Introduction
Leiomyosarcoma, a common subtype of soft tissue sarcoma (STS), accounts for up to 10% to 20% of all sarcomas.[1] Originating from either smooth muscle cells or their mesenchymal cell precursors, leiomyosarcoma primarily occurs in the retroperitoneum, uterus, and extremities, in descending order of frequency.[2] The genetic abnormalities in leiomyosarcoma are complex, and our current knowledge remains incomplete. The clinical presentation, signs, and symptoms vary depending on the site of origin. Prognosis is related to the tumor's location, size, and grade, with the tumor grade significantly impacting overall survival. With higher-grade lesions, the rate of distant metastasis increases. Treatment can be nuanced and often requires an interdisciplinary approach. The current standard of care involves upfront surgery for resectable tumors, with chemotherapy and radiation typically serving as adjuncts.[3] Treatment at high-volume sarcoma centers is preferred and improves patient outcomes.[4] The advent of targeted agents and immunotherapy can potentially improve patient outcomes in the future.
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
Although there are no definite causative factors for leiomyosarcoma, certain risk factors have been associated with developing STSs, including leiomyosarcoma. The significant risk factor for STS is a history of radiation exposure or radiotherapy, particularly at a young age.[5] STSs may also be part of genetic syndromes, such as retinoblastoma (due to RB1 gene deletion) and Li-Fraumeni syndrome (caused by a mutation in the TP53 gene).[6]
Epidemiology
Leiomyosarcoma accounts for 10% to 20% of all newly diagnosed STS.[7] In recent years, advancements in molecular diagnostics have enhanced the accuracy of diagnosis. Leiomyosarcoma most commonly occurs in the retroperitoneum, followed by the uterus, extremities, and trunk. Uterine sarcomas comprise about 3% to 7% of all uterine malignancies, with leiomyosarcoma being the most common subtype, accounting for nearly 80% of all uterine sarcomas.[8][9]
The incidence of leiomyosarcoma increases with age, reaching its peak in the seventh decade of life. The exception to this is uterine leiomyosarcoma, which occurs most commonly in perimenopausal women. Tumors associated with genetic syndromes occur earlier in life. Retroperitoneal leiomyosarcoma and tumors arising from visceral blood vessels are more common in women, whereas the disease at other sites is more common in men.[6]
Pathophysiology
Leiomyosarcomas are primarily associated with RB1 and PTEN tumor suppressor gene mutations. RB1 gene mutations are observed in 90% of patients. The cytogenetic and molecular changes in leiomyosarcoma are inconsistent, contributing to its highly heterogeneous nature. More recent data from The Cancer Genome Atlas revealed that leiomyosarcoma, like other STSs, is associated with a lower tumor mutational burden compared to other types of cancers.[10]
Histopathology
Histology
Leiomyosarcomas originate from the smooth muscle cells or their mesenchymal precursors.[11] Grossly, they are typically solitary, well-circumscribed lesions that often have areas of cystic degeneration and necrosis. Classic leiomyosarcoma is characterized by spindle-shaped cells arranged in intersecting fascicles, similar to smooth muscle. The nuclei are typically elongated and hyperchromatic with abundant eosinophilic cytoplasm.[2] Varying degrees of pleomorphism can occur. When poorly differentiated, leiomyosarcoma can resemble any undifferentiated STSs.[2]. Other histologic variants include pleiomorphic, myxoid, and undifferentiated leiomyosarcoma.
Uterine leiomyosarcomas are typically intramural and solitary and rarely arise from the cervix. Unlike leiomyomas, which have a well-defined, non-infiltrating border, leiomyosarcomas are often irregular and infiltrating. On gross inspection, most uterine leiomyosarcomas appear as large, solitary lesions with irregular and infiltrative borders.[12] On microscopy, the hallmarks of leiomyosarcoma include nuclear atypia, >5 to 10 mitoses per high-power field (HPF), and tumor necrosis (see Image. Leiomyosarcoma of the Uterus).[13] Tumor necrosis is typically coagulative, with viable tumor cells sharply demarcated from areas of necrosis. This is in contrast to the infarctive necrosis typically seen in benign tumors such as leiomyomas, where an area of regeneration surrounds the zone of necrosis. The pattern and type of necrosis can help differentiate benign from malignant lesions. Uterine leiomyosarcoma can further be divided into spindle cells (classic type), epitheloid (more than 50% epithelial cells), myxoid (hypocellular with basophilic stroma), and other rare types. Lesions that arise from smooth muscle cells but lack the definitive characteristics of leiomyosarcoma are labeled smooth muscle tumors of uncertain malignant potential (STUMP).[12]
Immunohistochemistry
Leiomyosarcoma stains positive for smooth muscle–specific markers such as muscle-specific actin, desmin, and h-caldesmon. Immunohistochemical stains are typically used for confirmation or in undifferentiated tumors where the cell of origin is unclear. Subtypes of leiomyosarcoma may require additional stains. In particular, epitheloid leiomyosarcoma can be confused for a carcinoma due to its tendency to stain positive for epithelial elements. In such cases, histone deacetylase-8 and myocardin are additional stains that can help confirm the diagnosis. Immunopositivity for p16 and p53 with a high Ki-67 proliferation index has also demonstrated high sensitivity and specificity for differentiating leiomyosarcoma and leiomyomas.
Compared to leiomyoma, leiomyosarcoma has a lower expression of estrogen (40% in leiomyosarcoma versus 70% in leiomyoma) and progesterone receptors (38% in leiomyosarcoma versus 88% in leiomyoma).[14].
History and Physical
The clinical presentation of leiomyosarcoma depends on the location and is highly variable. Symptoms are often due to compression of surrounding organs. In locations such as the retroperitoneum, tumors can grow very large before becoming symptomatic. Uterine lesions are often diagnosed through pathological examination after a hysterectomy for a suspected leiomyoma.[6][8]
Evaluation
The appropriate workup of a suspected sarcoma is critical. Initial cross-sectional imaging using either a computed tomography (CT) scan or magnetic resonance imaging (MRI) helps identify the extent of the lesion, its relationship to surrounding structures, and potential targets for biopsy. Although CT excels at assessing retroperitoneal and visceral lesions, MRI is effective for evaluating tumors originating in the extremities and head and neck.[15] As part of the initial workup, patients often obtain the imaging modality that is more readily available. Once a sarcoma is suspected, an image-guided core needle biopsy is required for diagnosis; fine-needle aspiration is insufficient for establishing a diagnosis.[16] For palpable lesions, image guidance improves the accuracy of core needle biopsies.[17] For abdominopelvic lesions, using a retroperitoneal approach is crucial to avoid peritoneal seeding and potential sarcomatosis. For superficial lesions, the biopsy must be conducted with consideration of potential surgical incision, ensuring that the biopsy tract is included in any surgical specimen. Incisional and excisional biopsies should be avoided, and with current techniques, an open surgical biopsy should rarely be necessary. Referring suspected STS cases to a high-volume center is advisable because inappropriate diagnostic workups, particularly inappropriate surgical procedures, can lead to distortion of tissue planes, seedling of sarcomatous tissue, and alteration of subsequent surgical approach.
A chest and abdominal CT scan is mandatory, given the risk of hematogenous metastasis. In cases of uterine leiomyosarcoma, an endometrial biopsy may yield a diagnosis; however, a negative biopsy does not rule out leiomyosarcoma. Any leiomyoma that continues to grow beyond menopause should be evaluated to rule out leiomyosarcoma.[6][8]
Treatment / Management
The management of leiomyosarcoma depends on the disease site, tumor size, grade, and patient-dependent factors. The potential options include surgical resection, radiation, and chemotherapy. A multidisciplinary approach at high-volume sarcoma centers is highly recommended. The primary goals of treatment with curative intent include achieving surgical resection with negative margins, reducing local recurrence, improving functional outcomes, and reducing the risk of distant metastasis.[6] Radiotherapy (RT) in STS improves local control, preserves function, and reduces local recurrence but does not improve survival.[18] The timing of radiotherapy also remains a topic of debate for both retroperitoneal and extremity or trunk leiomyosarcoma as major trials continue to accrue patients.[19] Details of treatment are provided in the following sections.(A1)
Differential Diagnosis
The clinical presentation of a patient with STS is vague and nonspecific. The morphological diagnosis based on microscopic examination remains the gold standard. Ancillary testing, including immunohistochemistry, classical cytogenetics, and molecular testing, aids in diagnosis. The World Health Organization (WHO) recognizes more than 70 different subtypes of sarcoma.[7] Diagnoses to consider due to similar presentation or histopathological similarity include meningioma, gastrointestinal stromal tumors, leiomyoma, dedifferentiated liposarcoma, endometrial stromal sarcoma, STUMP, inflammatory myofibroblastic tumor, and perivascular epithelioid cell tumor.
Surgical Oncology
The general surgery principles applicable to any sarcoma also pertain to patients diagnosed with leiomyosarcoma.
Leiomyosarcoma of the Extremity or Trunk
The primary goal of surgery is to perform an R0 resection while making every attempt to preserve critical neurovascular structures. Historically, amputation was often used in cases where negative margins could not be obtained. However, to maintain limb function or preserve critical structures, for example, sciatic nerve), an R1 or an R2 resection is acceptable.[20][21] While positive margins are associated with higher local recurrence rates, overall survival is determined by distant metastases. Available data suggest that a microscopically positive margin treated with appropriate adjuvant therapy does not significantly increase the rate of distant metastasis or reduce overall survival.[21] In cases where a close margin is anticipated preoperatively, radiation before surgery increases the likelihood of a negative margin. When a positive margin is encountered postoperatively, options may include re-excision, adjuvant radiation, or close observation. (see Image. Excision of Leiomyosarcoma of the Rectum)
Leiomyosarcoma of Retroperitoneum
Sarcomas of the retroperitoneum often present as large tumors and commonly originate from the smooth muscle lining of medium to large visceral veins, such as the inferior vena cava and the gonadal veins. The challenge in these cases is attaining a negative margin. An en bloc resection of the leiomyosarcoma along with any involved adjacent organs is associated with the best outcome for resectable tumors.[22] Because leiomyosarcomas are usually well-defined, compression of adjacent organs, rather than invasion, is a common intraoperative finding and sometimes allows for the preservation of surrounding organs. Tumor rupture and spillage are uniformly associated with poor outcomes and should be avoided.[23] Preoperative radiation is considered ineffective in reducing local recurrence and improving survival in retroperitoneal leiomyosarcoma. However, it may be used to facilitate resection in cases where preoperative imaging is concerning for a close margin.[24] (see Image. Excision of Leiomyosarcoma of the Rectum)
Uterine Leiomyosarcoma
Hysterectomy and en bloc resection of the leiomyosarcoma are the surgical standards for uterine leiomyosarcoma. Bilateral oophorectomy is recommended for all patients; however, ovarian preservation may be considered in some premenopausal patients.[25] Unlike carcinomas, the rate of lymph node involvement in leiomyosarcoma is low (5%-11%), and lymph node dissection is unnecessary.[26] When the preoperative workup is concerning for a uterine leiomyosarcoma, every effort should be made to perform an en bloc resection without tumor spillage. Extrauterine disease, if resectable, should be completely excised. However, the diagnosis is often made on an incidental myomectomy or hysterectomy specimen, which may require a second-look operation for complete removal.[25]
Metastatic and Recurrent Disease
The current National Comprehensive Cancer Network (NCCN) guidelines recommend pulmonary metastasectomy for oligometastatic disease in highly selected patients with no other sites of distant disease.[27] Local recurrences should be managed with surgical resection if amenable to complete tumor extirpation.[27]
Radiation Oncology
The general principles of radiotherapy for any STS also apply to patients diagnosed with non-uterine leiomyosarcoma. Uterine leiomyosarcoma is a separate subgroup of patients diagnosed with leiomyosarcoma, covered in a separate section.
Perioperative radiotherapy for STS is the gold standard of treatment for localized disease in extremities, trunk, and head or neck regions.[6] Two prospective randomized trials evaluating external beam radiotherapy and postoperative brachytherapy demonstrated better local control rates by adding adjuvant radiotherapy to surgery in patients with STS of the extremities and trunk.[18][28] Although the benefit of adjuvant radiotherapy is apparent in high-grade and low-grade STS, high-grade tumors derive a greater degree of benefit. Interstitial brachytherapy and intensity-modulated radiotherapy are two additional approaches for delivering radiotherapy to the extremity or trunk STS. They have never been compared to external beam radiotherapy in prospective trials for patients with STS.[29]
The timing of radiotherapy (preoperative and postoperative) is a topic of debate. Preoperative radiotherapy has the benefit of delivering a lower total dose with a shorter course of treatment. The treatment field is smaller, which leads to less radiation toxicity and improved extremity function. There is also potential downstaging of a borderline resectable sarcoma of an extremity with the possibility of salvaging the limb. However, preoperative radiotherapy is associated with a higher rate of wound healing complications (35% for preoperative radiotherapy compared to 17% with postoperative radiotherapy). On the other hand, postoperative radiotherapy allows for a definitive assessment of the tumor (including grade and margin status) and carries a lower rate of postoperative wound healing complications. However, it is associated with higher rates of fibrosis, edema, and joint stiffness.[30][31]
Patients with superficial or contained STS up to a size of 5 cm who undergo complete excision of the tumor with wide margins (more than 1 cm clean margin) can be monitored clinically without the need for postoperative radiotherapy. No evidence supporting this approach exists, and it is recommended that an interprofessional sarcoma group be involved before considering such an approach.[31] Adding adjuvant radiotherapy is superior to surgery alone in patients with positive margins.[20] Two prospective studies from Memorial-Sloan Kettering[28] and the National Cancer Institute (NCI)[18] demonstrate a low local recurrence rate if adjuvant radiotherapy is in the treatment plan for patients with positive surgical margins.
Although adding perioperative radiotherapy improves the local outcomes, it has not shown any benefit in terms of overall survival or distant recurrence-free survival.[31]
Considerations in Retroperitoneal Sarcoma
At this point, no consensus exists on the timing or benefit of perioperative radiotherapy for patients diagnosed with retroperitoneal STS. The first randomized trial conducted by the NCI, published in 1993, clearly demonstrated that intraoperative radiotherapy improved local control but did not show a difference in overall survival and distant metastasis.[19] A retrospective National Cancer Database (NCDB) study reported an overall survival benefit of preoperative and postoperative radiotherapy compared to patients who received surgery alone. However, this study has limitations due to a lack of specific information concerning factors such as dose, complications, and other pertinent details.[32] A phase 1 to 2 trial led by the Italian-Spanish sarcoma group enrolled 86 patients with retroperitoneal STS to receive ifosfamide at a dose of 14 g/m² with radiotherapy at a dose of 50.4 Gy. The study reported local and distant recurrence in 37% and 26% of patients, respectively, at the 5-year follow-up. The study reported disease-free survival and overall survival of 44% and 59%, respectively.[33] Results of the European Organization for Research and Treatment of Cancer (EORTC)-Soft Tissue and Bone Sarcoma Group (STBSG) trial (NCT01344018; EORTC 62092-22092) were posted on clinicaltrials.gov in April 2023. For up to 7 years, this phase 3 randomized clinical trial compared abdominal recurrence-free survival between two groups: one receiving preoperative radiotherapy plus surgery (n=133) and the other undergoing surgery alone (n=133), in patients with untreated nonmetastatic retroperitoneal sarcoma. Abdominal recurrence or death occurred in 45.9% of patients treated with surgery alone and in 45.1% of patients treated with preoperative radiotherapy plus surgery.
Considerations in Uterine Leiomyosarcoma
The role of radiotherapy in uterine leiomyosarcoma is different from that in extrauterine leiomyosarcoma. A phase 3 trial conducted by the EORTC (protocol 55874) evaluated the role of adjuvant external beam radiotherapy in patients with stage I and II uterine leiomyosarcoma. The results showed no significant difference between the two groups in local recurrence rates, distant recurrence rates, or overall survival. Although the results did not achieve statistical significance, researchers observed a trend toward reducing overall survival in the radiotherapy arm.[34] The French sarcoma group study (SARCGYN study) tested the combination of chemotherapy followed by radiotherapy versus radiotherapy alone. In the cohort of 81 patients, 53 patients had leiomyosarcoma. The chemotherapy included a combination of doxorubicin, cisplatin, and ifosfamide (4 cycles to be given after radiotherapy). The trial demonstrated a 3-year disease-free survival benefit in the combination arm, although no overall survival benefit was shown at the 3-year and 5-year benchmarks.[35] Adjuvant radiotherapy is not recommended in an optimally resected uterine leiomyosarcoma due to the lack of obvious benefit. In the advanced stage, incompletely resected, or metastatic disease, radiotherapy can be an option on a case-to-case basis.[8]
Neoadjuvant Chemoradiation for Extremity or Trunk Soft Tissue Sarcoma
Although perioperative radiotherapy is the standard of care for STS of the extremity or trunk, the combination of chemotherapy with radiotherapy is still a topic of debate. The experience from non-sarcoma tumors has shown the positive radio-sensitizing effect of combining chemotherapy with radiotherapy. The Radiation Therapy Oncology Group (RTOG) 9154 conducted a prospective phase 2 trial to evaluate the benefit of combining chemotherapy with radiotherapy in patients with STS of the extremity and trunk. Although grade 3 toxicity was relatively high in these patients, 5-year distant disease-free survival and overall survival were 64% and 71%, respectively.[36] Many other chemotherapy regimens have undergone testing since then. Among all of them, ifosfamide is the most effective drug. Patients who developed tumor necrosis on ifosfamide had the lowest local recurrence rates. Similarly, patients with STS of the extremity or trunk received an increasing dose of gemcitabine and ifosfamide concurrently with preoperative radiotherapy (50 Gy). This approach resulted in 5-year local control, distant metastasis-free, and overall survival rates of 85%, 80%, and 86%, respectively.[37] Despite encouraging results, neoadjuvant chemoradiation for STS or leiomyosarcoma remains experimental.
Medical Oncology
Leiomyosarcoma is a tumor with complex and unbalanced karyotypes characterized by severe genomic instability, which results in multiple genetic aberrations. As a result, leiomyosarcoma is considered moderately sensitive to chemotherapy.[38]
Adjuvant Chemotherapy after Surgery
After surgery, adjuvant chemotherapy for a patient diagnosed with STS has primarily been tested in patients with extremity and truncal STS.[39] Although adjuvant therapy has proven to benefit the pediatric age group, the same has been controversial for adult STS. The Sarcoma Meta-Analysis Collaboration (SMAC) published its first meta-analysis in 1997, which included 14 trials investigating the role of adjuvant chemotherapy in STS. Notably, the chemotherapy offered in these trials had doxorubicin as the only active component when combined with another drug. The analysis showed a statistically significant improvement in both local and distant recurrence-free survival, but the benefit in overall survival could not reach statistical significance. Despite this, the analysis demonstrated a 6% absolute benefit over 10 years.[40] The SMAC published an updated meta-analysis in 2008, including four more trials that included patients treated with the combination of ifosfamide and doxorubicin. This analysis again showed the same benefit in local recurrence-free survival, distant recurrence-free survival, and overall recurrence-free survival. However, this analysis showed a statistically significant overall survival benefit (odds ratio (OR) for death 0.56; 95% CI, 0.36 to 0.85). Subgroup analysis of patients receiving the combination of doxorubicin and ifosfamide showed an absolute risk reduction of 11%, which was not observed in the doxorubicin-only group.[41]
The Italian Sarcoma Group (ISG) conducted a randomized trial to assess the role of adjuvant chemotherapy in high-risk patients with spindle cell STS of extremities or pelvis. The group receiving adjuvant chemotherapy (epirubicin or ifosfamide with mesna) showed a better distant metastasis recurrence rate (45% versus 28%) and a better overall survival at 4 years (69% versus 50%). However, at a 7-year follow-up, the statistical significance was lost, and the local and distant relapse rates also became similar (44% versus 45%).[42] The EORTC conducted a randomized trial (ERTOC 62931) to investigate the role of adjuvant doxorubicin and ifosfamide in localized STS. All patients with positive margins also underwent adjuvant radiotherapy. This study did not report any difference in the recurrence-free survival or overall survival among the two groups. A significant limitation of this study is the use of ifosfamide at a lower dose (5 g/m2), which might have led to inferior results.[43]
Current guidelines suggest that in patients with high-grade or intermediate-grade STS, which is more than 5 cm in size, doxorubicin- and ifosfamide-based adjuvant chemotherapy can be considered as a viable option (Category 2B, NCCN Version 4.2019).
Neoadjuvant Chemotherapy
In theory, neoadjuvant chemotherapy can help shrink the tumor, thereby improving resectability, achieving negative margins, and enabling earlier control of microscopic disease, both local and distant. In addition, it can provide an essential clue in terms of the responsiveness of the tumor to chemotherapy. In a retrospective study examining STS and bone sarcoma, neoadjuvant chemotherapy was not associated with worse outcomes.[44] One of the early intergroup phase 3 trials evaluating the role of neoadjuvant chemotherapy (MAID regimen) in patients with large high-grade extremity STS and bone sarcomas reported a CR/PR rate of 32%. This study demonstrated a benefit in high-grade, borderline resectable lesions, or those tumors with pulmonary metastases, particularly in younger patients.[45] A European phase 2 to 3 trial in patients with STS of the extremity or trunk reported no difference in disease-free survival (56% and 52%) in patients receiving surgery with or without neoadjuvant chemotherapy. The trial's limitations include the use of a lower dose of chemotherapy and an inconsistent definition of high-risk sarcomas, where low-grade tumors larger than 8 cm are categorized as high-risk sarcoma.[46]
The ISG enrolled 252 patients to receive neoadjuvant chemotherapy, followed by surgery and then randomized them to receive two more postoperative cycles (epirubicin and ifosfamide). The patients with a positive or a negative margin on surgery had a similar overall survival. Patients who had a positive surgical margin and received adjuvant radiotherapy along with neoadjuvant chemotherapy had a cumulative local recurrence rate of zero. This study concluded that neoadjuvant chemotherapy could offset the negative impact of positive margins upon surgical resection and improve local control and survival.[47]
A histology-tailored approach was evaluated in a multicenter study, including the Italian, Spanish, French, and Polish Sarcoma Groups. The standard regimen of epirubicin and ifosfamide was compared to histology-tailored neoadjuvant treatment. The trial was stopped early due to overwhelming evidence in favor of standard chemotherapy with epirubicin and ifosfamide. The disease-free survival (62% versus 38%) and overall survival (89% versus 64%) favored the standard chemotherapy. Except for the myxoid liposarcoma subgroup, where trabectedin showed equal efficacy compared to standard chemotherapy, all other disease subgroups exhibited poor outcomes with histology-tailored therapy. This trial showed that a histology-tailored approach is not necessary when considering neoadjuvant chemotherapy.[48]
Chemotherapy in Metastatic or Unresectable Soft Tissue Sarcoma
First-line treatment: No established best first-line chemotherapy regimen exists for metastatic leiomyosarcoma. In a patient diagnosed with unresectable metastatic disease, therapy aims to palliate the symptoms and improve the quality of life.
Anthracycline-Based Regimens
Anthracyclines are usually the first choice of treatment for patients with metastatic STS. A response rate of 12% to 24% has been reported in the literature, although cardiotoxicity is a limiting factor in the use of doxorubicin.[49] Over many years, various chemotherapeutic agents have been combined with anthracyclines (doxorubicin or epirubicin) to achieve a better outcome. Although none of the regimens have ever demonstrated an improved overall survival, the combination arms have improved progression-free survival and response rates. The phase 3 trial EORTC STBSG 62012 was a pivotal trial comparing single-agent doxorubicin with a combination of doxorubicin and ifosfamide. Although the response rate with combination therapy doubled (26.5% versus 13.6%), and progression-free survival improved (7.4 months versus 4.6 months), the overall survival benefit at 1 year (60% versus 51% patients; P = .76 and median survival of 14.3 months versus 12.8 months in the single-agent arm) did not reach statistical significance. Although this was a negative trial, the combination of doxorubicin and ifosfamide is still considered for patients requiring tumor shrinkage before the surgery or when the tumor is close to a critical structure.[49]
The drug olaratumab, a platelet-derived growth factor receptor alpha-blocking antibody, was granted accelerated approval by the FDA in June 2016. This decision was based on the results of a phase 2 JGDG study that included patients with locally advanced unresectable or metastatic STS. However, the phase 3 trial (ANNOUNCE trial) reported at the American Society of Clinical Oncology (ASCO) 2018 annual meeting did not demonstrate the same overall survival benefit, which led to the suspension of the drug by the FDA.
Gemcitabine-Based Regimens
Gemcitabine has shown activity in STS as a single agent and in combination with other chemotherapy agents. The infusion rate, fixed at 10 mg/m2/min, is superior to the 30-minute infusion, which is more common.[50] The phase 2 study, Comparison of Gemcitabine Versus Gemcitabine Plus Docetaxel in Unresectable Soft Tissue Sarcoma (SARC002), showed an improved objective response, progression-free survival, and overall survival with gemcitabine plus docetaxel compared to gemcitabine alone in advanced, previously treated STS.[51] The subsets of both uterine leiomyosarcoma and extrauterine leiomyosarcoma were sensitive to the combination of gemcitabine and docetaxel. Following the success of this study, the combination of gemcitabine and docetaxel was compared with single-agent doxorubicin in the phase 3 UK-GeDDiS trial. No statistical difference was found between the two groups of doxorubicin and gemcitabine or docetaxel in the proportion of patients alive at 24 weeks. The median progression-free survival also did not differ between the two groups.[52] Notably, this trial used a lower dose of gemcitabine (675 mg/m2) and a regular dose of docetaxel (75 mg/m2), possibly contributing to the lower efficacy and higher toxicity, respectively. The trial also recommends using doxorubicin-based regimens as the first line of treatment in patients with metastatic STS.[52]
Ifosfamide Monotherapy
Ifosfamide monotherapy is also active in patients with metastatic STS, resulting in a response rate of 25% and a median overall survival of 1 year. The drug has a dose-response relationship, where higher doses result in a better response rate, albeit at the cost of higher toxicity. However, this has never translated into a better survival rate. Ifosfamide should be given at 9 to 10 g/m² with each cycle, repeated every 3 weeks. A dose beyond 12 g/m2 saturates the enzymes and only adds to toxicity. Hemorrhagic cystitis, myelotoxicity, nephrotoxicity, and neurotoxicity are the limiting toxicities of ifosfamide.[53]
A phase 3 randomized trial evaluated the effectiveness of administering a high dose of ifosfamide with stem cell rescue in patients with STS. No overall survival benefit was demonstrated in the trial.[54] An older meta-analysis of 1337 patients enrolled in EORTC-STBSG trials, evaluating the role of ifosfamide in the first-line setting, reported lower drug activity in patients with non-uterine leiomyosarcoma.[55] Hence, adding ifosfamide to doxorubicin is not routinely recommended in patients with non-uterine leiomyosarcoma unless the tumor is close to a critical organ or vessel.
Second-line treatment: Multiple drugs have been evaluated as a second-line treatment for STS or leiomyosarcoma. A gemcitabine-based regimen or an anthracycline-based regimen is an option in the second line, depending on the agents used in the first-line treatment. However, new targeted agents have provided a lot of options. Monotherapy with trabectedin is the most promising of these and has been approved by the FDA for patients with leiomyosarcoma and liposarcoma. Pazopanib, eribulin, liposomal doxorubicin, dacarbazine, and hormonal therapy (only for uterine leiomyosarcoma) are also active in leiomyosarcoma.
Trabectedin (ET-743)
Trabectedin is approved in the United States and European Union to treat patients with unresectable or metastatic liposarcoma or leiomyosarcoma who have progressed after receiving a first-line anthracycline-based regimen or were ineligible for such a regimen. The approval basis was the progression-free survival benefit (4.2 months versus 1.5 months; CI, 0.44 to 0.70; P < .01) demonstrated in the phase 3 trial, where trabectedin was compared with dacarbazine. Although no overall survival benefit was observed in the trial, patients in the trabectedin arm had stable disease for a significantly longer time (6.01 months versus 4.17 months; P < 0.001) and had a higher clinical benefit ratio (34% versus 19%; hazard ratio (HR), 2.3; CI, 1.45 to 3.7; P < .001) compared to those in the dacarbazine arm. The adverse effect profile is quite manageable with myelosuppression. Hepatic toxicity was reported as the most common grade 3/4 toxicity in the phase 3 trial.[56] The French sarcoma group recently concluded a phase 2 trial to evaluate if trabectedin can be stopped after six cycles. After six cycles, the patients who discontinued therapy experienced a rapid progression of the disease and a significantly low progression-free survival.[57] Currently, trabectedin is being tested in combination with immunotherapy and chemotherapy to improve treatment efficacy.
Pazopanib
Multiple anti-angiogenic agents, such as bevacizumab, sorafenib, sunitinib, pazopanib, vandetanib, DC-101, and TNP-470, have shown anti-sarcoma activity in mouse models. Few of these drugs showed clinical activity in phase 1 trials; however, the success of most of these drugs could not be replicated in phase 2 trials.[58]
Pazopanib is an oral multitarget tyrosine kinase inhibitor approved for use in advanced STS, regardless of histology, for patients who have previously received chemotherapy. Pazopanib is administered at an oral dosage of 800 mg daily. Pazopanib is an inhibitor of vascular endothelial growth factor receptor (VEGF)-mediated angiogenesis and blocks the growth-promoting receptor tyrosine kinase (RTKs), including platelet-derived growth factor receptor, fibroblast growth factor receptor, and KIT-1. The FDA approved the drug based on the progression-free survival benefit (4.6 months versus 1.6 months; HR, 0.35, 95% CI; 0.26 to 0.48; P < .001) observed in the phase 3 trial (PALETTE) where the study compared pazopanib to placebo. The pazopanib arm had no observable overall survival benefit compared to the placebo arm. Of note, liposarcoma was excluded from the phase 3 trial due to poor response in the phase 2 trial.[59] Hepatotoxicity, cardiotoxicity, and thyroid dysfunction are some of the most common adverse effects. Rarely, reports of reversible posterior leukoencephalopathy syndrome have been documented.[60][61]
Eribulin (E7389)
Eribulin mesylate (E7389) is an analog of halichondrin B approved for metastatic breast cancer. Eribulin mesylate blocks the G2 phase and the M phase of the cell cycle through a tubulin-based antimitotic mechanism. This blockage inhibits the spindle formation in the cell cycle, leading to cancer cell apoptosis.[62] The FDA has approved eribulin for patients with unresectable or metastatic liposarcoma whose tumor progressed after anthracycline chemotherapy, based on the results of the phase 3 trial (E7389-G000-309).[63] In a pre-planned subset analysis of the phase 3 trial results, the patients diagnosed with leiomyosarcoma showed comparable efficacy to single-agent dacarbazine. The median overall survival for eribulin versus dacarbazine (12.7 months versus 13.0 months, respectively (HR, 0.93 [95% CI, 0.71 to 1.20]; P = .57) and the median progression-free survival (2.2 months versus 2.6 months, respectively, (HR, 1.07 [95% CI, 0.84 to 1.38]; P = .58) and objective response rate (5% versus 7%) were not significantly different.[64] The FDA has not yet approved the drug for leiomyosarcoma. However, it provides a reasonable option for patients with leiomyosarcoma. Eribulin is administered intravenously at a dosage of 1.4 mg/m² over 2 to 5 minutes on Days 1 and 8 of a 21-day cycle. Close monitoring of liver and renal function is necessary with appropriate dose adjustments in patients with impairment of either of the two. Adverse effects of eribulin include neutropenia, peripheral nerve neuropathy, and QTc prolongation.
Immunotherapy
Immunotherapy has not shown much promise in the STS subgroup overall. Except for a few case reports, there is a scarcity of evidence of immunotherapy working for patients diagnosed with STS. Two trials evaluating nivolumab and pembrolizumab did not demonstrate any benefit in the leiomyosarcoma subgroup.[65][66] The phase 2 trial, ALLIANCE A091401, evaluated the role of single-agent nivolumab versus the combination of nivolumab and ipilimumab in patients with heavily treated, unselected, metastatic sarcoma. A total of 38 patients were evaluable in each arm. One-third of all the patients had uterine or extrauterine leiomyosarcoma. Out of the 8 patients who achieved a response in either arm, 3 had leiomyosarcoma. The objective response rate for single-agent nivolumab was 8% compared to a 16% response in the arm treated with nivolumab and ipilimumab. The median overall survival was 14.3 months in the combination arm, but the rate of grade 3 and 4 events was 14%. The lower rate of adverse events was attributed to the lower dose of ipilimumab (used at 1 mg/kg). Arguing that a high tumor mutational burden, characteristic of leiomyosarcoma, may have led to a higher activity of the combination of nivolumab and ipilimumab in leiomyosarcoma.[65] Currently, multiple agents are under investigation in combination with chemotherapy and radiotherapy, which are in phase 1 to 2 studies. The FDA has not yet approved the combination of ipilimumab and nivolumab in STS.
Tumors that Exhibit Specific Gene Mutations
Microsatellite Instability-High
- The FDA granted accelerated approval for pembrolizumab in May 2017 for adult and pediatric patients with unresectable or metastatic solid tumors exhibiting microsatellite instability-high (MSI-H) or mismatch repair deficiency (dMMR). The eligible patients included those whose tumors progressed after prior treatment and those with no satisfactory alternative treatment options.[67] The approval was granted based on the 149 patients included in the KEYNOTE-016, KEYNOTE-164, KEYNOTE-012, KEYNOTE-028, and KEYNOTE-158 trials.[67]
Neurotrophic Tyrosine Kinase Inhibitor - Entrectinib[68]
- Entrectinib has approval for patients who have a solid tumor that harbors the NTRK gene fusion without a known acquired resistance mutation, are metastatic, or where surgical resection is likely to result in severe morbidity, and have progressed following treatment or have no satisfactory alternative therapy. The FDA approved Entrectinib in April 2019 based on an integrated analysis of phase 2 STARTRK-2, phase 1 STARTRK-1, and the phase 1 ALKA-372-001 trials. These trials were conducted across 15 countries and 150 clinical trial sites. Entrectinib demonstrated an objective response rate of 57%.
Dacarbazine
Dacarbazine is an alkylating agent that has activity in STS. The drug has shown a response rate of 30% in combination with doxorubicin versus doxorubicin alone and a response rate of 49% in combination with gemcitabine versus dacarbazine alone.[53][69] A recent phase 2 trial evaluating the combination of sorafenib and dacarbazine in patients with metastatic leiomyosarcoma (approximately 60% of patients), synovial sarcoma, and malignant peripheral nerve sheath tumor achieved its primary endpoint of disease control rate of 46% at 18 weeks.[70] The dose depends on the protocol used. Myelosuppression and hepatotoxicity are significant adverse events.
Pegylated Liposomal Doxorubicin
Pegylated liposomal doxorubicin is a formulation of doxorubicin in poly(ethylene glycol)-coated (stealth) liposomes. The drug has a prolonged circulation time and does not cause cardiotoxicity like doxorubicin. Dermatologic toxicity is the main adverse effect of pegylated liposomal doxorubicin.[71] Multiple studies have demonstrated the activity of pegylated liposomal doxorubicin in metastatic STS either alone or combined with ifosfamide.[72][73] Pegylated liposomal doxorubicin was also tested against single-agent doxorubicin in an EORTC-SSTSBG phase 2 trial.[73] Although comparable response rates were reported amongst the two groups, as expected, cardiotoxicity was significantly lower in the pegylated liposomal doxorubicin arm. The Food and Drug Administration (FDA) has not yet approved pegylated liposomal doxorubicin for patients with leiomyosarcoma.
Hormonal Therapy
As with other gynecological malignancies, uterine leiomyosarcoma exhibits estrogen receptors in 7% to 70% of patients and progesterone receptors in 17% to 60% of patients. Retrospective studies have shown aromatase inhibitor activity with letrozole, anastrozole, or exemestane in uterine leiomyosarcoma, with partial response reported in 9% to 12% of patients.[74][75]
Staging
Leiomyosarcoma staging is according to the organ of origin. Uterine leiomyosarcoma staging follows the Federation of Gynecology and Obstetrics (FIGO) staging system. According to the American Joint Committee of Cancer (AJCC) staging, an extrauterine leiomyosarcoma gets staged. However, the 8th edition of AJCC has resolved a long-standing variability in STS staging. In the latest edition, AJCC has provided a separate staging system for STS of the retroperitoneum, head or neck region, and extremities or trunk region.[76] Notably, despite the AJCC 8th edition addressing some significant issues of previous editions, the staging system still does not translate well into survival.[76]
Grading of Soft Tissue Sarcoma or Leiomyosarcoma
The College of American Pathologists and the AJCC recommend the 3-tiered system of the French Federation of Cancer Centres/Fédération Nationale des Centres de Lutte Contre le Cancer (FNCLCC). The relative ease of use and well-balanced components, including a score for dedifferentiation (scored from 1 to 3), mitoses (scored from 1 to 3), and necrosis (scored from 0 to 2), make the French system a suitable scoring system.[77]
- Tumor differentiation
- 1: Closely resemble normal adult mesenchymal tissue
- 2: Histologic typing is uncertain
- 3: Embryonal or undifferentiated sarcoma, sarcoma of doubtful type, synovial sarcoma, soft tissue osteosarcoma, and Ewing sarcoma or primitive neuroectodermal tumor of soft tissue
- Mitotic count: measured in the most mitotically active area of sarcoma, 10 successive HPFs are assessed using a 40x objective.
- 1: 0-9 mitosis/HPF
- 2: 10-19 mitosis/HPF
- 3: >20 mitosis/HPF
- Tumor necrosis
- 0: No necrosis
- 1: <50% necrosis
- 2: >50% necrosis
Grading System
The grading system entails assigning a cumulative score to the sarcoma based on evaluations of tumor differentiation, mitotic count, and tumor necrosis. For instance, a leiomyosarcoma with a differentiation score of 1, a mitotic count score of 1, and a tumor necrosis score of 0 would be classified as grade 2 (1 + 1 + 0).
- GX: Grade cannot be assessed
- G1: Total score = 2–3
- G2: Total score = 4–5
- G3: Total score = 6–8
AJCC 8th edition staging for soft tissue sarcoma of extremities and trunk:[76]
TNM staging:
- Tumor staging (T):
- T1: Tumor less than or equal to 5 cm in greatest dimension
- T2: Tumor over 5 cm and less than or equal to 10 cm in greatest dimension
- T3: Tumor over 10 cm and less than or equal to 15 cm in greatest dimension
- T4: Tumor over 15 cm in greatest dimension
- Node staging (N):
- N0: No regional lymph node metastasis or unknown lymph node status
- N1: Regional lymph node metastasis
- Metastasis staging (M):
- M0: No distant metastasis
- M1: Distant metastasis
Stage groups:
- Stage I:
- Stage IA: T1; N0; M0; G1
- Stage IB: T2, T3, T4; N0; M0; G1
- Stage II: T1; N0; M0; G2/3
- Stage III
- Stage IIIA: T2; N0; M0; G2/3
- Stage IIIB: T3, T4; N0; M0; G2/3
- Stage IV: Any T; N1; M0; any G Any T; any N; M1; any G
AJCC 8th edition staging for retroperitoneum: The staging of STS of the retroperitoneum is similar to the staging system used for extremities and trunk. The only difference is that in the patients with STS of the retroperitoneum, the N1 node-positive patients are classified under Stage IIIB. In contrast, in the STS of the extremities and trunk, they fall under Stage IV.[78]
AJCC 8th edition staging for head and neck: There are no staging groups in the head and neck region in AJCC 8th edition as it would need the application of the French grading system, and very extensive lesions (T4) remain unclassified.[77]
- T category
- T1: less than or equal to 2 cm
- T2: greater than 2 but less than or equal to 4 cm
- T3: greater than 4 cm
- T4: Invasion of adjoining structures
- T4a: Invasion of the orbit, skull base, dura, central compartment viscera, pterygoid muscles, or facial skeletal involvement
- T4b: Invasion of brain parenchyma, involvement of the central nervous system through the perineural spread, invasion of prevertebral muscle, or carotid artery encasement
- N category
- N0: No regional lymph node metastasis
- N1: Regional lymph node metastasis
- M category
- M0: No distant Metastasis
- M1: Distant metastasis
FIGO staging for uterine leiomyosarcoma (2009):[8]
- Stage I: Tumor limited to the uterus
- IA: Less than 5 cm in greatest dimension
- IB: More than 5 cm in greatest dimension
- Stage II: Tumor extends beyond the uterus, within the pelvis
- IIA: Adnexal involvement
- IIB: Involvement of other pelvic tissues
- Stage III: Tumor invades abdominal tissues
- IIIA: 1 site
- IIIB: more than 1 site
- IIIC: Involves pelvic and para-aortic lymph nodes
- Stage IV: Tumor invades pelvic organs and distant metastasis
- IVA: Invasion of bladder or rectum
- IVB: Distant metastases
Prognosis
The three most important prognostic factors are histologic grade, tumor size, and tumor depth. Tumor size, bone, or neurovascular involvement, together with the grade of the tumor, are significantly associated with poor outcomes, specifically in leiomyosarcoma.[79] Available calculators and normograms have been developed to help determine prognosis.[80]
Histologic grade is the most predictive risk factor. This factor can independently estimate the aggressiveness of cancer, the probability of distant metastasis, and disease-specific survival.[79][81][82] Leiomyosarcoma is inherently an aggressive malignancy, with 90% of patients diagnosed with grade 2 to 3 cancer, and as a result, are at a higher risk of distant recurrence and decreased disease-specific survival compared to other histologies.[81] Larger tumors have a worse outcome, especially with tumors over 10 cm.[83]
Tumor location is an independent prognostic risk factor. Leiomyosarcoma of the extremities has a better outcome compared to retroperitoneal leiomyosarcoma.[82] Uterine tumors and those arising from large visceral blood vessels have been reported to have worse outcomes, although the data are debatable. Tumor depth is an important prognostic factor independent of tumor size and histologic grade, directly correlating with a worse outcome.[76][84][85]
Uterine leiomyosarcoma also follows the same pattern in prognostic features. Various studies have reported age, disease stage, surgical margins, tumor size, cellular atypia, mitotic rate, the involvement of lymphovascular channels, lymph node positivity, oophorectomy, or presence or absence of necrosis as factors that can determine prognosis.[8] Memorial Sloan Kettering has developed a clinical nomogram that utilizes age, grade, tumor size, mitotic rate, presence of cervical invasion, locoregional metastasis, and distant metastasis to assess the 5-year overall survival. The nomogram performed better compared to more traditional staging systems, such as the FIGO and AJCC classifications, in predicting the overall survival.[86].
In a recent meta-analysis of 580 patients with metastatic STS and lung-only metastasis recorded in the EORTC-STSBG trials, age, time between initial diagnosis and treatment, performance status, and involvement of the primary site were reported to help determine the prognosis of this subgroup of patients. The analysis showed that patients with a nontarget pulmonary lesion (pleural involvement) had the worst outcomes. The study also confirmed that patients treated with the combination of doxorubicin and ifosfamide in the first-line setting, followed by monotherapy with anthracycline, ifosfamide, and trabectedin or brostallicin, had the best overall survival and progression-free survival.[87]
Complications
Complications of leiomyosarcoma are site-specific. Common issues include external compression due to mass effect and early metastasis due to aggressive tumors.
Deterrence and Patient Education
Leiomyosarcoma is a rare but aggressive STS with no clear etiology in most patients and is generally seen in older patients. Patients with soft tissue masses of the trunk or extremity should seek evaluation by a specialist, particularly if the mass is increasing in size. For abdominopelvic tumors, symptoms are often vague and depend on the site of the lesion. Diagnosis and treatment at a center of excellence is highly recommended. The mainstay of treatment is surgical excision, with radiation and chemotherapy serving as adjuncts. Metastatic disease is fatal and generally shows poor response to systemic therapy. Participation in clinical trials is crucial to better understanding this disease.
Pearls and Other Issues
Key facts to keep in mind for healthcare professionals managing patients with leiomyosarcoma are as follows:
- Patients diagnosed with leiomyosarcoma should be treated at centers that experience a high volume of such patients.
- All patient cases should be discussed at tumor boards, including surgical oncologists or orthopedic oncologists, radiation oncologists, and pathologists trained in diagnosing sarcoma, medical oncologists, and radiologists.
- Gynecologists and oncologists should collaborate with the surgical oncologists and the team listed above to diagnose uterine leiomyosarcoma patients.
- The three most important prognostic factors are histologic grade, tumor depth, and tumor size.
- Surgical resection with a negative margin leads to the best outcomes in terms of overall survival.
- Perioperative radiotherapy decreases the local recurrence rate and increases local disease-free survival but has not shown a benefit in distant relapse or overall survival.
- Preoperative radiotherapy is associated with fewer complications compared to postoperative radiotherapy, although wound complications are higher with preoperative radiotherapy. A gap of 4 to 5 weeks between surgery and radiotherapy appears to reduce wound complications with preoperative radiotherapy.
- Neoadjuvant chemotherapy has proven effective in high-risk STS or leiomyosarcoma of the extremity or trunk; however, the evidence is limited.
- If neoadjuvant therapy is chosen, then a standard chemotherapy regimen with anthracycline and ifosfamide should be used rather than tailoring it according to the histology.
- No evidence exists to support the regular use of adjuvant therapy; however, if adjuvant therapy is administered, then anthracycline and ifosfamide should be used.
- Anthracycline-based regimens are usually the first-line regimens for metastatic STS or leiomyosarcoma.
- A gemcitabine-docetaxel combination can be the first-line regimen for uterine leiomyosarcoma.
- Ifosfamide is less effective in patients with extrauterine leiomyosarcoma compared to those with uterine leiomyosarcoma.
- Although the GeDDis trial did not demonstrate a difference between the 2 arms, doxorubicin remains the first treatment choice.
- Trabectedin is an effective treatment for leiomyosarcoma. Clinicians should not discontinue the drug in patients who respond to trabectedin unless the patient develops toxicity or progression of the tumor.
- Pazopanib, anlotinib, eribulin, dacarbazine, and pegylated liposomal doxorubicin are effective as single agents in leiomyosarcoma.
Enhancing Healthcare Team Outcomes
Leiomyosarcoma is one of the most common subtypes of soft tissue sarcoma. The clinical presentation may be nonspecific and depends on the site of origin. A mass suspected to be a sarcoma should have multidisciplinary teams involved from the beginning. An image-guided core needle biopsy is critical, keeping fascial and peritoneal planes intact. Treatment relies on surgery, but chemotherapy and radiation are commonly used. The timing and sequence of therapies must be carefully planned. Patient outcomes are dependent on timely, evidence-based treatment. Given the rarity of most individual sarcomas, enrolling patients in clinical trials and keeping up-to-date with developments aimed at improving patient outcomes are critical.
Ultimately, patient outcomes rely on interprofessional communication, cooperation, and action. Inappropriate management of sarcomas often begins even before diagnosis, with poorly planned or conducted biopsies. Early involvement of a dedicated, multidisciplinary team is crucial.
Media
(Click Image to Enlarge)
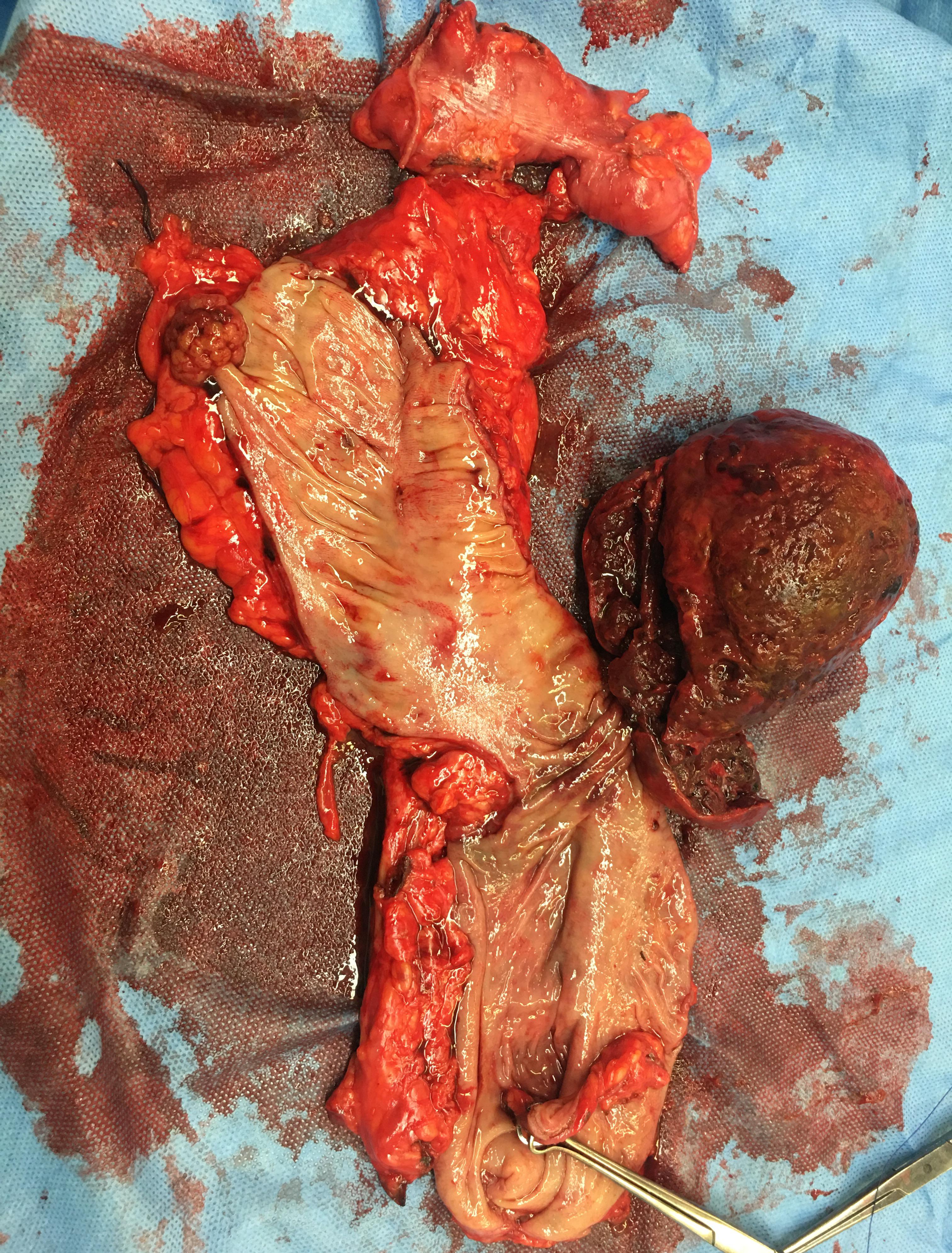
Excision of Leiomyosarcoma of the Rectum. The image illustrates the surgical excision of a leiomyosarcoma in the rectum. In cases where a positive margin is detected following the surgery, treatment options include reexcision, adjuvant radiation therapy, or close observation.
Contributed by F Mulita, MD, and I Kehagias, MD
(Click Image to Enlarge)
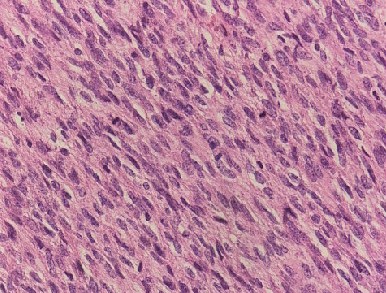
Leiomyosarcoma of the Uterus. A microscopic view of leiomyosarcoma of the uterus showing nuclear atypia, >5 to 10 mitoses per high-power field, and tumor necrosis.
Contributed by SR Zaidi, MD
References
Devaud N, Vornicova O, Abdul Razak AR, Khalili K, Demicco EG, Mitric C, Bernardini MQ, Gladdy RA. Leiomyosarcoma: Current Clinical Management and Future Horizons. Surgical oncology clinics of North America. 2022 Jul:31(3):527-546. doi: 10.1016/j.soc.2022.03.011. Epub [PubMed PMID: 35715148]
Serrano C, George S. Leiomyosarcoma. Hematology/oncology clinics of North America. 2013 Oct:27(5):957-74. doi: 10.1016/j.hoc.2013.07.002. Epub 2013 Aug 26 [PubMed PMID: 24093170]
Swallow CJ, Strauss DC, Bonvalot S, Rutkowski P, Desai A, Gladdy RA, Gonzalez R, Gyorki DE, Fairweather M, van Houdt WJ, Stoeckle E, Park JB, Albertsmeier M, Nessim C, Cardona K, Fiore M, Hayes A, Tzanis D, Skoczylas J, Ford SJ, Ng D, Mullinax JE, Snow H, Haas RL, Callegaro D, Smith MJ, Bouhadiba T, Stacchiotti S, Jones RL, DeLaney T, Roland CL, Raut CP, Gronchi A, Transatlantic Australasian RPS Working Group (TARPSWG). Management of Primary Retroperitoneal Sarcoma (RPS) in the Adult: An Updated Consensus Approach from the Transatlantic Australasian RPS Working Group. Annals of surgical oncology. 2021 Nov:28(12):7873-7888. doi: 10.1245/s10434-021-09654-z. Epub 2021 Apr 14 [PubMed PMID: 33852100]
Level 3 (low-level) evidenceAdam MA, Moris D, Behrens S, Nussbaum DP, Jawitz O, Turner M, Lidsky M, Blazer D 3rd. Hospital Volume Threshold for the Treatment of Retroperitoneal Sarcoma. Anticancer research. 2019 Apr:39(4):2007-2014. doi: 10.21873/anticanres.13311. Epub [PubMed PMID: 30952744]
Robinson E, Neugut AI, Wylie P. Clinical aspects of postirradiation sarcomas. Journal of the National Cancer Institute. 1988 Apr 20:80(4):233-40 [PubMed PMID: 3280809]
George S, Serrano C, Hensley ML, Ray-Coquard I. Soft Tissue and Uterine Leiomyosarcoma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2018 Jan 10:36(2):144-150. doi: 10.1200/JCO.2017.75.9845. Epub 2017 Dec 8 [PubMed PMID: 29220301]
Guo X, Jo VY, Mills AM, Zhu SX, Lee CH, Espinosa I, Nucci MR, Varma S, Forgó E, Hastie T, Anderson S, Ganjoo K, Beck AH, West RB, Fletcher CD, van de Rijn M. Clinically Relevant Molecular Subtypes in Leiomyosarcoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2015 Aug 1:21(15):3501-11. doi: 10.1158/1078-0432.CCR-14-3141. Epub 2015 Apr 20 [PubMed PMID: 25896974]
Roberts ME, Aynardi JT, Chu CS. Uterine leiomyosarcoma: A review of the literature and update on management options. Gynecologic oncology. 2018 Dec:151(3):562-572. doi: 10.1016/j.ygyno.2018.09.010. Epub 2018 Sep 21 [PubMed PMID: 30244960]
Cui RR, Wright JD, Hou JY. Uterine leiomyosarcoma: a review of recent advances in molecular biology, clinical management and outcome. BJOG : an international journal of obstetrics and gynaecology. 2017 Jun:124(7):1028-1037. doi: 10.1111/1471-0528.14579. Epub 2017 Apr 1 [PubMed PMID: 28128524]
Level 3 (low-level) evidenceCancer Genome Atlas Research Network. Electronic address: elizabeth.demicco@sinaihealthsystem.ca, Cancer Genome Atlas Research Network. Comprehensive and Integrated Genomic Characterization of Adult Soft Tissue Sarcomas. Cell. 2017 Nov 2:171(4):950-965.e28. doi: 10.1016/j.cell.2017.10.014. Epub [PubMed PMID: 29100075]
Martin-Liberal J. Leiomyosarcoma: Principles of management. Intractable & rare diseases research. 2013 Nov:2(4):127-9. doi: 10.5582/irdr.2013.v2.4.127. Epub [PubMed PMID: 25343116]
Ip PP, Cheung AN. Pathology of uterine leiomyosarcomas and smooth muscle tumours of uncertain malignant potential. Best practice & research. Clinical obstetrics & gynaecology. 2011 Dec:25(6):691-704. doi: 10.1016/j.bpobgyn.2011.07.003. Epub 2011 Aug 23 [PubMed PMID: 21865091]
Bell SW, Kempson RL, Hendrickson MR. Problematic uterine smooth muscle neoplasms. A clinicopathologic study of 213 cases. The American journal of surgical pathology. 1994 Jun:18(6):535-58 [PubMed PMID: 8179071]
Level 3 (low-level) evidenceLeitao MM, Soslow RA, Nonaka D, Olshen AB, Aghajanian C, Sabbatini P, Dupont J, Hensley M, Sonoda Y, Barakat RR, Anderson S. Tissue microarray immunohistochemical expression of estrogen, progesterone, and androgen receptors in uterine leiomyomata and leiomyosarcoma. Cancer. 2004 Sep 15:101(6):1455-62 [PubMed PMID: 15316901]
Demas BE, Heelan RT, Lane J, Marcove R, Hajdu S, Brennan MF. Soft-tissue sarcomas of the extremities: comparison of MR and CT in determining the extent of disease. AJR. American journal of roentgenology. 1988 Mar:150(3):615-20 [PubMed PMID: 3257620]
Strauss DC, Qureshi YA, Hayes AJ, Thway K, Fisher C, Thomas JM. The role of core needle biopsy in the diagnosis of suspected soft tissue tumours. Journal of surgical oncology. 2010 Oct 1:102(5):523-9. doi: 10.1002/jso.21600. Epub [PubMed PMID: 20872955]
Narvani AA, Tsiridis E, Saifuddin A, Briggs T, Cannon S. Does image guidance improve accuracy of core needle biopsy in diagnosis of soft tissue tumours? Acta orthopaedica Belgica. 2009 Apr:75(2):239-44 [PubMed PMID: 19492564]
Yang JC, Chang AE, Baker AR, Sindelar WF, Danforth DN, Topalian SL, DeLaney T, Glatstein E, Steinberg SM, Merino MJ, Rosenberg SA. Randomized prospective study of the benefit of adjuvant radiation therapy in the treatment of soft tissue sarcomas of the extremity. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 1998 Jan:16(1):197-203 [PubMed PMID: 9440743]
Level 1 (high-level) evidenceHaas RL, Baldini EH, Chung PW, van Coevorden F, DeLaney TF. Radiation therapy in retroperitoneal sarcoma management. Journal of surgical oncology. 2018 Jan:117(1):93-98. doi: 10.1002/jso.24892. Epub 2017 Nov 22 [PubMed PMID: 29165813]
Byerly S, Chopra S, Nassif NA, Chen P, Sener SF, Eisenberg BL, Tseng WW. The role of margins in extremity soft tissue sarcoma. Journal of surgical oncology. 2016 Mar:113(3):333-8. doi: 10.1002/jso.24112. Epub 2015 Dec 10 [PubMed PMID: 26662660]
Kandel R, Coakley N, Werier J, Engel J, Ghert M, Verma S, Sarcoma Disease Site Group of Cancer Care Ontario’s Program in Evidence-Based Care. Surgical margins and handling of soft-tissue sarcoma in extremities: a clinical practice guideline. Current oncology (Toronto, Ont.). 2013 Jun:20(3):e247-54. doi: 10.3747/co.20.1308. Epub [PubMed PMID: 23737694]
Level 1 (high-level) evidenceBonvalot S, Miceli R, Berselli M, Causeret S, Colombo C, Mariani L, Bouzaiene H, Le Péchoux C, Casali PG, Le Cesne A, Fiore M, Gronchi A. Aggressive surgery in retroperitoneal soft tissue sarcoma carried out at high-volume centers is safe and is associated with improved local control. Annals of surgical oncology. 2010 Jun:17(6):1507-14. doi: 10.1245/s10434-010-1057-5. Epub [PubMed PMID: 20393803]
MacNeill AJ, Fiore M. Surgical morbidity in retroperitoneal sarcoma resection. Journal of surgical oncology. 2018 Jan:117(1):56-61. doi: 10.1002/jso.24902. Epub 2018 Jan 3 [PubMed PMID: 29314041]
Bonvalot S, Gronchi A, Le Péchoux C, Swallow CJ, Strauss D, Meeus P, van Coevorden F, Stoldt S, Stoeckle E, Rutkowski P, Rastrelli M, Raut CP, Hompes D, De Paoli A, Sangalli C, Honoré C, Chung P, Miah A, Blay JY, Fiore M, Stelmes JJ, Dei Tos AP, Baldini EH, Litière S, Marreaud S, Gelderblom H, Haas RL. Preoperative radiotherapy plus surgery versus surgery alone for patients with primary retroperitoneal sarcoma (EORTC-62092: STRASS): a multicentre, open-label, randomised, phase 3 trial. The Lancet. Oncology. 2020 Oct:21(10):1366-1377. doi: 10.1016/S1470-2045(20)30446-0. Epub 2020 Sep 14 [PubMed PMID: 32941794]
Level 1 (high-level) evidenceSeagle BL, Sobecki-Rausch J, Strohl AE, Shilpi A, Grace A, Shahabi S. Prognosis and treatment of uterine leiomyosarcoma: A National Cancer Database study. Gynecologic oncology. 2017 Apr:145(1):61-70. doi: 10.1016/j.ygyno.2017.02.012. Epub 2017 Mar 15 [PubMed PMID: 28317559]
Mbatani N, Olawaiye AB, Prat J. Uterine sarcomas. International journal of gynaecology and obstetrics: the official organ of the International Federation of Gynaecology and Obstetrics. 2018 Oct:143 Suppl 2():51-58. doi: 10.1002/ijgo.12613. Epub [PubMed PMID: 30306577]
von Mehren M, Kane JM, Agulnik M, Bui MM, Carr-Ascher J, Choy E, Connelly M, Dry S, Ganjoo KN, Gonzalez RJ, Holder A, Homsi J, Keedy V, Kelly CM, Kim E, Liebner D, McCarter M, McGarry SV, Mesko NW, Meyer C, Pappo AS, Parkes AM, Petersen IA, Pollack SM, Poppe M, Riedel RF, Schuetze S, Shabason J, Sicklick JK, Spraker MB, Zimel M, Hang LE, Sundar H, Bergman MA. Soft Tissue Sarcoma, Version 2.2022, NCCN Clinical Practice Guidelines in Oncology. Journal of the National Comprehensive Cancer Network : JNCCN. 2022 Jul:20(7):815-833. doi: 10.6004/jnccn.2022.0035. Epub [PubMed PMID: 35830886]
Level 1 (high-level) evidencePisters PW, Harrison LB, Leung DH, Woodruff JM, Casper ES, Brennan MF. Long-term results of a prospective randomized trial of adjuvant brachytherapy in soft tissue sarcoma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 1996 Mar:14(3):859-68 [PubMed PMID: 8622034]
Level 1 (high-level) evidenceHaas RL, Delaney TF, O'Sullivan B, Keus RB, Le Pechoux C, Olmi P, Poulsen JP, Seddon B, Wang D. Radiotherapy for management of extremity soft tissue sarcomas: why, when, and where? International journal of radiation oncology, biology, physics. 2012 Nov 1:84(3):572-80. doi: 10.1016/j.ijrobp.2012.01.062. Epub 2012 Apr 18 [PubMed PMID: 22520481]
Griffin AM, Dickie CI, Catton CN, Chung PW, Ferguson PC, Wunder JS, O'Sullivan B. The influence of time interval between preoperative radiation and surgical resection on the development of wound healing complications in extremity soft tissue sarcoma. Annals of surgical oncology. 2015 Sep:22(9):2824-30. doi: 10.1245/s10434-015-4631-z. Epub 2015 May 28 [PubMed PMID: 26018726]
Tiong SS, Dickie C, Haas RL, O'Sullivan B. The role of radiotherapy in the management of localized soft tissue sarcomas. Cancer biology & medicine. 2016 Sep:13(3):373-383 [PubMed PMID: 27807504]
Nussbaum DP, Rushing CN, Lane WO, Cardona DM, Kirsch DG, Peterson BL, Blazer DG 3rd. Preoperative or postoperative radiotherapy versus surgery alone for retroperitoneal sarcoma: a case-control, propensity score-matched analysis of a nationwide clinical oncology database. The Lancet. Oncology. 2016 Jul:17(7):966-975. doi: 10.1016/S1470-2045(16)30050-X. Epub 2016 May 17 [PubMed PMID: 27210906]
Level 2 (mid-level) evidenceGronchi A, De Paoli A, Dani C, Merlo DF, Quagliuolo V, Grignani G, Bertola G, Navarria P, Sangalli C, Buonadonna A, De Sanctis R, Sanfilippo R, Dei Tos AP, Stacchiotti S, Giorello L, Fiore M, Bruzzi P, Casali PG. Preoperative chemo-radiation therapy for localised retroperitoneal sarcoma: a phase I-II study from the Italian Sarcoma Group. European journal of cancer (Oxford, England : 1990). 2014 Mar:50(4):784-92. doi: 10.1016/j.ejca.2013.11.021. Epub 2013 Dec 5 [PubMed PMID: 24316063]
Reed NS, Mangioni C, Malmström H, Scarfone G, Poveda A, Pecorelli S, Tateo S, Franchi M, Jobsen JJ, Coens C, Teodorovic I, Vergote I, Vermorken JB, European Organisation for Research and Treatment of Cancer Gynaecological Cancer Group. Phase III randomised study to evaluate the role of adjuvant pelvic radiotherapy in the treatment of uterine sarcomas stages I and II: an European Organisation for Research and Treatment of Cancer Gynaecological Cancer Group Study (protocol 55874). European journal of cancer (Oxford, England : 1990). 2008 Apr:44(6):808-18. doi: 10.1016/j.ejca.2008.01.019. Epub 2008 Apr 2 [PubMed PMID: 18378136]
Level 1 (high-level) evidencePautier P, Floquet A, Gladieff L, Bompas E, Ray-Coquard I, Piperno-Neumann S, Selle F, Guillemet C, Weber B, Largillier R, Bertucci F, Opinel P, Duffaud F, Reynaud-Bougnoux A, Delcambre C, Isambert N, Kerbrat P, Netter-Pinon G, Pinto N, Duvillard P, Haie-Meder C, Lhommé C, Rey A. A randomized clinical trial of adjuvant chemotherapy with doxorubicin, ifosfamide, and cisplatin followed by radiotherapy versus radiotherapy alone in patients with localized uterine sarcomas (SARCGYN study). A study of the French Sarcoma Group. Annals of oncology : official journal of the European Society for Medical Oncology. 2013 Apr:24(4):1099-104. doi: 10.1093/annonc/mds545. Epub 2012 Nov 8 [PubMed PMID: 23139262]
Level 1 (high-level) evidenceKraybill WG, Harris J, Spiro IJ, Ettinger DS, DeLaney TF, Blum RH, Lucas DR, Harmon DC, Letson GD, Eisenberg B. Long-term results of a phase 2 study of neoadjuvant chemotherapy and radiotherapy in the management of high-risk, high-grade, soft tissue sarcomas of the extremities and body wall: Radiation Therapy Oncology Group Trial 9514. Cancer. 2010 Oct 1:116(19):4613-21. doi: 10.1002/cncr.25350. Epub [PubMed PMID: 20572040]
Tseng WW, Zhou S, To CA, Thall PF, Lazar AJ, Pollock RE, Lin PP, Cormier JN, Lewis VO, Feig BW, Hunt KK, Ballo MT, Patel S, Pisters PW. Phase 1 adaptive dose-finding study of neoadjuvant gemcitabine combined with radiation therapy for patients with high-risk extremity and trunk soft tissue sarcoma. Cancer. 2015 Oct 15:121(20):3659-67. doi: 10.1002/cncr.29544. Epub 2015 Jul 15 [PubMed PMID: 26177983]
Grimer R, Judson I, Peake D, Seddon B. Guidelines for the management of soft tissue sarcomas. Sarcoma. 2010:2010():506182. doi: 10.1155/2010/506182. Epub 2010 May 31 [PubMed PMID: 20634933]
Loong HH, Wong KH, Tse T. Controversies and consensus of neoadjuvant chemotherapy in soft-tissue sarcomas. ESMO open. 2018:3(Suppl 1):e000293. doi: 10.1136/esmoopen-2017-000293. Epub 2018 Jan 3 [PubMed PMID: 29333281]
Level 3 (low-level) evidence. Adjuvant chemotherapy for localised resectable soft-tissue sarcoma of adults: meta-analysis of individual data. Sarcoma Meta-analysis Collaboration. Lancet (London, England). 1997 Dec 6:350(9092):1647-54 [PubMed PMID: 9400508]
Level 1 (high-level) evidencePervaiz N, Colterjohn N, Farrokhyar F, Tozer R, Figueredo A, Ghert M. A systematic meta-analysis of randomized controlled trials of adjuvant chemotherapy for localized resectable soft-tissue sarcoma. Cancer. 2008 Aug 1:113(3):573-81. doi: 10.1002/cncr.23592. Epub [PubMed PMID: 18521899]
Level 1 (high-level) evidenceFrustaci S, Gherlinzoni F, De Paoli A, Bonetti M, Azzarelli A, Comandone A, Olmi P, Buonadonna A, Pignatti G, Barbieri E, Apice G, Zmerly H, Serraino D, Picci P. Adjuvant chemotherapy for adult soft tissue sarcomas of the extremities and girdles: results of the Italian randomized cooperative trial. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2001 Mar 1:19(5):1238-47 [PubMed PMID: 11230464]
Level 1 (high-level) evidenceWoll PJ, Reichardt P, Le Cesne A, Bonvalot S, Azzarelli A, Hoekstra HJ, Leahy M, Van Coevorden F, Verweij J, Hogendoorn PC, Ouali M, Marreaud S, Bramwell VH, Hohenberger P, EORTC Soft Tissue and Bone Sarcoma Group and the NCIC Clinical Trials Group Sarcoma Disease Site Committee. Adjuvant chemotherapy with doxorubicin, ifosfamide, and lenograstim for resected soft-tissue sarcoma (EORTC 62931): a multicentre randomised controlled trial. The Lancet. Oncology. 2012 Oct:13(10):1045-54. doi: 10.1016/S1470-2045(12)70346-7. Epub 2012 Sep 4 [PubMed PMID: 22954508]
Level 1 (high-level) evidenceBiau DJ, Weiss KR, Bhumbra RS, Davidson D, Brown C, Griffin A, Wunder JS, Ferguson PC. Monitoring the adequacy of surgical margins after resection of bone and soft-tissue sarcoma. Annals of surgical oncology. 2013 Jun:20(6):1858-64. doi: 10.1245/s10434-012-2863-8. Epub 2013 Jan 31 [PubMed PMID: 23370669]
Antman K, Crowley J, Balcerzak SP, Rivkin SE, Weiss GR, Elias A, Natale RB, Cooper RM, Barlogie B, Trump DL. An intergroup phase III randomized study of doxorubicin and dacarbazine with or without ifosfamide and mesna in advanced soft tissue and bone sarcomas. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 1993 Jul:11(7):1276-85 [PubMed PMID: 8315425]
Level 1 (high-level) evidenceGortzak E, Azzarelli A, Buesa J, Bramwell VH, van Coevorden F, van Geel AN, Ezzat A, Santoro A, Oosterhuis JW, van Glabbeke M, Kirkpatrick A, Verweij J, E.O.R.T.C. Soft Tissue Bone Sarcoma Group and the National Cancer Institute of Canada Clinical Trials Group/Canadian Sarcoma Group. A randomised phase II study on neo-adjuvant chemotherapy for 'high-risk' adult soft-tissue sarcoma. European journal of cancer (Oxford, England : 1990). 2001 Jun:37(9):1096-103 [PubMed PMID: 11378339]
Level 1 (high-level) evidenceGronchi A, Verderio P, De Paoli A, Ferraro A, Tendero O, Majò J, Martin J, Comandone A, Grignani G, Pizzamiglio S, Quagliuolo V, Picci P, Frustaci S, Dei Tos AP, Palassini E, Stacchiotti S, Ferrari S, Fiore M, Casali PG. Quality of surgery and neoadjuvant combined therapy in the ISG-GEIS trial on soft tissue sarcomas of limbs and trunk wall. Annals of oncology : official journal of the European Society for Medical Oncology. 2013 Mar:24(3):817-23. doi: 10.1093/annonc/mds501. Epub 2012 Oct 30 [PubMed PMID: 23110811]
Level 2 (mid-level) evidenceGronchi A, Ferrari S, Quagliuolo V, Broto JM, Pousa AL, Grignani G, Basso U, Blay JY, Tendero O, Beveridge RD, Ferraresi V, Lugowska I, Merlo DF, Fontana V, Marchesi E, Donati DM, Palassini E, Palmerini E, De Sanctis R, Morosi C, Stacchiotti S, Bagué S, Coindre JM, Dei Tos AP, Picci P, Bruzzi P, Casali PG. Histotype-tailored neoadjuvant chemotherapy versus standard chemotherapy in patients with high-risk soft-tissue sarcomas (ISG-STS 1001): an international, open-label, randomised, controlled, phase 3, multicentre trial. The Lancet. Oncology. 2017 Jun:18(6):812-822. doi: 10.1016/S1470-2045(17)30334-0. Epub 2017 May 9 [PubMed PMID: 28499583]
Level 1 (high-level) evidenceLinch M, Miah AB, Thway K, Judson IR, Benson C. Systemic treatment of soft-tissue sarcoma-gold standard and novel therapies. Nature reviews. Clinical oncology. 2014 Apr:11(4):187-202. doi: 10.1038/nrclinonc.2014.26. Epub 2014 Mar 18 [PubMed PMID: 24642677]
Patel SR, Gandhi V, Jenkins J, Papadopolous N, Burgess MA, Plager C, Plunkett W, Benjamin RS. Phase II clinical investigation of gemcitabine in advanced soft tissue sarcomas and window evaluation of dose rate on gemcitabine triphosphate accumulation. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2001 Aug 1:19(15):3483-9 [PubMed PMID: 11481354]
Maki RG, Wathen JK, Patel SR, Priebat DA, Okuno SH, Samuels B, Fanucchi M, Harmon DC, Schuetze SM, Reinke D, Thall PF, Benjamin RS, Baker LH, Hensley ML. Randomized phase II study of gemcitabine and docetaxel compared with gemcitabine alone in patients with metastatic soft tissue sarcomas: results of sarcoma alliance for research through collaboration study 002 [corrected]. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2007 Jul 1:25(19):2755-63 [PubMed PMID: 17602081]
Level 1 (high-level) evidenceSeddon B, Strauss SJ, Whelan J, Leahy M, Woll PJ, Cowie F, Rothermundt C, Wood Z, Benson C, Ali N, Marples M, Veal GJ, Jamieson D, Küver K, Tirabosco R, Forsyth S, Nash S, Dehbi HM, Beare S. Gemcitabine and docetaxel versus doxorubicin as first-line treatment in previously untreated advanced unresectable or metastatic soft-tissue sarcomas (GeDDiS): a randomised controlled phase 3 trial. The Lancet. Oncology. 2017 Oct:18(10):1397-1410. doi: 10.1016/S1470-2045(17)30622-8. Epub 2017 Sep 4 [PubMed PMID: 28882536]
Level 1 (high-level) evidenceSleijfer S, Seynaeve C, Verweij J. Using single-agent therapy in adult patients with advanced soft tissue sarcoma can still be considered standard care. The oncologist. 2005 Nov-Dec:10(10):833-41 [PubMed PMID: 16314294]
Bui-Nguyen B, Ray-Coquard I, Chevreau C, Penel N, Bay JO, Coindre JM, Cupissol D, Italiano A, Bonichon F, Lotz JP, Thyss A, Jimenez M, Mathoulin-Pélissier S, Blay JY, GSF-GETO French Sarcoma Group. High-dose chemotherapy consolidation for chemosensitive advanced soft tissue sarcoma patients: an open-label, randomized controlled trial. Annals of oncology : official journal of the European Society for Medical Oncology. 2012 Mar:23(3):777-784. doi: 10.1093/annonc/mdr282. Epub 2011 Jun 7 [PubMed PMID: 21652583]
Level 1 (high-level) evidenceSleijfer S, Ouali M, van Glabbeke M, Krarup-Hansen A, Rodenhuis S, Le Cesne A, Hogendoorn PC, Verweij J, Blay JY. Prognostic and predictive factors for outcome to first-line ifosfamide-containing chemotherapy for adult patients with advanced soft tissue sarcomas: an exploratory, retrospective analysis on large series from the European Organization for Research and Treatment of Cancer-Soft Tissue and Bone Sarcoma Group (EORTC-STBSG). European journal of cancer (Oxford, England : 1990). 2010 Jan:46(1):72-83. doi: 10.1016/j.ejca.2009.09.022. Epub [PubMed PMID: 19853437]
Level 2 (mid-level) evidenceDemetri GD, von Mehren M, Jones RL, Hensley ML, Schuetze SM, Staddon A, Milhem M, Elias A, Ganjoo K, Tawbi H, Van Tine BA, Spira A, Dean A, Khokhar NZ, Park YC, Knoblauch RE, Parekh TV, Maki RG, Patel SR. Efficacy and Safety of Trabectedin or Dacarbazine for Metastatic Liposarcoma or Leiomyosarcoma After Failure of Conventional Chemotherapy: Results of a Phase III Randomized Multicenter Clinical Trial. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2016 Mar 10:34(8):786-93. doi: 10.1200/JCO.2015.62.4734. Epub 2015 Sep 14 [PubMed PMID: 26371143]
Level 1 (high-level) evidenceLe Cesne A, Blay JY, Domont J, Tresch-Bruneel E, Chevreau C, Bertucci F, Delcambre C, Saada-Bouzid E, Piperno-Neumann S, Bay JO, Mir O, Ray-Coquard I, Ryckewaert T, Valentin T, Isambert N, Italiano A, Clisant S, Penel N. Interruption versus continuation of trabectedin in patients with soft-tissue sarcoma (T-DIS): a randomised phase 2 trial. The Lancet. Oncology. 2015 Mar:16(3):312-9. doi: 10.1016/S1470-2045(15)70031-8. Epub 2015 Feb 11 [PubMed PMID: 25680558]
Level 1 (high-level) evidenceVo KT, Matthay KK, DuBois SG. Targeted antiangiogenic agents in combination with cytotoxic chemotherapy in preclinical and clinical studies in sarcoma. Clinical sarcoma research. 2016:6():9. doi: 10.1186/s13569-016-0049-z. Epub 2016 Jun 7 [PubMed PMID: 27274393]
van der Graaf WT, Blay JY, Chawla SP, Kim DW, Bui-Nguyen B, Casali PG, Schöffski P, Aglietta M, Staddon AP, Beppu Y, Le Cesne A, Gelderblom H, Judson IR, Araki N, Ouali M, Marreaud S, Hodge R, Dewji MR, Coens C, Demetri GD, Fletcher CD, Dei Tos AP, Hohenberger P, EORTC Soft Tissue and Bone Sarcoma Group, PALETTE study group. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet (London, England). 2012 May 19:379(9829):1879-86. doi: 10.1016/S0140-6736(12)60651-5. Epub 2012 May 16 [PubMed PMID: 22595799]
Level 1 (high-level) evidenceLee ATJ, Jones RL, Huang PH. Pazopanib in advanced soft tissue sarcomas. Signal transduction and targeted therapy. 2019:4():16. doi: 10.1038/s41392-019-0049-6. Epub 2019 May 17 [PubMed PMID: 31123606]
Deguchi S, Mitsuya K, Nakasu Y, Hayashi N, Katagiri H, Murata H, Wasa J, Takahashi M, Endo M. Posterior reversible encephalopathy syndrome (PRES) induced by pazopanib, a multi-targeting tyrosine kinase inhibitor, in a patient with soft-tissue sarcoma: case report and review of the literature. Investigational new drugs. 2018 Apr:36(2):346-349. doi: 10.1007/s10637-017-0521-5. Epub 2017 Oct 25 [PubMed PMID: 29067537]
Level 3 (low-level) evidenceMcBride A, Butler SK. Eribulin mesylate: a novel halichondrin B analogue for the treatment of metastatic breast cancer. American journal of health-system pharmacy : AJHP : official journal of the American Society of Health-System Pharmacists. 2012 May 1:69(9):745-55. doi: 10.2146/ajhp110237. Epub [PubMed PMID: 22517020]
Schöffski P, Chawla S, Maki RG, Italiano A, Gelderblom H, Choy E, Grignani G, Camargo V, Bauer S, Rha SY, Blay JY, Hohenberger P, D'Adamo D, Guo M, Chmielowski B, Le Cesne A, Demetri GD, Patel SR. Eribulin versus dacarbazine in previously treated patients with advanced liposarcoma or leiomyosarcoma: a randomised, open-label, multicentre, phase 3 trial. Lancet (London, England). 2016 Apr 16:387(10028):1629-37. doi: 10.1016/S0140-6736(15)01283-0. Epub 2016 Feb 10 [PubMed PMID: 26874885]
Level 1 (high-level) evidenceBlay JY, Schöffski P, Bauer S, Krarup-Hansen A, Benson C, D'Adamo DR, Jia Y, Maki RG. Eribulin versus dacarbazine in patients with leiomyosarcoma: subgroup analysis from a phase 3, open-label, randomised study. British journal of cancer. 2019 May:120(11):1026-1032. doi: 10.1038/s41416-019-0462-1. Epub 2019 May 8 [PubMed PMID: 31065111]
Level 1 (high-level) evidenceD'Angelo SP, Mahoney MR, Van Tine BA, Atkins J, Milhem MM, Jahagirdar BN, Antonescu CR, Horvath E, Tap WD, Schwartz GK, Streicher H. Nivolumab with or without ipilimumab treatment for metastatic sarcoma (Alliance A091401): two open-label, non-comparative, randomised, phase 2 trials. The Lancet. Oncology. 2018 Mar:19(3):416-426. doi: 10.1016/S1470-2045(18)30006-8. Epub 2018 Jan 19 [PubMed PMID: 29370992]
Level 2 (mid-level) evidenceTaube JM, Klein A, Brahmer JR, Xu H, Pan X, Kim JH, Chen L, Pardoll DM, Topalian SL, Anders RA. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clinical cancer research : an official journal of the American Association for Cancer Research. 2014 Oct 1:20(19):5064-74. doi: 10.1158/1078-0432.CCR-13-3271. Epub 2014 Apr 8 [PubMed PMID: 24714771]
Marcus L, Lemery SJ, Keegan P, Pazdur R. FDA Approval Summary: Pembrolizumab for the Treatment of Microsatellite Instability-High Solid Tumors. Clinical cancer research : an official journal of the American Association for Cancer Research. 2019 Jul 1:25(13):3753-3758. doi: 10.1158/1078-0432.CCR-18-4070. Epub 2019 Feb 20 [PubMed PMID: 30787022]
Liu D, Offin M, Harnicar S, Li BT, Drilon A. Entrectinib: an orally available, selective tyrosine kinase inhibitor for the treatment of NTRK, ROS1, and ALK fusion-positive solid tumors. Therapeutics and clinical risk management. 2018:14():1247-1252. doi: 10.2147/TCRM.S147381. Epub 2018 Jul 20 [PubMed PMID: 30050303]
García-Del-Muro X, López-Pousa A, Maurel J, Martín J, Martínez-Trufero J, Casado A, Gómez-España A, Fra J, Cruz J, Poveda A, Meana A, Pericay C, Cubedo R, Rubió J, De Juan A, Laínez N, Carrasco JA, de Andrés R, Buesa JM, Spanish Group for Research on Sarcomas. Randomized phase II study comparing gemcitabine plus dacarbazine versus dacarbazine alone in patients with previously treated soft tissue sarcoma: a Spanish Group for Research on Sarcomas study. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2011 Jun 20:29(18):2528-33. doi: 10.1200/JCO.2010.33.6107. Epub 2011 May 23 [PubMed PMID: 21606430]
Level 1 (high-level) evidenceD'Adamo DR, Dickson MA, Keohan ML, Carvajal RD, Hensley ML, Hirst CM, Ezeoke MO, Ahn L, Qin LX, Antonescu CR, Lefkowitz RA, Maki RG, Schwartz GK, Tap WD. A Phase II Trial of Sorafenib and Dacarbazine for Leiomyosarcoma, Synovial Sarcoma, and Malignant Peripheral Nerve Sheath Tumors. The oncologist. 2019 Jun:24(6):857-863. doi: 10.1634/theoncologist.2018-0160. Epub 2018 Aug 20 [PubMed PMID: 30126857]
Level 2 (mid-level) evidenceAnsari L, Shiehzadeh F, Taherzadeh Z, Nikoofal-Sahlabadi S, Momtazi-Borojeni AA, Sahebkar A, Eslami S. The most prevalent side effects of pegylated liposomal doxorubicin monotherapy in women with metastatic breast cancer: a systematic review of clinical trials. Cancer gene therapy. 2017 May:24(5):189-193. doi: 10.1038/cgt.2017.9. Epub 2017 Apr 14 [PubMed PMID: 28409561]
Level 1 (high-level) evidenceGrenader T, Goldberg A, Hadas-Halperin I, Gabizon A. Long-term response to pegylated liposomal doxorubicin in patients with metastatic soft tissue sarcomas. Anti-cancer drugs. 2009 Jan:20(1):15-20. doi: 10.1097/CAD.0b013e3283198058. Epub [PubMed PMID: 19342997]
Level 2 (mid-level) evidenceJudson I, Radford JA, Harris M, Blay JY, van Hoesel Q, le Cesne A, van Oosterom AT, Clemons MJ, Kamby C, Hermans C, Whittaker J, Donato di Paola E, Verweij J, Nielsen S. Randomised phase II trial of pegylated liposomal doxorubicin (DOXIL/CAELYX) versus doxorubicin in the treatment of advanced or metastatic soft tissue sarcoma: a study by the EORTC Soft Tissue and Bone Sarcoma Group. European journal of cancer (Oxford, England : 1990). 2001 May:37(7):870-7 [PubMed PMID: 11313175]
Level 1 (high-level) evidenceO'Cearbhaill R, Zhou Q, Iasonos A, Soslow RA, Leitao MM, Aghajanian C, Hensley ML. Treatment of advanced uterine leiomyosarcoma with aromatase inhibitors. Gynecologic oncology. 2010 Mar:116(3):424-9. doi: 10.1016/j.ygyno.2009.10.064. Epub 2009 Nov 24 [PubMed PMID: 19932916]
Level 2 (mid-level) evidenceThanopoulou E, Thway K, Khabra K, Judson I. Treatment of hormone positive uterine leiomyosarcoma with aromatase inhibitors. Clinical sarcoma research. 2014:4():5. doi: 10.1186/2045-3329-4-5. Epub 2014 Jun 26 [PubMed PMID: 25018868]
Cates JMM. The AJCC 8th Edition Staging System for Soft Tissue Sarcoma of the Extremities or Trunk: A Cohort Study of the SEER Database. Journal of the National Comprehensive Cancer Network : JNCCN. 2018 Feb:16(2):144-152. doi: 10.6004/jnccn.2017.7042. Epub [PubMed PMID: 29439175]
Hahn E, Huang SH, Hosni A, Razak AA, Jones RL, Dickson BC, Sturgis EM, Patel SG, O'Sullivan B. Ending 40 years of silence: Rationale for a new staging system for soft tissue sarcoma of the head and neck. Clinical and translational radiation oncology. 2019 Feb:15():13-19. doi: 10.1016/j.ctro.2018.11.009. Epub 2018 Nov 27 [PubMed PMID: 30582016]
Tanaka K, Tsumura H. Eighth edition of the American Joint Committee on Cancer staging system for soft tissue sarcoma of the trunk and extremity: in search of a better staging system. Annals of translational medicine. 2019 Mar:7(Suppl 1):S11. doi: 10.21037/atm.2019.01.31. Epub [PubMed PMID: 31032292]
Coindre JM, Terrier P, Guillou L, Le Doussal V, Collin F, Ranchère D, Sastre X, Vilain MO, Bonichon F, N'Guyen Bui B. Predictive value of grade for metastasis development in the main histologic types of adult soft tissue sarcomas: a study of 1240 patients from the French Federation of Cancer Centers Sarcoma Group. Cancer. 2001 May 15:91(10):1914-26 [PubMed PMID: 11346874]
Lu YJ, Wang H, Fang LY, Wang WJ, Song W, Wang Y, Huang YQ, Din ZL. A nomogram for predicting overall survival in patients with uterine leiomyosarcoma: a SEER population-based study. Future oncology (London, England). 2020 Apr:16(10):573-584. doi: 10.2217/fon-2019-0674. Epub 2020 Mar 6 [PubMed PMID: 32141309]
Pisters PW, Leung DH, Woodruff J, Shi W, Brennan MF. Analysis of prognostic factors in 1,041 patients with localized soft tissue sarcomas of the extremities. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 1996 May:14(5):1679-89 [PubMed PMID: 8622088]
Zagars GK, Ballo MT, Pisters PW, Pollock RE, Patel SR, Benjamin RS, Evans HL. Prognostic factors for patients with localized soft-tissue sarcoma treated with conservation surgery and radiation therapy: an analysis of 1225 patients. Cancer. 2003 May 15:97(10):2530-43 [PubMed PMID: 12733153]
Level 2 (mid-level) evidenceD'Angelo E, Espinosa I, Ali R, Gilks CB, Rijn Mv, Lee CH, Prat J. Uterine leiomyosarcomas: tumor size, mitotic index, and biomarkers Ki67, and Bcl-2 identify two groups with different prognosis. Gynecologic oncology. 2011 May 1:121(2):328-33. doi: 10.1016/j.ygyno.2011.01.022. Epub 2011 Feb 12 [PubMed PMID: 21316747]
Maki RG, Moraco N, Antonescu CR, Hameed M, Pinkhasik A, Singer S, Brennan MF. Toward better soft tissue sarcoma staging: building on american joint committee on cancer staging systems versions 6 and 7. Annals of surgical oncology. 2013 Oct:20(11):3377-83. doi: 10.1245/s10434-013-3052-0. Epub 2013 Jun 18 [PubMed PMID: 23775410]
Brennan MF, Antonescu CR, Moraco N, Singer S. Lessons learned from the study of 10,000 patients with soft tissue sarcoma. Annals of surgery. 2014 Sep:260(3):416-21; discussion 421-2. doi: 10.1097/SLA.0000000000000869. Epub [PubMed PMID: 25115417]
Zivanovic O, Jacks LM, Iasonos A, Leitao MM Jr, Soslow RA, Veras E, Chi DS, Abu-Rustum NR, Barakat RR, Brennan MF, Hensley ML. A nomogram to predict postresection 5-year overall survival for patients with uterine leiomyosarcoma. Cancer. 2012 Feb 1:118(3):660-9. doi: 10.1002/cncr.26333. Epub 2011 Jul 12 [PubMed PMID: 21751199]
Lindner LH, Litière S, Sleijfer S, Benson C, Italiano A, Kasper B, Messiou C, Gelderblom H, Wardelmann E, Le Cesne A, Blay JY, Marreaud S, Hindi N, Desar IME, Gronchi A, van der Graaf WTA. Prognostic factors for soft tissue sarcoma patients with lung metastases only who are receiving first-line chemotherapy: An exploratory, retrospective analysis of the European Organization for Research and Treatment of Cancer-Soft Tissue and Bone Sarcoma Group (EORTC-STBSG). International journal of cancer. 2018 Jun 15:142(12):2610-2620. doi: 10.1002/ijc.31286. Epub 2018 Feb 14 [PubMed PMID: 29383713]
Level 2 (mid-level) evidence