected by MPA.
Media
(Click Image to Enlarge)
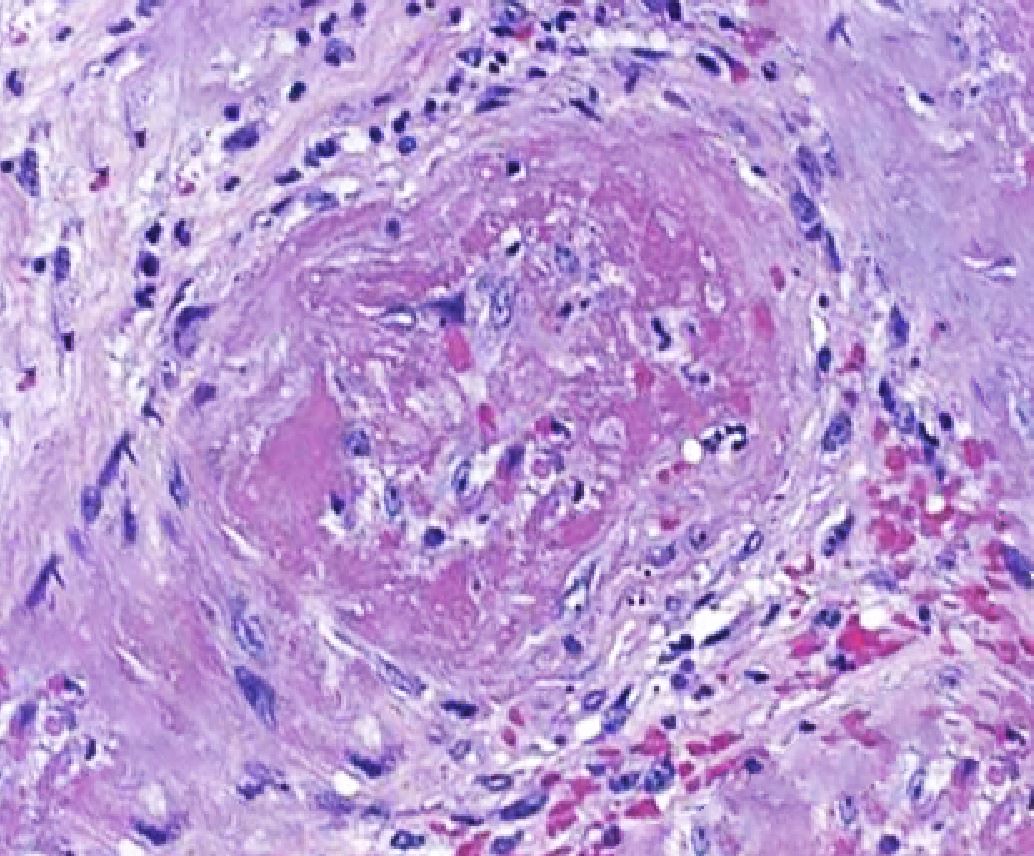
Histopathological Changes in Microscopic Polyangiitis Showing Fibrosis.
Contributed by Rian Kabir, MD
(Click Image to Enlarge)
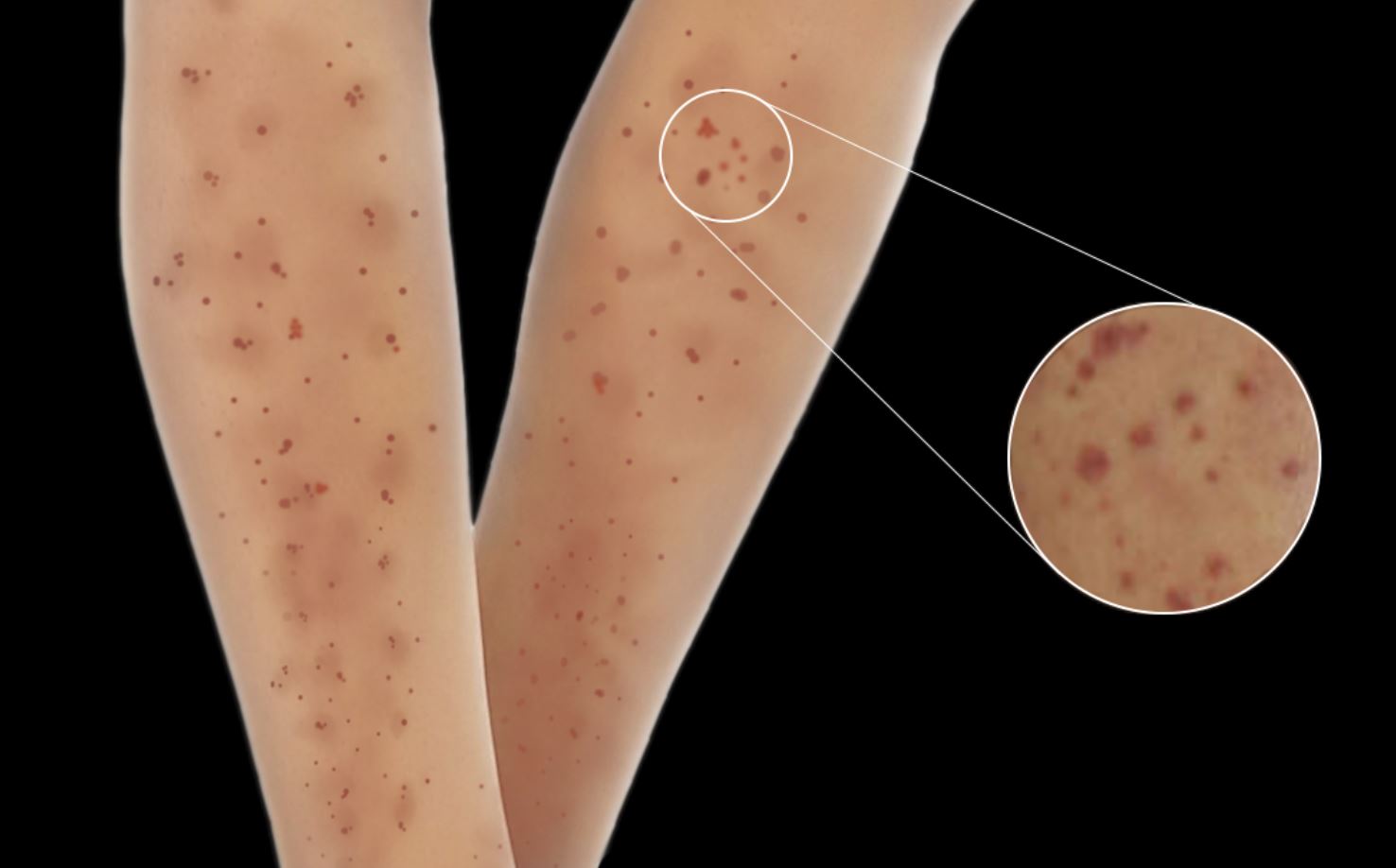
Typical Skin Manifestations Observed in Microscopic Polyangiitis.
Contributed by R Kabir, MD
References
Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F, Flores-Suarez LF, Gross WL, Guillevin L, Hagen EC, Hoffman GS, Jayne DR, Kallenberg CG, Lamprecht P, Langford CA, Luqmani RA, Mahr AD, Matteson EL, Merkel PA, Ozen S, Pusey CD, Rasmussen N, Rees AJ, Scott DG, Specks U, Stone JH, Takahashi K, Watts RA. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis and rheumatism. 2013 Jan:65(1):1-11. doi: 10.1002/art.37715. Epub [PubMed PMID: 23045170]
Level 3 (low-level) evidenceChung SA, Seo P. Microscopic polyangiitis. Rheumatic diseases clinics of North America. 2010 Aug:36(3):545-58. doi: 10.1016/j.rdc.2010.04.003. Epub 2010 Jun 11 [PubMed PMID: 20688249]
Greco A, Rizzo MI, De Virgilio A, Gallo A, Fusconi M, Pagliuca G, Martellucci S, Turchetta R, Longo L, De Vincentiis M. Goodpasture's syndrome: a clinical update. Autoimmunity reviews. 2015 Mar:14(3):246-53. doi: 10.1016/j.autrev.2014.11.006. Epub 2014 Nov 15 [PubMed PMID: 25462583]
Kallenberg CG, Heeringa P, Stegeman CA. Mechanisms of Disease: pathogenesis and treatment of ANCA-associated vasculitides. Nature clinical practice. Rheumatology. 2006 Dec:2(12):661-70 [PubMed PMID: 17133251]
de Lind van Wijngaarden RA, van Rijn L, Hagen EC, Watts RA, Gregorini G, Tervaert JW, Mahr AD, Niles JL, de Heer E, Bruijn JA, Bajema IM. Hypotheses on the etiology of antineutrophil cytoplasmic autoantibody associated vasculitis: the cause is hidden, but the result is known. Clinical journal of the American Society of Nephrology : CJASN. 2008 Jan:3(1):237-52 [PubMed PMID: 18077783]
Walulik A, Łysak K, Błaszkiewicz M, Górecki I, Gomułka K. The Role of Neutrophils in ANCA-Associated Vasculitis: The Pathogenic Role and Diagnostic Utility of Autoantibodies. International journal of molecular sciences. 2023 Dec 7:24(24):. doi: 10.3390/ijms242417217. Epub 2023 Dec 7 [PubMed PMID: 38139045]
Kain R, Tadema H, McKinney EF, Benharkou A, Brandes R, Peschel A, Hubert V, Feenstra T, Sengölge G, Stegeman C, Heeringa P, Lyons PA, Smith KG, Kallenberg C, Rees AJ. High prevalence of autoantibodies to hLAMP-2 in anti-neutrophil cytoplasmic antibody-associated vasculitis. Journal of the American Society of Nephrology : JASN. 2012 Mar:23(3):556-66. doi: 10.1681/ASN.2011090920. Epub 2012 Feb 9 [PubMed PMID: 22323643]
Level 2 (mid-level) evidenceJennette JC, Nachman PH. ANCA Glomerulonephritis and Vasculitis. Clinical journal of the American Society of Nephrology : CJASN. 2017 Oct 6:12(10):1680-1691. doi: 10.2215/CJN.02500317. Epub 2017 Aug 25 [PubMed PMID: 28842398]
Lyons PA, Rayner TF, Trivedi S, Holle JU, Watts RA, Jayne DR, Baslund B, Brenchley P, Bruchfeld A, Chaudhry AN, Cohen Tervaert JW, Deloukas P, Feighery C, Gross WL, Guillevin L, Gunnarsson I, Harper L, Hrušková Z, Little MA, Martorana D, Neumann T, Ohlsson S, Padmanabhan S, Pusey CD, Salama AD, Sanders JS, Savage CO, Segelmark M, Stegeman CA, Tesař V, Vaglio A, Wieczorek S, Wilde B, Zwerina J, Rees AJ, Clayton DG, Smith KG. Genetically distinct subsets within ANCA-associated vasculitis. The New England journal of medicine. 2012 Jul 19:367(3):214-23. doi: 10.1056/NEJMoa1108735. Epub [PubMed PMID: 22808956]
Level 2 (mid-level) evidenceWakamatsu K, Mitsuhashi Y, Yamamoto T, Tsuboi R. Propylthiouracil-induced antineutrophil cytoplasmic antibody positive vasculitis clinically mimicking pyoderma gangrenosum. The Journal of dermatology. 2012 Aug:39(8):736-8. doi: 10.1111/j.1346-8138.2011.01399.x. Epub 2011 Dec 2 [PubMed PMID: 22132781]
Level 3 (low-level) evidencePendergraft WF 3rd, Niles JL. Trojan horses: drug culprits associated with antineutrophil cytoplasmic autoantibody (ANCA) vasculitis. Current opinion in rheumatology. 2014 Jan:26(1):42-9. doi: 10.1097/BOR.0000000000000014. Epub [PubMed PMID: 24276086]
Level 3 (low-level) evidenceBerti A, Cornec D, Crowson CS, Specks U, Matteson EL. The Epidemiology of Antineutrophil Cytoplasmic Autoantibody-Associated Vasculitis in Olmsted County, Minnesota: A Twenty-Year US Population-Based Study. Arthritis & rheumatology (Hoboken, N.J.). 2017 Dec:69(12):2338-2350. doi: 10.1002/art.40313. Epub 2017 Nov 9 [PubMed PMID: 28881446]
Massicotte-Azarniouch D, Herrera CA, Jennette JC, Falk RJ, Free ME. Mechanisms of vascular damage in ANCA vasculitis. Seminars in immunopathology. 2022 May:44(3):325-345. doi: 10.1007/s00281-022-00920-0. Epub 2022 Mar 7 [PubMed PMID: 35254509]
Frankel SK, Schwarz MI. The pulmonary vasculitides. American journal of respiratory and critical care medicine. 2012 Aug 1:186(3):216-24. doi: 10.1164/rccm.201203-0539CI. Epub 2012 Jun 7 [PubMed PMID: 22679011]
Sun L, Wang H, Jiang X, Mo Y, Yue Z, Huang L, Liu T. Clinical and pathological features of microscopic polyangiitis in 20 children. The Journal of rheumatology. 2014 Aug:41(8):1712-9. doi: 10.3899/jrheum.131300. Epub 2014 Jul 1 [PubMed PMID: 24986845]
Level 2 (mid-level) evidenceHauer HA, Bajema IM, van Houwelingen HC, Ferrario F, Noël LH, Waldherr R, Jayne DR, Rasmussen N, Bruijn JA, Hagen EC, European Vasculitis Study Group (EUVAS). Renal histology in ANCA-associated vasculitis: differences between diagnostic and serologic subgroups. Kidney international. 2002 Jan:61(1):80-9 [PubMed PMID: 11786087]
Lababidi MH, Odigwe C, Okolo C, Elhassan A, Iroegbu N. Microscopic polyangiitis causing diffuse alveolar hemorrhage and rapidly progressive glomerulonephritis. Proceedings (Baylor University. Medical Center). 2015 Oct:28(4):469-71 [PubMed PMID: 26424944]
Wang H, Sun L, Tan W. Clinical features of children with pulmonary microscopic polyangiitis: report of 9 cases. PloS one. 2015:10(4):e0124352. doi: 10.1371/journal.pone.0124352. Epub 2015 Apr 29 [PubMed PMID: 25923706]
Level 3 (low-level) evidenceWilke L, Prince-Fiocco M, Fiocco GP. Microscopic polyangiitis: a large single-center series. Journal of clinical rheumatology : practical reports on rheumatic & musculoskeletal diseases. 2014 Jun:20(4):179-82. doi: 10.1097/RHU.0000000000000108. Epub [PubMed PMID: 24847742]
Level 2 (mid-level) evidenceKarras A. Microscopic Polyangiitis: New Insights into Pathogenesis, Clinical Features and Therapy. Seminars in respiratory and critical care medicine. 2018 Aug:39(4):459-464. doi: 10.1055/s-0038-1673387. Epub 2018 Nov 7 [PubMed PMID: 30404112]
Barowka SE, Perrine TR, Hughes K. "I'm coughing up blood". Journal of the Mississippi State Medical Association. 2016 Nov:57(11):354-356 [PubMed PMID: 30281235]
Kluger N, Pagnoux C, Guillevin L, Francès C, French Vasculitis Study Group. Comparison of cutaneous manifestations in systemic polyarteritis nodosa and microscopic polyangiitis. The British journal of dermatology. 2008 Sep:159(3):615-20. doi: 10.1111/j.1365-2133.2008.08725.x. Epub 2008 Jul 19 [PubMed PMID: 18647311]
Lhote F, Cohen P, Guillevin L. Polyarteritis nodosa, microscopic polyangiitis and Churg-Strauss syndrome. Lupus. 1998:7(4):238-58 [PubMed PMID: 9643314]
Gibson LE. Cutaneous manifestations of antineutrophil cytoplasmic antibody-associated vasculitis (AAV): a concise review with emphasis on clinical and histopathologic correlation. International journal of dermatology. 2022 Dec:61(12):1442-1451. doi: 10.1111/ijd.16247. Epub 2022 May 22 [PubMed PMID: 35599359]
Eriksson P, Segelmark M, Hallböök O. Frequency, Diagnosis, Treatment, and Outcome of Gastrointestinal Disease in Granulomatosis with Polyangiitis and Microscopic Polyangiitis. The Journal of rheumatology. 2018 Apr:45(4):529-537. doi: 10.3899/jrheum.170249. Epub 2018 Feb 1 [PubMed PMID: 29419474]
Hattori N, Mori K, Misu K, Koike H, Ichimura M, Sobue G. Mortality and morbidity in peripheral neuropathy associated Churg-Strauss syndrome and microscopic polyangiitis. The Journal of rheumatology. 2002 Jul:29(7):1408-14 [PubMed PMID: 12136898]
Level 2 (mid-level) evidenceXu J, Ding Y, Qu Z, Yu F. Posterior Reversible Encephalopathy Syndrome in a Patient With Microscopic Polyangiitis: A Case Report and Literature Review. Frontiers in medicine. 2021:8():792744. doi: 10.3389/fmed.2021.792744. Epub 2021 Dec 24 [PubMed PMID: 35071272]
Level 3 (low-level) evidenceKu BD, Shin HY. Multiple bilateral non-hemorrhagic cerebral infarctions associated with microscopic polyangiitis. Clinical neurology and neurosurgery. 2009 Dec:111(10):904-6. doi: 10.1016/j.clineuro.2009.08.008. Epub 2009 Sep 3 [PubMed PMID: 19733002]
Level 3 (low-level) evidenceEschun GM, Mink SN, Sharma S. Pulmonary interstitial fibrosis as a presenting manifestation in perinuclear antineutrophilic cytoplasmic antibody microscopic polyangiitis. Chest. 2003 Jan:123(1):297-301 [PubMed PMID: 12527637]
Level 3 (low-level) evidenceIida T, Adachi T, Tabeya T, Nakagaki S, Yabana T, Goto A, Kondo Y, Kasai K. Rare type of pancreatitis as the first presentation of anti-neutrophil cytoplasmic antibody-related vasculitis. World journal of gastroenterology. 2016 Feb 21:22(7):2383-90. doi: 10.3748/wjg.v22.i7.2383. Epub [PubMed PMID: 26900301]
Sassi SB, Ghorbel IB, Mizouni H, Houman MH, Hentati F. Microscopic polyangiitis presenting with peripheral and central neurological manifestations. Neurological sciences : official journal of the Italian Neurological Society and of the Italian Society of Clinical Neurophysiology. 2011 Aug:32(4):727-9. doi: 10.1007/s10072-011-0653-x. Epub 2011 Jun 17 [PubMed PMID: 21681367]
Level 3 (low-level) evidenceHellmich B, Sanchez-Alamo B, Schirmer JH, Berti A, Blockmans D, Cid MC, Holle JU, Hollinger N, Karadag O, Kronbichler A, Little MA, Luqmani RA, Mahr A, Merkel PA, Mohammad AJ, Monti S, Mukhtyar CB, Musial J, Price-Kuehne F, Segelmark M, Teng YKO, Terrier B, Tomasson G, Vaglio A, Vassilopoulos D, Verhoeven P, Jayne D. EULAR recommendations for the management of ANCA-associated vasculitis: 2022 update. Annals of the rheumatic diseases. 2024 Jan 2:83(1):30-47. doi: 10.1136/ard-2022-223764. Epub 2024 Jan 2 [PubMed PMID: 36927642]
Schirmer JH, Sanchez-Alamo B, Hellmich B, Jayne D, Monti S, Luqmani RA, Tomasson G. Systematic literature review informing the 2022 update of the EULAR recommendations for the management of ANCA-associated vasculitis (AAV): part 1-treatment of granulomatosis with polyangiitis and microscopic polyangiitis. RMD open. 2023 Jul:9(3):. doi: 10.1136/rmdopen-2023-003082. Epub [PubMed PMID: 37479496]
Level 1 (high-level) evidenceJayne DRW, Merkel PA, Schall TJ, Bekker P, ADVOCATE Study Group. Avacopan for the Treatment of ANCA-Associated Vasculitis. The New England journal of medicine. 2021 Feb 18:384(7):599-609. doi: 10.1056/NEJMoa2023386. Epub [PubMed PMID: 33596356]
Casal Moura M, Gauckler P, Anders HJ, Bruchfeld A, Fernandez-Juarez GM, Floege J, Frangou E, Goumenos D, Segelmark M, Turkmen K, van Kooten C, Tesar V, Geetha D, Fervenza FC, Jayne DRW, Stevens KI, Kronbichler A. Management of antineutrophil cytoplasmic antibody-associated vasculitis with glomerulonephritis as proposed by the ACR 2021, EULAR 2022 and KDIGO 2021 guidelines/recommendations. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 2023 Oct 31:38(11):2637-2651. doi: 10.1093/ndt/gfad090. Epub [PubMed PMID: 37164940]
Walsh M, Collister D, Zeng L, Merkel PA, Pusey CD, Guyatt G, Au Peh C, Szpirt W, Ito-Hara T, Jayne DRW, Plasma exchange and glucocorticoid dosing for patients with ANCA-associated vasculitis BMJ Rapid Recommendations Group. The effects of plasma exchange in patients with ANCA-associated vasculitis: an updated systematic review and meta-analysis. BMJ (Clinical research ed.). 2022 Feb 25:376():e064604. doi: 10.1136/bmj-2021-064604. Epub 2022 Feb 25 [PubMed PMID: 35217545]
Level 1 (high-level) evidenceWard ND, Cosner DE, Lamb CA, Li W, Macknis JK, Rooney MT, Zhang PL. Top Differential Diagnosis Should Be Microscopic Polyangiitis in ANCA-Positive Patient with Diffuse Pulmonary Hemorrhage and Hemosiderosis. Case reports in pathology. 2014:2014():286030. doi: 10.1155/2014/286030. Epub 2014 Nov 30 [PubMed PMID: 25525543]
Level 3 (low-level) evidenceChotiyarnwong P, McCloskey EV. Pathogenesis of glucocorticoid-induced osteoporosis and options for treatment. Nature reviews. Endocrinology. 2020 Aug:16(8):437-447. doi: 10.1038/s41574-020-0341-0. Epub 2020 Apr 14 [PubMed PMID: 32286516]
Haldar S, Dru C, Bhowmick NA. Mechanisms of hemorrhagic cystitis. American journal of clinical and experimental urology. 2014:2(3):199-208 [PubMed PMID: 25374922]
Kitching AR, Anders HJ, Basu N, Brouwer E, Gordon J, Jayne DR, Kullman J, Lyons PA, Merkel PA, Savage COS, Specks U, Kain R. ANCA-associated vasculitis. Nature reviews. Disease primers. 2020 Aug 27:6(1):71. doi: 10.1038/s41572-020-0204-y. Epub 2020 Aug 27 [PubMed PMID: 32855422]
Robson J, Doll H, Suppiah R, Flossmann O, Harper L, Höglund P, Jayne D, Mahr A, Westman K, Luqmani R. Glucocorticoid treatment and damage in the anti-neutrophil cytoplasm antibody-associated vasculitides: long-term data from the European Vasculitis Study Group trials. Rheumatology (Oxford, England). 2015 Mar:54(3):471-81. doi: 10.1093/rheumatology/keu366. Epub 2014 Sep 8 [PubMed PMID: 25205825]
Level 2 (mid-level) evidenceRobson J, Doll H, Suppiah R, Flossmann O, Harper L, Höglund P, Jayne D, Mahr A, Westman K, Luqmani R. Damage in the anca-associated vasculitides: long-term data from the European vasculitis study group (EUVAS) therapeutic trials. Annals of the rheumatic diseases. 2015 Jan:74(1):177-84. doi: 10.1136/annrheumdis-2013-203927. Epub 2013 Nov 15 [PubMed PMID: 24243925]
Stone JH, Hoffman GS, Merkel PA, Min YI, Uhlfelder ML, Hellmann DB, Specks U, Allen NB, Davis JC, Spiera RF, Calabrese LH, Wigley FM, Maiden N, Valente RM, Niles JL, Fye KH, McCune JW, St Clair EW, Luqmani RA, International Network for the Study of the Systemic Vasculitides (INSSYS). A disease-specific activity index for Wegener's granulomatosis: modification of the Birmingham Vasculitis Activity Score. International Network for the Study of the Systemic Vasculitides (INSSYS). Arthritis and rheumatism. 2001 Apr:44(4):912-20 [PubMed PMID: 11318006]