Introduction
Primary ciliary dysfunction (PCD), first described in 1976, is a disorder of the structure and function of motile cilia that results in chronic oto-sinopulmonary disease.[1] Cilia are hair-like structures that exist on the surface of cells.[2] Motile cilia exist throughout the upper and lower respiratory tract, with each respiratory epithelial cell containing hundreds of cilia. Clearance of the airway and movement of respiratory secretions depends on the cilia beating in a coordinated fashion.[2][3] PCD is a heterogeneous genetic condition that causes various abnormalities in the axonemal structures that comprise the cilia. This subsequently results in abnormal cilia structure and function.[3][4]
This abnormal function impairs the ability of cilia to function in a coordinated manner, impairing mucociliary clearance and causing chronic upper and lower respiratory inflammation.[5] Primary ciliary dysfunction typically presents with neonatal respiratory distress, early-onset year-round cough, nasal congestion, and laterality defect (situs inversus).[6] Primary ciliary dysfunction diagnosis is challenging, given the lack of a single diagnostic test and the many conditions resulting in similar symptoms. Kartagener syndrome, which occurs in about 50% of PCD patients, is a triad of chronic sinusitis, bronchiectasis, and situs inversus resulting from embryonic (nodal) ciliary dyskinesia.[1][7]
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
Primary ciliary dysfunction is a genetic disorder that results in abnormal ciliary ultrastructure or function. Currently, 40 known genes are associated with primary ciliary dysfunction, with the majority following autosomal recessive inheritance.[4] DNAI1 and DNAH5, which encode components of the outer dynein arm complex in cilia, are the 2 most common genes associated with PCD.[4] Two rare X-linked genes, RPGR and OFD1, have also been identified.[8] Different mutations, even to the same gene, can result in varying degrees of loss of function of the cilia, further contributing to the heterogeneity of the disease.[9]
Epidemiology
The reported prevalence of primary ciliary dysfunction is estimated to be between 1:10,000 to 1:20,000 newborns.[6] Given difficulties in diagnosing primary ciliary dysfunction, misdiagnosis or delayed diagnosis is common. Therefore, primary ciliary dysfunction may be more prevalent than reported.[4] Studies are likely to underestimate due to misdiagnosis.[10][4]
In the United States, the prevalence of PCD is around 1:16,000 live births. These numbers may be affected by geographic area and consanguinity. Based on the autosomal recessive type of inheritance, the chance of having children with PCD is 1:4. No age, sex, or racial predilection has been noted.
Pathophysiology
Cilia exist on epithelial cells lining the nasopharynx, lower respiratory tract, paranasal sinuses, middle ear, and reproductive tract.[2][4] In primary ciliary dysfunction, mutations in genes encoding for axonemal structures of motile cilia result in cilia that are abnormal in structure and function. Defects can cause outer dynein arm defects, inner dynein arm defects, central microtubular abnormalities, radial spoke defects, and other ultrastructural abnormalities.[2][3][9][11][12]
Given the wide range of possible mutations, there is significant variation in clinical presentations.[13] In the respiratory tract, abnormal ciliary movements cause impaired mucociliary clearance. This results in chronic oto-sinopulmonary infections.[6][13][14]
The sperm tail and fimbriae of fallopian tubes also have motile cilia. Therefore, male and female infertility often occurs. Situs abnormalities occur in about 50% of cases of PCD. Normal ciliary movement is needed for visceral rotation. In the absence of normal ciliary function during embryogenesis, abnormal orientation can result. Kartagener syndrome is a triad of chronic sinusitis, bronchiectasis, and situs inversus resulting from ciliary dysfunction.[14]
History and Physical
Clinical presentations of ciliary dysfunction are highly variable. Most newborns with primary ciliary dyskinesia develop neonatal respiratory distress with atelectasis on chest radiographs. Unlike other causes of respiratory distress in newborns, which occur in the first few hours after birth, respiratory distress from primary ciliary dyskinesia occurs 12 to 24 hours after birth in full-term infants (see Image. Ciliary Dyskinesia).[12]
Four main clinical features have been described in primary ciliary dyskinesia, including unexplained neonatal respiratory distress, early-onset year-round wet cough, early-onset year-round nasal congestion, and laterality defects.[12][15] If 3 or more of these features are met, the specificity for primary ciliary dyskinesia diagnosis is more than 96%.[16]
Patients typically present in childhood, with a median age of diagnosis around 5 years. Typically, they present due to year-round respiratory infections with mucopurulent sputum and chronic daily cough.[12] The physical exam may be notable for crackles and intermittent wheezing. Clubbing can be seen in severe cases.[17] Similarly, year-round nasal congestion and recurrent sinusitis are often seen. Nasal polyposis may be present on the physical exam.[18] Hearing loss can occur and is thought to be related to recurrent otitis media, often present in childhood and adolescence.[14] Typically, this resolves by about age 12.[19]
About half of the patients with primary ciliary dyskinesia also have situs inversus totalis, including the right and left lung transposition.[14] Male infertility is common and occurs in nearly 100% of males.[12] Females can present with reduced fertility or ectopic pregnancy due to abnormal fallopian tube transit of oocytes.[12]
Physical examination may reveal the following signs:
- Inflammation of tympanic membranes
- Perforation with hearing loss
- Right-sided apex beat and heart sounds if associated with dextrocardia
- Evidence of situs inversus, such as the spleen and liver on the incorrect side
- Digital clubbing due to chronic and recurrent lower respiratory infections
Evaluation
Diagnosing primary ciliary dyskinesia is challenging as it results from various defects in cilia, and no single diagnostic test can detect all these defects (Figure).[20][21] Diagnosis is typically achieved with clinical features, namely unexplained neonatal respiratory distress, early onset chronic cough, chronic nasal congestion, and defect, in conjunction with diagnostic testing to demonstrate abnormal ciliary ultrastructure and motility.[16][22]
In patients older than 5, nasal nitric oxide measurements can be used as a screening or diagnostic tool for diagnosing primary ciliary dyskinesia.[23] Paranasal sinus epithelium produces nitric oxide, and nasal nitric oxide measurements are typically extremely low (less than 100 nL/min) in primary ciliary dyskinesia patients. Decreased nasal nitric oxide values can also be seen in acute viral respiratory infections and patients with cystic fibrosis. Therefore, patients should fully recover from viral illnesses, and cystic fibrosis should be ruled out before performing this test.[24] No absolute threshold has been validated as a cut-off value for nasal nitric oxide, which makes it somewhat challenging to standardize across centers. Sensitivity and specificity vary depending on the cut-off value used. One study, which used 30 nL/min as a cut-off, reported a sensitivity of 91% and specificity of 96% in diagnosis.[21] Therefore, while this is a good screening test, confirmation testing is required to characterize the abnormal ciliary ultrastructure and function further.
Per guidelines outlined in the European Respiratory Society, high-speed videomicroscopy analysis (HSVA) for the evaluation of ciliary motility is extremely sensitive (100% sensitivity) and specific (93% specificity) and should be conducted as well.[21] If both the HSVA and inhaled nitric oxide results in equivocal values, electron microscopy of a nasal or bronchial biopsy should be performed as this has 100% specificity.[21] Various ciliary defects, such as the absence of dynein arms, can be observed on electron microscopic examinations of the samples obtained by ciliary biopsy. Immotile cilia, poor motility, and ineffective and slow beat patterns have also been seen.[5]
Advances in electron microscopy have enabled a deeper understanding of the structural consequences of genetic variations.[5] Although electron microscopy (EM) analysis can aid the diagnosis, a subset of patients with primary ciliary dyskinesia with no demonstrable ultra-structural abnormalities were identified. As such, electron microscopy can have a false negative rate of 20%-30%. Negative results need to be reviewed in the context of the entire clinical picture.[12][21]
Additionally, false positives can occur in all the above testing methods due to secondary loss of cilia function. This can occur, for example, if the sample was taken during an acute viral illness that caused transient loss of function. Cell culture can be used to minimize this impact.[25] Genetic testing should also be considered, as 33 genes have been associated with primary ciliary dyskinesia. However, not all of them are included in the commercially available primary ciliary dyskinesia genetic testing panel. As genetic panels continue to develop and advance, genetic testing continues to be a valued test for diagnosis and research. Currently, however, there remain limitations as known PCD patients can have normal genetic testing.[16] In the United States, a combination of ciliary biopsy to demonstrate ciliary ultrastructural abnormalities and genetic testing is used for the diagnosis. In contrast, a combination of genetic testing and high-speed video microscopy is used in Europe.
Treatment / Management
Similar to diagnosis, there is no current 'gold standard' treatment or management of primary ciliary dysfunction. Given that no curative therapy exists, treatment primarily relies on symptom management and varies based on the clinical presentation. Most current treatment options are largely extrapolated from other diseases that cause bronchiectasis, such as cystic fibrosis.[6] Primary ciliary dysfunction patients should be managed in centers specializing in primary and chronic pulmonary diseases.[6][26] Primary ciliary dysfunction patients should be evaluated by a pulmonologist 2 to 4 times per year with spirometry and surveillance cultures of sputum. A chest radiograph should be completed at the time of diagnosis and during acute respiratory exacerbations. At least once after diagnosis, a chest CT, preferably a high-resolution CT scan, to evaluate for bronchiectasis should be considered.[23](B3)
Unlike cystic fibrosis, the lung clearance index does not seem to be a helpful monitoring tool for patients with PCD. Additionally, children with primary ciliary dysfunction should be evaluated by an ear-nose-throat specialist 1 to 2 times per year and as needed in adults. An audiology assessment should be performed since there is a high incidence of recurrent otitis media with chronic middle ear effusions and associated complications of conductive hearing loss.[14] Patients should be monitored for chronic rhinosinusitis and nasal polyps. Prevention of acute exacerbations of bronchiectasis from bacterial infections is a mainstay of treatment as it slows down the progression of the disease.[6] Prevention of exacerbations is achieved by aiding the body in clearing secretions, thus reducing the microbial load. This is obtained by daily chest physiotherapy and effective airway hydration.[23](B3)
While there are many similarities between the treatment of cystic fibrosis (CF) and PCD, it has been found that certain mucolytic agents, such as DNase, while effective in CF, are ineffective for PCD. When prevention fails, and acute exacerbations occur, antibiotics are the mainstay of treatment and should be started even when symptoms appear mild. Obtaining regular sputum samples helps guide antibiotic choice for empiric coverage during an acute exacerbation. Pseudomonas aeruginosa should always be considered, and patients may require IV antibiotics for such infections. For recurrent exacerbations, prevention with chronic antibiotic suppression, such as azithromycin, can effectively reduce the number of infections.[27] Primary ciliary dysfunction patients should receive influenza vaccination and pneumococcal vaccination as indicated.(A1)
A bilateral lung transplant may be necessary for patients with end-stage respiratory disease.[28] Symptomatic treatment of chronic rhinosinusitis is characterized by nasal saline lavage and intranasal glucocorticoids. Endoscopic sinus surgery has been shown to improve chronic rhinosinusitis.[18] The use of tympanostomy tubes in these patients as a management strategy for recurrent otitis media continues to be debated, as many patients improve symptoms with conservative therapy.[19] It is crucial to provide patients with counseling on impaired fertility. Gene editing has been proposed as a possible future treatment for primary ciliary dysfunction. Ex vivo gene repair by site-specific recombination has been found to rescue ciliary beating, which could have tremendous application in primary ciliary dysfunction patients.[6](B2)
Differential Diagnosis
Some important differential diagnoses are to be considered with the pretext of primary ciliary dysfunction.[29] These include the following:
- Cystic fibrosis
- Foreign body aspiration
- Idiopathic interstitial pneumonia
- Idiopathic nasal polyposis
- Immunosuppression
- Malignancy
- Post-infectious bronchiectasis
- Severe atopy
- Tracheobronchomegaly
Prognosis
While many similarities exist in the clinical presentation and management between patients with cystic fibrosis and primary ciliary dysfunction, 1 notable difference is that the rate of decline of lung function is typically much slower in PCD than in cystic fibrosis.[30] As such, patients with primary ciliary dysfunction can often continue to live an active lifestyle, particularly when compliant with the management guidelines outlined above. Given the high variability of the disease, it is difficult to predict the type of decline in lung function and patient experiences. However, some studies suggest aggressive airway clearance and the use of antibiotics early may slow the course of the disease. One study found that about 60% of the patients had stable pulmonary function tests (PFTs) in a 30-year longitudinal study. About 30% of the patients in this study suffered from a decrease in lung function.[31]
Complications
Frequent bronchiectasis exacerbations with recurrent infections are a complication for patients with primary ciliary dysfunction. Other complications include recurrent ear infections, chronic sinusitis, and infertility.[12][15]
Consultations
A pulmonologist for routine PFTS and airway clearance management should follow patients diagnosed with primary ciliary dysfunction. Additionally, patients should be evaluated at least initially by ENT providers if suffering from chronic sinusitis and recurrent otitis media. Genetic consultation can be helpful for patients seeking additional information. Lastly, fertility consultation may be required for adults with PCD requesting information on family planning.[26]
Deterrence and Patient Education
To limit potential complications, patients should be educated on routine vaccinations and smoking cessation. Parental genetic counseling should be offered for newly diagnosed infants and children. The significance of regular health monitoring must be emphasized. Counsel patients to avoid allergens, exposure to respiratory pathogens, and environmental irritants.
Enhancing Healthcare Team Outcomes
Primary ciliary dysfunction is managed by an interprofessional team of healthcare workers, including clinicians, nurses, pharmacists, and respiratory therapists. Primary ciliary dysfunction patients should be managed in centers specializing in primary and chronic pulmonary diseases. Patients should be evaluated by a pulmonologist 2 to 4 times per year. Spirometry and surveillance cultures of sputum should be done during these visits. Additionally, children with primary ciliary dysfunction should be evaluated by an ear-nose-throat specialist 1 to 2 times per year and as needed in adults. An audiology assessment should be performed since there is a high incidence of recurrent otitis media with chronic middle ear effusions and associated complications of conductive hearing loss. The primary care provider should monitor for chronic rhinosinusitis and nasal polyps. These patients should receive daily chest physiotherapy, influenza, and pneumococcal vaccinations, as indicated. Antibiotics should be prescribed for acute respiratory exacerbations.[19][26]
Media
(Click Image to Enlarge)
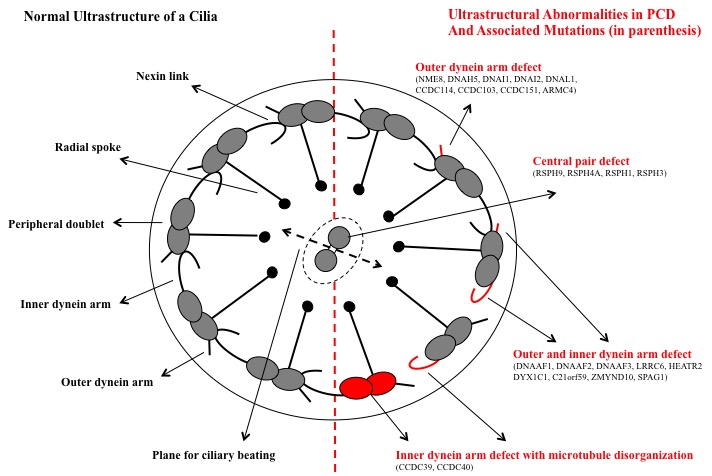
Ciliary Dyskinesia. Normal ultrastructure of a cilia, ultrastructure abnormalities in primary ciliary dysfunction, and associated mutations.
Contributed by G Sharma, MD, FCCP, FAAP
References
Tadesse A, Alemu H, Silamsaw M, Gebrewold Y. Kartagener's syndrome: a case report. Journal of medical case reports. 2018 Jan 10:12(1):5. doi: 10.1186/s13256-017-1538-2. Epub 2018 Jan 10 [PubMed PMID: 29316973]
Level 3 (low-level) evidenceHorani A, Ferkol TW. Understanding Primary Ciliary Dyskinesia and Other Ciliopathies. The Journal of pediatrics. 2021 Mar:230():15-22.e1. doi: 10.1016/j.jpeds.2020.11.040. Epub 2020 Nov 23 [PubMed PMID: 33242470]
Level 3 (low-level) evidenceAntony D, Brunner HG, Schmidts M. Ciliary Dyneins and Dynein Related Ciliopathies. Cells. 2021 Jul 25:10(8):. doi: 10.3390/cells10081885. Epub 2021 Jul 25 [PubMed PMID: 34440654]
Zariwala MA, Knowles MR, Omran H. Genetic defects in ciliary structure and function. Annual review of physiology. 2007:69():423-50 [PubMed PMID: 17059358]
Level 3 (low-level) evidenceShoemark A. Applications of emerging transmission electron microscopy technology in PCD research and diagnosis. Ultrastructural pathology. 2017 Nov-Dec:41(6):408-414. doi: 10.1080/01913123.2017.1365789. Epub 2017 Sep 18 [PubMed PMID: 28922052]
Mirra V, Werner C, Santamaria F. Primary Ciliary Dyskinesia: An Update on Clinical Aspects, Genetics, Diagnosis, and Future Treatment Strategies. Frontiers in pediatrics. 2017:5():135. doi: 10.3389/fped.2017.00135. Epub 2017 Jun 9 [PubMed PMID: 28649564]
Nonaka S, Tanaka Y, Okada Y, Takeda S, Harada A, Kanai Y, Kido M, Hirokawa N. Randomization of left-right asymmetry due to loss of nodal cilia generating leftward flow of extraembryonic fluid in mice lacking KIF3B motor protein. Cell. 1998 Dec 11:95(6):829-37 [PubMed PMID: 9865700]
Level 3 (low-level) evidenceNarayan D, Krishnan SN, Upender M, Ravikumar TS, Mahoney MJ, Dolan TF Jr, Teebi AS, Haddad GG. Unusual inheritance of primary ciliary dyskinesia (Kartagener's syndrome). Journal of medical genetics. 1994 Jun:31(6):493-6 [PubMed PMID: 8071978]
Level 3 (low-level) evidenceHornef N, Olbrich H, Horvath J, Zariwala MA, Fliegauf M, Loges NT, Wildhaber J, Noone PG, Kennedy M, Antonarakis SE, Blouin JL, Bartoloni L, Nüsslein T, Ahrens P, Griese M, Kuhl H, Sudbrak R, Knowles MR, Reinhardt R, Omran H. DNAH5 mutations are a common cause of primary ciliary dyskinesia with outer dynein arm defects. American journal of respiratory and critical care medicine. 2006 Jul 15:174(2):120-6 [PubMed PMID: 16627867]
Kuehni CE, Frischer T, Strippoli MP, Maurer E, Bush A, Nielsen KG, Escribano A, Lucas JS, Yiallouros P, Omran H, Eber E, O'Callaghan C, Snijders D, Barbato A, ERS Task Force on Primary Ciliary Dyskinesia in Children. Factors influencing age at diagnosis of primary ciliary dyskinesia in European children. The European respiratory journal. 2010 Dec:36(6):1248-58. doi: 10.1183/09031936.00001010. Epub 2010 Jun 7 [PubMed PMID: 20530032]
Level 2 (mid-level) evidenceDavis SD, Ferkol TW, Rosenfeld M, Lee HS, Dell SD, Sagel SD, Milla C, Zariwala MA, Pittman JE, Shapiro AJ, Carson JL, Krischer JP, Hazucha MJ, Cooper ML, Knowles MR, Leigh MW. Clinical features of childhood primary ciliary dyskinesia by genotype and ultrastructural phenotype. American journal of respiratory and critical care medicine. 2015 Feb 1:191(3):316-24. doi: 10.1164/rccm.201409-1672OC. Epub [PubMed PMID: 25493340]
Knowles MR, Daniels LA, Davis SD, Zariwala MA, Leigh MW. Primary ciliary dyskinesia. Recent advances in diagnostics, genetics, and characterization of clinical disease. American journal of respiratory and critical care medicine. 2013 Oct 15:188(8):913-22. doi: 10.1164/rccm.201301-0059CI. Epub [PubMed PMID: 23796196]
Level 3 (low-level) evidenceGuo Z, Chen W, Wang L, Qian L. Clinical and Genetic Spectrum of Children with Primary Ciliary Dyskinesia in China. The Journal of pediatrics. 2020 Oct:225():157-165.e5. doi: 10.1016/j.jpeds.2020.05.052. Epub 2020 Jun 2 [PubMed PMID: 32502479]
Level 2 (mid-level) evidencePiatti G, De Santi MM, Torretta S, Pignataro L, Soi D, Ambrosetti U. Cilia and Ear. The Annals of otology, rhinology, and laryngology. 2017 Apr:126(4):322-327. doi: 10.1177/0003489417691299. Epub 2017 Feb 12 [PubMed PMID: 28290230]
Lucas JS, Burgess A, Mitchison HM, Moya E, Williamson M, Hogg C, National PCD Service, UK. Diagnosis and management of primary ciliary dyskinesia. Archives of disease in childhood. 2014 Sep:99(9):850-6. doi: 10.1136/archdischild-2013-304831. Epub 2014 Apr 25 [PubMed PMID: 24771309]
Leigh MW, Ferkol TW, Davis SD, Lee HS, Rosenfeld M, Dell SD, Sagel SD, Milla C, Olivier KN, Sullivan KM, Zariwala MA, Pittman JE, Shapiro AJ, Carson JL, Krischer J, Hazucha MJ, Knowles MR. Clinical Features and Associated Likelihood of Primary Ciliary Dyskinesia in Children and Adolescents. Annals of the American Thoracic Society. 2016 Aug:13(8):1305-13. doi: 10.1513/AnnalsATS.201511-748OC. Epub [PubMed PMID: 27070726]
Yiallouros PK, Kouis P, Middleton N, Nearchou M, Adamidi T, Georgiou A, Eleftheriou A, Ioannou P, Hadjisavvas A, Kyriacou K. Clinical features of primary ciliary dyskinesia in Cyprus with emphasis on lobectomized patients. Respiratory medicine. 2015 Mar:109(3):347-56. doi: 10.1016/j.rmed.2015.01.015. Epub 2015 Jan 31 [PubMed PMID: 25698650]
Level 2 (mid-level) evidenceAlanin MC, Aanaes K, Høiby N, Pressler T, Skov M, Nielsen KG, Johansen HK, von Buchwald C. Sinus surgery can improve quality of life, lung infections, and lung function in patients with primary ciliary dyskinesia. International forum of allergy & rhinology. 2017 Mar:7(3):240-247. doi: 10.1002/alr.21873. Epub 2016 Nov 23 [PubMed PMID: 27879058]
Level 2 (mid-level) evidenceMajithia A, Fong J, Hariri M, Harcourt J. Hearing outcomes in children with primary ciliary dyskinesia--a longitudinal study. International journal of pediatric otorhinolaryngology. 2005 Aug:69(8):1061-4 [PubMed PMID: 16005347]
Level 2 (mid-level) evidenceWahl GM, Spike BT. Cell state plasticity, stem cells, EMT, and the generation of intra-tumoral heterogeneity. NPJ breast cancer. 2017:3():14. doi: 10.1038/s41523-017-0012-z. Epub 2017 Apr 19 [PubMed PMID: 28649654]
Jackson CL, Behan L, Collins SA, Goggin PM, Adam EC, Coles JL, Evans HJ, Harris A, Lackie P, Packham S, Page A, Thompson J, Walker WT, Kuehni C, Lucas JS. Accuracy of diagnostic testing in primary ciliary dyskinesia. The European respiratory journal. 2016 Mar:47(3):837-48. doi: 10.1183/13993003.00749-2015. Epub 2015 Dec 2 [PubMed PMID: 26647444]
Lucas JS, Barbato A, Collins SA, Goutaki M, Behan L, Caudri D, Dell S, Eber E, Escudier E, Hirst RA, Hogg C, Jorissen M, Latzin P, Legendre M, Leigh MW, Midulla F, Nielsen KG, Omran H, Papon JF, Pohunek P, Redfern B, Rigau D, Rindlisbacher B, Santamaria F, Shoemark A, Snijders D, Tonia T, Titieni A, Walker WT, Werner C, Bush A, Kuehni CE. European Respiratory Society guidelines for the diagnosis of primary ciliary dyskinesia. The European respiratory journal. 2017 Jan:49(1):. doi: 10.1183/13993003.01090-2016. Epub 2017 Jan 4 [PubMed PMID: 27836958]
Barbato A, Frischer T, Kuehni CE, Snijders D, Azevedo I, Baktai G, Bartoloni L, Eber E, Escribano A, Haarman E, Hesselmar B, Hogg C, Jorissen M, Lucas J, Nielsen KG, O'Callaghan C, Omran H, Pohunek P, Strippoli MP, Bush A. Primary ciliary dyskinesia: a consensus statement on diagnostic and treatment approaches in children. The European respiratory journal. 2009 Dec:34(6):1264-76. doi: 10.1183/09031936.00176608. Epub [PubMed PMID: 19948909]
Level 3 (low-level) evidenceNyilas S, Schlegtendal A, Singer F, Goutaki M, Kuehni CE, Casaulta C, Latzin P, Koerner-Rettberg C. Alternative inert gas washout outcomes in patients with primary ciliary dyskinesia. The European respiratory journal. 2017 Jan:49(1):. pii: 1600466. doi: 10.1183/13993003.00466-2016. Epub 2017 Jan 25 [PubMed PMID: 28122863]
Hirst RA, Jackson CL, Coles JL, Williams G, Rutman A, Goggin PM, Adam EC, Page A, Evans HJ, Lackie PM, O'Callaghan C, Lucas JS. Culture of primary ciliary dyskinesia epithelial cells at air-liquid interface can alter ciliary phenotype but remains a robust and informative diagnostic aid. PloS one. 2014:9(2):e89675. doi: 10.1371/journal.pone.0089675. Epub 2014 Feb 25 [PubMed PMID: 24586956]
Level 2 (mid-level) evidenceLobo LJ, Zariwala MA, Noone PG. Primary ciliary dyskinesia. QJM : monthly journal of the Association of Physicians. 2014 Sep:107(9):691-9. doi: 10.1093/qjmed/hcu063. Epub 2014 Mar 19 [PubMed PMID: 24652656]
Kobbernagel HE, Buchvald FF, Haarman EG, Casaulta C, Collins SA, Hogg C, Kuehni CE, Lucas JS, Moser CE, Quittner AL, Raidt J, Rosthøj S, Sørensen AL, Thomsen K, Werner C, Omran H, Nielsen KG. Efficacy and safety of azithromycin maintenance therapy in primary ciliary dyskinesia (BESTCILIA): a multicentre, double-blind, randomised, placebo-controlled phase 3 trial. The Lancet. Respiratory medicine. 2020 May:8(5):493-505. doi: 10.1016/S2213-2600(20)30058-8. Epub [PubMed PMID: 32380069]
Level 1 (high-level) evidenceTkebuchava T, Niederhäuser U, Weder W, von Segesser LK, Bauersfeld U, Felix H, Lachat M, Turina MI. Kartagener's syndrome: clinical presentation and cardiosurgical aspects. The Annals of thoracic surgery. 1996 Nov:62(5):1474-9 [PubMed PMID: 8893586]
Level 3 (low-level) evidenceBush A, Payne D, Pike S, Jenkins G, Henke MO, Rubin BK. Mucus properties in children with primary ciliary dyskinesia: comparison with cystic fibrosis. Chest. 2006 Jan:129(1):118-23 [PubMed PMID: 16424421]
Davis SD, Rosenfeld M, Lee HS, Ferkol TW, Sagel SD, Dell SD, Milla C, Pittman JE, Shapiro AJ, Sullivan KM, Nykamp KR, Krischer JP, Zariwala MA, Knowles MR, Leigh MW. Primary Ciliary Dyskinesia: Longitudinal Study of Lung Disease by Ultrastructure Defect and Genotype. American journal of respiratory and critical care medicine. 2019 Jan 15:199(2):190-198. doi: 10.1164/rccm.201803-0548OC. Epub [PubMed PMID: 30067075]
Marthin JK, Petersen N, Skovgaard LT, Nielsen KG. Lung function in patients with primary ciliary dyskinesia: a cross-sectional and 3-decade longitudinal study. American journal of respiratory and critical care medicine. 2010 Jun 1:181(11):1262-8. doi: 10.1164/rccm.200811-1731OC. Epub 2010 Feb 18 [PubMed PMID: 20167855]
Level 2 (mid-level) evidence