Introduction
Neonatal jaundice is a clinical manifestation of elevated total serum bilirubin (TSB), termed neonatal hyperbilirubinemia, which results from bilirubin that is deposited into an infant's skin. The characteristic features of neonatal jaundice include yellowish skin, sclerae, and mucous membranes. Jaundice derives from the French word jaune, meaning yellow. Neonatal jaundice is the most frequently encountered medical condition in the first 2 weeks of life and a common cause of readmission to the hospital after birth.[1] Approximately 60% of term and 80% of preterm newborns develop clinical jaundice in the first week after birth.[2] Neonatal jaundice is usually a mild, transient, and self-limiting condition known as physiologic jaundice. However, this should be distinguished from the more severe pathologic jaundice. The two types of neonatal hyperbilirubinemia are unconjugated hyperbilirubinemia (UHB) and conjugated hyperbilirubinemia (CHB).
When neonatal jaundice is clinically identified, the underlying etiology of neonatal hyperbilirubinemia must be determined. In most neonates, unconjugated hyperbilirubinemia is the cause of clinical jaundice. However, some infants have conjugated hyperbilirubinemia, which is always pathologic and signifies an underlying medical or surgical etiology. Failure to identify and treat pathologic jaundice may result in bilirubin encephalopathy and associated neurological sequelae. The causes of pathologic UHB and CHB are numerous and varied. Preterm infants and those with congenital enzyme deficiencies are particularly prone to the harmful effects of unconjugated bilirubin on the central nervous system.[3][4]
Unconjugated hyperbilirubinemia is diagnosed by assessing bilirubin levels with a transcutaneous measurement device or blood samples for total serum bilirubin. Conjugated hyperbilirubinemia is typically diagnosed through laboratory studies, including serum aminotransferase, prothrombin time, urine cultures, tests for inborn errors of metabolism, and, in some cases, imaging studies. Severe hyperbilirubinemia can cause bilirubin-induced neurological dysfunction (BIND) and, if not treated adequately, may lead to acute and chronic bilirubin encephalopathy.[5] Phototherapy and exchange transfusions are the mainstays of treatment of UHB, and a subset of patients also respond to intravenous immunoglobulin (IVIG). Treatment of CHB is more complex and depends on the etiology of the jaundice. Despite advances in the care and management of hyperbilirubinemia, it remains a significant cause of neonatal morbidity and mortality.[6]
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
The underlying etiology of neonatal jaundice is neonatal hyperbilirubinemia, which has 2 distinct types: unconjugated and conjugated hyperbilirubinemia, also known as indirect and direct hyperbilirubinemia, respectively. (See Image. Metabolic Pathway for Bilirubin in the Hepatocyte).
Unconjugated Hyperbilirubinemia
Unconjugated (ie, indirect) hyperbilirubinemia (UHB) is the more common type and is either physiologic or pathologic. Physiologic jaundice accounts for 75% of neonatal hyperbilirubinemia and results from a physiologic alteration in neonatal bilirubin metabolism. Healthy adults have a total serum bilirubin (TSB) level of less than 1mg/dL. In neonates, normal TSB levels are comparatively higher, with age-dependant levels. Even healthy full-term newborns have an increased bilirubin load due to higher red blood cell (RBC) mass and decreased RBC lifespan. Metabolic bilirubin clearance is also compromised due to impaired activity of uridine diphosphate glucuronosyltransferase (UGT), the enzyme needed for bilirubin conjugation. The activity level of the newborn UGT enzyme is approximately 1% that of an adult.[7] Moreover, neonates also have increased enterohepatic circulation, further contributing to elevated TSB levels. Physiologic jaundice typically appears in full-term infants 24 hours after birth, peaks at around 48 to 96 hours, and resolves by 2 to 3 weeks.[2] Conversely, pathologic unconjugated hyperbilirubinemia occurs within the first 24 hours after birth when the TSB level is >95% on age-specific bilirubin nomograms or increases ≥5 mg/dL/day or >0.2 mg/dL/hour.[8] Based on the mechanism of bilirubin elevation, the etiology of unconjugated hyperbilirubinemia can be subdivided into the following 3 categories: increased bilirubin production, decreased bilirubin clearance, and miscellaneous.
Increased Bilirubin Production
The production of bilirubin can increase secondary to immune-mediated hemolysis caused by blood group incompatibilities (eg, such as ABO and Rhesus (Rh) incompatibility) and nonimmune mediated hemolysis, which is caused by RBC membrane defects (eg, hereditary spherocytosis and elliptocytosis), RBC enzyme defects (eg, glucose-6-phosphate dehydrogenase [G6PD], pyruvate kinase deficiencies), sequestration-like cephalohematoma, subgaleal hemorrhage, intracranial hemorrhage, polycythemia, and sepsis. Exaggerated immune or nonimmune mediated hemolysis is the most common cause of pathologic hyperbilirubinemia. Immune-mediated hemolysis occurs with blood group incompatibility as ABO/Rh incompatibility and causes hemolytic disease of the newborn (HDN). In HDN from ABO incompatibility, preformed maternal anti-A and anti-B antibodies of the immunoglobulin G (IgG) subclass cross the placenta and cause hemolysis and UHB in newborns with blood types A, B, or AB. Although the direct antiglobulin test (DAT) aids diagnosis, the sensitivity and positive predictive value for severe UHB are low.[9] ABO incompatibility between mother and fetus exists in about 15% of pregnancies, but HDN occurs in only about 4% of newborns with ABO incompatibility.[10]
In Rh incompatibility, an Rh-negative mother, exposed to Rh-positive RBCs during a previous pregnancy, becomes sensitized and develops antibodies against Rh antigen. Initially, sensitization produces IgM antibodies that cannot cross the placenta. However, during subsequent pregnancies, IgG antibodies cross the placenta, causing RBC hemolysis in a fetus with Rh-positive blood. The Rh antigen is very immunogenic, and the resultant HDN is usually severe, often leading to hydrops fetalis or severe UHB in newborns. Prevention of neonatal UHB caused by immune-mediated hemolysis begins in pregnancy with the recognition of mothers at risk for developing Rh antibodies. The American College of Obstetricians and Gynecologists (ACOG) recommends that all Rh-negative pregnant women receive anti-D immune globulin at 28 weeks gestation and again after delivery if the infant is Rh-positive or the blood type is unknown.[11]
Nonimmune causes of UHB include RBC enzyme defects, RBC membrane defects, hemoglobinopathies, sepsis, sequestration, and polycythemia. Glucose-6 phosphatase dehydrogenase (G6PD) deficiency is the most common RBC enzyme defect, transmitted as an X-linked recessive trait. G6PD protects RBCs against oxidative damage by generating NADPH (nicotinamide adenine dinucleotide phosphate hydrogenase) from NADP (nicotinamide adenine dinucleotide phosphate). When exposed to oxidant stressors like illness, certain medications, dyes, and foods such as fava beans, G6PD deficient RBCs are hemolyzed, causing anemia and hyperbilirubinemia. More than 200 different mutations cause G6PD deficiency.[12] The clinical presentation differs depending on the variant, and some newborns may develop severe hyperbilirubinemia and bilirubin encephalopathy. Pyruvate kinase deficiency (PKD) is another condition that causes hemolysis and may present as UHB in newborns. PKD is an autosomal recessive (AR) disorder interfering with glycolysis and cellular energy production. In PKD, RBCs have shortened life spans, resulting in hemolytic anemia and UHB.[13]
Conditions causing UHB due to RBC membrane defects include hereditary spherocytosis (HS) and hereditary elliptocytosis (HE). HS (ie, Minkowski Chauffard disease) is the most common RBC membrane defect caused by RBC membrane protein mutations.[14] Most cases are transmitted as an autosomal dominant (AD) trait and can present in the neonatal period with UHB.[15] Hereditary elliptocytosis is another type of RBC membrane defect caused by structural membrane protein mutations in which elliptical-shaped RBCs are trapped in the spleen, leading to extravascular hemolysis and elevated TSB. HE is usually asymptomatic but may cause UHB in the neonatal period.[16]
Etiologies that cause RBC sequestration (eg, cephalohematoma, subgaleal hemorrhage, and intracranial hemorrhage) are also risk factors for neonatal UHB due to increased bilirubin load. Polycythemia is another condition associated with an increased risk of UHB in newborns, associated with intrauterine growth restriction (IUGR), infants of diabetic mothers (IDM), large for gestational age (LGA) infants, maternal smoking, high altitude, twin to twin transfusions, and placental transfusion (eg, delayed cord clamping and umbilical cord milking). Studies show that delayed cord clamping reduces the incidence of postnatal anemia and leads to improved neurodevelopmental outcomes among term and preterm infants.[17][18] Delayed cord clamping has gained popularity but may also increase the risk of hyperbilirubinemia.[19][20]
Decreased Bilirubin Clearance
Indirect hyperbilirubinemia due to decreased bilirubin clearance usually results from quantitative or qualitative defects in the uridine diphosphate glucuronosyltransferase (UGT) enzyme. Gilbert syndrome and Crigler–Najjar syndrome type I and II are 3 disorders resulting from an abnormality of the UGT enzyme. Gilbert syndrome is the most common due to a mutation in the UGT1A1 gene leading to decreased UGT production and subsequent unconjugated hyperbilirubinemia.[21] Gilbert syndrome typically presents as mild jaundice at times of physiologic stress in the absence of hemolysis or liver dysfunction. However, presentations during the neonatal period are rare and usually associated with G6PD deficiency.[22][3] Crigler-Najjar syndrome type I is an AR disorder resulting from a complete absence of UGT activity. Affected patients present with severe hyperbilirubinemia in the first few days of life, often leading to bilirubin encephalopathy. Patients with Crigler-Najjar syndrome type II retain some of the activity of UGT enzymes; therefore, their TSB levels are not as elevated as in patients with the type I variant, and bilirubin encephalopathy rarely develops.[23]
Miscellaneous Causes
Other etiologies of UHG include congenital hypothyroidism, sulfa medications, ceftriaxone, penicillins, intestinal obstruction, pyloric stenosis, breast milk jaundice, and suboptimal intake with breastfeeding. Infants of mothers with diabetes are at higher risk of developing unconjugated hyperbilirubinemia. Jaundice secondary to breastfeeding and breast milk are 2 other common etiologies of UHB in newborns. Breastfeeding jaundice, known as suboptimal intake hyperbilirubinemia, occurs in the first week of life due to inadequate breast milk consumption, leading to dehydration and occasionally hypernatremia.[7] Poor intake decreases intestinal motility and elimination of bilirubin in the stool. Conversely, breast milk jaundice occurs late in the first week after birth, peaks in the second week, and usually resolves by 2 weeks of age, though the condition may persist for up to 3 months. Breast milk jaundice is rarely pathologic and is associated with adequate intake and good weight gain.[24] Inhibition of the UGT enzyme by pregnanediol and the deconjugation of conjugated bilirubin in the intestines by beta-glucuronidase present in breast milk is thought to be the primary underlying pathophysiology.[25][26]
Other miscellaneous causes of UHB include infants of mothers with diabetes (IDM), gastrointestinal obstruction, congenital hypothyroidism, and certain medications. IDM often have polycythemia, with an increased incidence of jaundice.[27] UHB in congenital hypothyroidism is related to decreased bilirubin hepatic uptake, impaired UGT activity, and sluggish gut motility, while gastrointestinal obstruction promotes increased bilirubin recycling by augmenting enterohepatic circulation. When prescribed in the neonatal period, certain medications may worsen UHB by displacing bilirubin from albumin, affecting albumin binding.[28] Sepsis can also predispose a newborn to UHB by causing oxidative damage to RBCs and increasing bilirubin load.[29]
Additionally, most infants with clinical UHB have a combination of 2 or more predisposing contributing factors, including prematurity, a history of jaundice requiring phototherapy in parents or siblings, Asian ethnicity, male gender, and exclusive breastfeeding.[2] Preterm infants are at increased risk of bilirubin encephalopathy and kernicterus and require close monitoring, although there is insufficient data and a lack of consensus guidelines on managing UHB in preterm infants.[30][31] Because of the increased risk of neurotoxicity, the TSB threshold for initiation of phototherapy is lower than for term infants. However, bilirubin is an antioxidant and may have a physiologic role in neonates.[32][33] Keeping TSB levels low by aggressive treatment in preterm infants may reduce this antioxidant level and potentially worsen retinopathy of prematurity. Moreover, reduced antioxidant status is also associated with chronic lung disease and neurological injury. Therefore, treating UHB in premature neonates is challenging without evidence-based guidelines. The most recent clinical practice guidelines for managing hyperbilirubinemia of the American Academy of Pediatrics in 2022 included only infants >35 weeks of gestation.[34][30]
Conjugated Hyperbilirubinemia
Conjugated hyperbilirubinemia (CHB), also referred to as neonatal cholestasis, is characterized by the elevation of serum conjugated (ie, direct) bilirubin (>1.0 mg/dL) due to impaired hepatobiliary function. Distinguishing CHB from UHB is critical because cholestatic neonatal jaundice is almost always pathologic and warrants prompt evaluation and treatment.[35] The causes of CHB are extensive and typically classified into the following categories:
- Infection: Congenital infections (eg, syphilis, toxoplasmosis, HIV, herpes virus, and rubella) should be included in the differential diagnosis of neonatal cholestasis, especially when stigmata of congenital infection, including growth restriction, coagulopathy, skin rash, or thrombocytopenia, are present. Cytomegalovirus (CMV) is also a common congenital infection with many manifestations. Most infected newborns are asymptomatic, but hepatomegaly and CHB may indicate hepatic involvement.[36] Carefully reviewing maternal history, specific serologies, and viral culture results aid diagnosis. Urine and blood cultures are also a component of the diagnostic evaluation, as urinary tract infections and septicemia can cause CHB in neonates. Additionally, microcirculatory changes in the liver, a direct effect of bacterial products, and toxins released by bacteria are thought to be the possible mechanisms of cholestasis in patients with UTI.[37]
- Obstruction of biliary flow: Conditions with this underlying pathophysiology include biliary atresia, choledochal cysts, neonatal sclerosing cholangitis, and neonatal cholelithiasis. Biliary atresia (BA) is the most common cause of conjugated hyperbilirubinemia in infants, with an incidence that varies by geographic location.[38] Taiwan, the region with the highest incidence, has a reported frequency of 1 in 6000 live births. In the United States, the incidence is about 1 in 12,000 live births.[39] The etiology of BA is not well understood, but genetic factors, viral infections, toxins, chronic inflammation, and autoimmune injury to bile ducts seem to play a role in the pathogenesis. The disease involves both intra-hepatic and extrahepatic bile ducts and classically presents around 2 to 4 weeks after birth with pale stools and jaundice. The initial ultrasound examination may reveal an absent gallbladder and the classic "triangular cord" sign, a ductal remnant of the extrahepatic bile duct.[40] Early diagnosis is critical to maximizing the response to the Kasai operation (ie, hepatic portoenterostomy).[41] If surgery is delayed until after 90 days of life, <25% of patients are likely to respond, compared to surgery performed within 60 days when >70% of patients will establish adequate bile flow.[42]
Choledochal cysts can also cause biliary flow obstruction. The intrahepatic and extrahepatic bile ducts are dilated when choledochal cysts are present, and ultrasonography can detect cysts with normal or dilated intrahepatic bile ducts as opposed to the sclerosed ducts of biliary atresia. However, cystic biliary atresia may resemble choledochal cysts.[43] Neonatal sclerosing cholangitis (NSC) is a rare form of cholangiopathy that presents in infancy with CHB, hepatosplenomegaly, pale stools, and high serum gamma-glutamyltransferase activity (GGT).[44] Neonatal cholelithiasis is another rare entity that causes significant direct hyperbilirubinemia.[45]
- Genetic: There are several genetic etiologies (eg, Alagille syndrome, alpha-1 anti-trypsin deficiency, galactosemia, fructosemia, Tyrosinemia type 1, cystic fibrosis, progressive familial intrahepatic cholestasis [PFIC], Aagenaes syndrome, Dubin-Johnson syndrome, bile acid synthesis disorders [BSAD]) that commonly result in CHB. Alagille syndrome (ALGS) is an AD disorder caused by mutations in the JAG1 or NOTCH2 genes, leading to a lack of interlobular bile ducts.[46] With an incidence of 1 in 30,000 live births, ALGS is the most common cause of familial intrahepatic cholestasis, though CHB in patients with ALGS may resolve with age.[47][35] Characteristic clinical features include butterfly vertebrae, congenital heart defects (eg, peripheral pulmonic stenosis), kidney involvement, dysmorphic features (eg, broad forehead and a small pointed chin), and posterior embryotoxon of the eye. Gamma-glutamyl transferase (GGT) levels are elevated, often up to 20 times the standard value. Occasionally, patients with cystic fibrosis (CF) present with cholestasis because of abnormal bile that plugs the bile ducts.[48] In developing nations where newborn screening with immunoreactive trypsinogen is unavailable, neonatal cholestasis may be the first clue to diagnosing CF.
Alpha-1-antitrypsin deficiency is the most common genetic cause of cholestasis and can mimic biliary atresia in early infancy. Accumulation of anti-trypsin polymers in the hepatocyte endoplasmic reticulum of a patient with the PIZZ genotype leads to apoptosis of hepatocytes, ultimately resulting in cholestasis and cirrhosis later in childhood.[49] As with ALGS, cholestasis may improve with age. Galactosemia, fructosemia, and tyrosinemia type 1 are a few of the inborn errors of metabolism known to cause cholestasis in neonates. Newborns with galactosemia can present with cholestatic jaundice, cataracts, hepatomegaly, failure to thrive, renal tubular acidosis, and Escherichia coli sepsis after ingesting galactose from milk.[50] Galactose-1-phosphate uridyl transferase (GALT) deficiency leads to the accumulation of toxic galactose metabolites in multiple organs. The presence of urine-reducing substances suggests galactosemia, but GALT activity in the liver or erythrocytes confirms the diagnosis. Neonatal cholestasis is a presenting feature in hereditary tyrosinemia type 1, another AR disorder caused by a deficiency of the enzyme fumarylacetoacetate hydroxylase. Other characteristics of this disorder include renal Fanconi syndrome, hepatomegaly, coagulation abnormality, and the risk of hepatocellular carcinoma in untreated older patients.[51]
Progressive familial intrahepatic cholestasis (PFIC) is a heterogeneous group of 3 genetic disorders involving canalicular hepatobiliary transport that presents with cholestasis.[52] Types 1 and 2 usually manifest in the neonatal period, while type 3 presents later in infancy. Affected patients with PFIC frequently develop cirrhosis and end-stage liver disease during childhood. In diagnostic studies, the GGT level is within the normal range in types 1 and 2 and elevated in type 3 patients. PFIC type 1 is caused by a mutation in the ATP8B1 gene, which encodes the FIC1 protein, whereas PFIC type 2 is caused by a mutation in the ABCB11 gene, which encodes for the bile salt excretory protein (BSEP). PFIC type 3 is caused by a mutation in the ABCB4 gene, which encodes for the multi-drug resistant-3 protein (MDR3).[53] Aagenaes syndrome, also known as lymphedema cholestasis syndrome (LCS), is another idiopathic familial intrahepatic cholestasis syndrome characterized by neonatal cholestasis and lymphedema in the lower extremities. Aagenaes syndrome is transmitted as an autosomal recessive (AR) trait usually seen in individuals of Norwegian descent.[54] Dubin-Johnson syndrome (DJS) is a rare AR disorder caused by a mutation in the ABCC2 gene, which codes for a non-biliary ion transporter in the liver. A unique feature of DJS is a black-colored liver and the excretion of coproporphyrin 1 in urine.[55] Bile acid synthesis disorder (BASD) results from a deficiency of an enzyme involved in synthesizing bile acids from cholesterol. BASDs are an uncommon cause of cholestasis, but many are curable with medical therapy.
- Miscellaneous: Other conditions that may cause CHD include idiopathic neonatal hepatitis, parenteral nutrition-induced cholestasis, gestational alloimmune liver disease, neonatal hemochromatosis, and hypotension. Parenteral nutrition-associated cholestasis (PNAC) is a significant iatrogenic cause of cholestasis in preterm infants managed with parenteral nutrition (PN). PNAC is present in about 20% of neonates receiving PN for 2 weeks or longer.[56] Duration of PN use and intestinal failure are 2 independent risk factors for PNAC. Though the mechanism is not entirely clear, likely being multifactorial, abnormal bile salt metabolism due to prematurity and the harmful effects of PN components are thought to be the main culprits.[57] Other factors, including sepsis and necrotizing enterocolitis, can also potentiate liver injury.[58] Gestational alloimmune liver disease (GALD), which causes almost all cases of neonatal hemochromatosis, is a fulminant alloimmune disorder and results from intrahepatic and extrahepatic iron deposition, leading to liver failure.[59] In GALD, maternal IgG immunoglobulin against fetal hepatocytes crosses the placenta, causing complement-mediated damage to fetal hepatocytes. Characteristic features involve signs of liver failure, including hypoglycemia, coagulopathy, hypoalbuminemia, cholestatic jaundice, edema, and elevated liver enzymes. GALD has a risk of recurrence in subsequent pregnancies of approximately 90% and can result in fetal or neonatal death.[60] The term idiopathic neonatal hepatitis is used when the etiology of neonatal cholestasis cannot be ascertained after an extensive diagnostic evaluation. Newer diagnostic tools enable more precise diagnoses, with fewer cases of neonatal cholestasis now classified as "idiopathic."[42]
Epidemiology
Unconjugated hyperbilirubinemia is frequently encountered in the neonatal period. About 80% of term and preterm newborns will present with clinical jaundice with a TSB >5 mg/dL.[2][61] However, only approximately 10% of neonates require phototherapy.[62] Physiologic jaundice is the most frequent cause of clinical jaundice after the first day of life, estimated to account for 50% of cases.[63] Approximately 15% of breastfed infants will develop physiologic UHB lasting >3 weeks.[64]
Only a minority of jaundiced newborns have pathologic hyperbilirubinemia. Severe hyperbilirubinemia, commonly defined as a TSB>25 mg/dL, occurs in approximately 1 out of 2500 live births. Among these, ABO incompatibility, followed by G6PD deficiency, is the most frequently identified cause.[65] Newborns with Southeast and East Asian ancestry have higher recorded TSB levels than Black or White infants.[66][67] Neonatal jaundice also appears more common in infants living at high altitudes and around the Mediterranean Sea, especially in Greece.[68][69]
Acute bilirubin encephalopathy occurs at a rate of approximately 1 in 10,000 live births, and the incidence of chronic bilirubin encephalopathy is comparitively lower, estimated at 1 in 50,000 to 100,000 live births.[70] However, developing nations report higher rates of kernicterus, a permanent neurologic condition.[71]
Conjugated hyperbilirubinemia is much less common than UHB, with an incidence of around 1 in 2500 term infants.[72] The most frequently identified cause of cholestatic jaundice in the neonatal period is biliary atresia, accounting for an estimated 25% to 40% of all cases, followed by infections and PN-induced cholestasis.[35][73] Approximately 60% to 70% of patients with BA will require liver transplantation in childhood, remaining the most common indication for a pediatric liver transplant.[74]
Pathophysiology
Bilirubin is produced from the catabolism of heme, a breakdown product of hemoglobin, in the reticuloendothelial system (RES). First, heme is converted to biliverdin, releasing iron and carbon monoxide via the action of the enzyme heme oxygenase.[75] Biliverdin is then converted to bilirubin by the enzyme biliverdin reductase. This hydrophobic unconjugated bilirubin binds to albumen, is transported to the liver, and is conjugated with glucuronic acid in the smooth endoplasmic reticulum by the enzyme uridine diphosphate-glucuronosyltransferase (UGT). Conjugated bilirubin is water soluble and is excreted in bile, passed into the gastrointestinal (GI) tract, and eliminated in feces. Some conjugated bilirubin is deconjugated in the GI tract by beta-glucuronidase and reabsorbed through the enterohepatic circulation.[76]
Newborn infants have higher TSB levels than adults due to higher hemoglobin levels at birth, a shorter RBC life span, and limited conjugating ability of the neonatal liver.[77] Healthy, full-term newborns typically have peak serum bilirubin concentrations of 5 to 6 mg/dL compared to adult levels of <1 mg/dL. Pathologic jaundice in neonates is related to increased production of bilirubin in the RES, impaired hepatic uptake, deficient conjugation of bilirubin, and enhanced enterohepatic circulation of bilirubin.[75]
In severe hyperbilirubinemia, unbound, unconjugated bilirubin crosses the blood-brain barrier and binds to the brainstem, hippocampus, cerebellum, globus pallidus, and subthalamic nuclei.[2] At the cellular level, bilirubin inhibits certain mitochondrial enzymes, interferes with DNA and protein synthesis, induces breaks in DNA strands, and hampers phosphorylation.[78] Bilirubin also impairs tyrosine uptake and alters the normal functioning of N-methyl-D-aspartate–receptor ion channels.[79][80] These mechanisms are implicated in the pathogenesis of bilirubin toxicity that clinically manifests as bilirubin-induced neurologic dysfunction (BIND) and bilirubin encephalopathy. The duration of exposure to bilirubin and the amount of bilirubin in the brain determines the severity of brain damage. However, the TSB level does not correlate well with bilirubin toxicity without hemolysis.[75] Preterm infants are more vulnerable to the toxic effects of free unconjugated bilirubin. This may be related to comparatively lower serum albumin levels, CNS immaturity, and comorbidities such as intraventricular hemorrhage, periventricular leukomalacia, sepsis, necrotizing enterocolitis, and bronchopulmonary dysplasia.[71]
Conjugated hyperbilirubinemia results from abnormalities in the uptake, metabolism, transport, and excretion of bile salts and bilirubin, which increases bile acid in the liver, promoting the proliferation of bile ducts and fibrosis.[81] Bile acid is also responsible for the inflammation and apoptosis of hepatocytes, culminating in hepatocellular injury and cirrhosis.[82] Deficient bile secretion in cholestasis results in malabsorption of fat and fat-soluble vitamins A, D, E, and K and may lead to inadequate nutrition and weight gain.[83]
Histopathology
Kernicterus is derived from the German word kern, meaning core, referring to the basal ganglia. Yellow staining of deeper brain nuclei is seen on autopsy specimens of infants with severe unconjugated hyperbilirubinemia. Histopathologic findings include nuclei that have undergone pyknosis, cytoplasm vacuolation, and fading of the Nissl substance in neurons.[84]
A liver biopsy is usually necessary for a definitive diagnosis of cholestasis as it helps to differentiate biliary atresia from idiopathic neonatal hepatitis. Histopathologic features of BA include the expansion of the hepatic portal tracts with edema, fibrodysplasia, bile ductular proliferation, and the presence of bile plugs in the ductal lumen. Multinucleate giant cells and hemopoiesis are also findings noted on histopathologic examinations of cholestatic liver samples.[85] Although not diagnostic of any particular disorder, the prominence of hepatic erythropoiesis is seen more frequently in cholestasis of infectious etiology. Pathognomonic histologic findings of other cholestatic conditions include periodic acid-Schiff (PAS) positive granules noted with alpha-1 antitrypsin deficiency, a paucity of bile ducts associated with Alagille syndrome, and periductal necrosis and inflammation seen in sclerosing cholangitis.[86] Among familial causes of cholestasis, canalicular cholestasis with a marked absence of ductular proliferation and isolated periportal biliary metaplasia of hepatocytes is commonly seen in patients with PFIC type 1. Though the histopathology of PFIC type 2 is similar, the altered liver architecture and extensive lobular and portal fibrosis with inflammation occur more frequently.[53]
History and Physical
The evaluation of the neonate with jaundice begins with a detailed history, including pregnancy complications, family history, delivery, the onset of associated symptoms, feeding adequacy, and maternal serologies. Stool and urine color may provide a clue about the type of jaundice. Minor risk factors are serum bilirubin in the high-intermediate range, macrosomic infants of diabetic mothers, polycythemia, male gender, and maternal age older than 25.[8] The American Academy of Pediatrics (AAP) recommends universal visual screening of all newborns for jaundice every 12 hours from the time of birth until the infant is discharged home. Furthermore, clinicians should identify risk factors for severe hyperbilirubinemia.[8] Major risk factors in newborns >35 weeks gestation include:[8]
- A bilirubin level in the high-risk zone before hospital discharge
- Jaundice in the first 24 hours after birth
- Maternal-fetal blood group incompatibility
- Gestational age <36 weeks [87]
- History of phototherapy in an immediate family member
- A cephalhematoma or significant bruising after delivery
- Exclusive breastfeeding
- Asian descent
To assess for jaundice, newborns should ideally be examined in daylight. However, clinical assessment is unreliable, especially if a newborn has received phototherapy or has dark skin.[88] Therefore, clinically significant jaundice should always be confirmed with a TSB or transcutaneous bilirubin (TcB). A focused physical examination may identify the cause of pathologic jaundice. Signs include pallor, petechiae, cephalohematoma, extensive bruising, hepatosplenomegaly, weight loss, and dehydration. All infants with jaundice should be assessed for clinical features of bilirubin encephalopathy, including poor feeding, lethargy, altered sleep, abnormal tone, and seizures. However, up to 15% of neonates with kernicterus are clinically asymptomatic in the newborn period.[75] Infants with signs and symptoms suggesting neonatal cholestasis often have multisystem involvement, which may guide diagnostic investigations.
Evaluation
Diagnosis Studies in Unconjugated Hyperbilirubinemia
The AAP recommends an infant's bilirubin levels should be assessed between 24 and 48 hours after birth. If an infant leaves the hospital prior to this time frame, a bilirubin level should be measured before discharge home. Bilirubin levels are initially evaluated with a transcutaneous measurement device or blood samples for total serum bilirubin.[89] Transcutaneous estimation of bilirubin can be used as a screening test for hyperbilirubinemia to reduce the frequency of blood tests, but the utility is limited in infants with dark skin and following phototherapy use.[90][91] The serum level should be measured when the transcutaneous bilirubin (TcB) level exceeds the 95th percentile on the transcutaneous nomogram or 75% of the TSB threshold for phototherapy. Another limitation of the TcB is the inability to detect the direct fraction of bilirubin required for diagnosing neonatal cholestasis.
Recommended studies to identify a hemolytic disease etiology of unconjugated hyperbilirubinemia include maternal and neonatal blood types, a direct antibody test (DAT), complete blood cell (CBC), reticulocyte count, blood smear, and G6PD testing. Serum albumin, considered a surrogate marker for free bilirubin, should be checked, especially if the TSB level approaches the level where exchange transfusion is indicated. Free bilirubin is the fraction responsible for bilirubin-induced toxicity.[92] The bilirubin-albumin ratio (B/A) ratio is an additional tool that may predict the risk of kernicterus and serve as a guide when considering the escalation of care and exchange transfusion.
Radiographic imaging is usually not required for most cases of UCH. Brain magnetic resonance imaging (MRI) findings have high sensitivity for bilirubin encephalopathy, with posteromedial borders of the globus pallidus being the most sensitive region for detecting signal changes. Infants with bilirubin encephalopathy demonstrate hyperintense signals on T1-weighted sequences in the acute stage that eventually become hyperintense on T2-weighted sequences as the condition evolves. Magnetic resonance spectroscopy (MRS) shows increased levels of glutamate and decreased levels of N-acetyl-aspartate and choline.[93] However, the absence of these findings does not exclude the risk of chronic bilirubin encephalopathy.
Diagnostic Studies in Conjugated Hyperbilirubinemia
Serum aminotransferases may provide evidence of hepatocellular injury in patients with conjugated hyperbilirubinemia. Alkaline phosphatase and elevated GGT levels may indicate an obstruction in biliary channels. The prothrombin time (PT), the international normalized ratio (INR), and serum albumin evaluate hepatic synthesis and function. Additional tests like TORCH titers for in-utero infections, urine cultures, viral cultures, serologic titers, newborn screening, tests for inborn errors of metabolism, alpha-1 antitrypsin phenotype, and genetic profiles may be diagnostic.
Radiology may be necessary to evaluate neonatal cholestasis. Hepatic ultrasonography can identify sludging in the biliary tree, gallstones, inspissated bile, and choledochal cysts. A triangular cord sign on hepatic ultrasound has high sensitivity and almost 100% specificity for biliary atresia.[81] Hepatobiliary scintigraphy is another modality utilized to evaluate neonatal cholestasis. Decreased excretion of the tracer 24 hours after introduction suggests obstruction and further differentiates obstructive and nonobstructive causes of cholestasis.[94] Prior treatment with phenobarbital improves the sensitivity of hepatobiliary scintigraphy. However, a liver biopsy is the gold standard for diagnosing neonatal cholestasis. Histopathologic interpretation by an experienced pathologist will identify the correct diagnosis in 90% to 95% of cases and may prevent unnecessary interventions in patients with intrahepatic cholestasis.[95]
Treatment / Management
Treatment of Unconjugated Hyperbilirubinemia
Phototherapy and exchange transfusion are the mainstays of treatment for newborns with unconjugated hyperbilirubinemia. Other treatment modalities may be utilized based on patient response to these therapies. The approach to treatment is determined by the neurotoxicity risk factors present, neonatal gestational age, and hour-specific TSB.[8](A1)
Phototherapy
Phototherapy (PT) remains the first-line treatment for managing pathologic unconjugated hyperbilirubinemia. PT reduces TSB to safe levels and decreases the risk of bilirubin toxicity and the need for exchange transfusion. The TSB level at which PT is indicated is determined based on the risk factors for neurotoxicity present, the infant's gestational age, and hour-specific TSB.[34][8] Risk factors for neurotoxicity that lower the TSB threshold to initiate PT include:[34][8] (A1)
- Neonatal gestational age <38 weeks
- Serum albumin <3.0 g/dL
- Positive DAT or diagnosis of G6PD deficiency or other hemolytic diseases
- Sepsis
- Significant clinical deterioration in the preceding 24 hours
However, for premature infants, few standardized guidelines exist, and most hospitals compile their own protocols for phototherapy and exchange transfusion in preterm infants based on birth weight and gestational age.[31] The efficacy of phototherapy depends on the dose and wavelength of light used, as well as the infant's exposed surface area. Increasing the dose of PT can be achieved by placing phototherapy units at a minimum safe distance from the infant and increasing the number of units used.
Bilirubin optimally absorbs light in the blue-green range (ie, 460-490 nm). The underlying mechanism of PT involves inducing photoisomerization and converting bilirubin into lumirubin, which is readily excreted into bile and urine.[96] During phototherapy, the neonate's maximum body surface area should be exposed to the light source while keeping the eyes covered to avoid retinal injury, and interruptions should be minimized. The maintenance of hydration is necessary to ensure adequate urine output, as most bilirubin is excreted in the urine as lumirubin, a structural isomer of bilirubin formed during phototherapy. Therefore, breastfeeding support should be offered to all nursing mothers as early initiation of breastfeeding and frequent, on-demand feeding decreases the likelihood of dehydration. Although supplemental oral water and dextrose water are not recommended, supplemental pumped breastmilk or infant formula can be considered for feeding issues, including infants with ineffective sucking or latching or inadequate maternal milk production.[34]
After phototherapy is discontinued, there may be an increase in the total serum bilirubin level, known as the rebound bilirubin. This level is usually lower than the pretreatment level and rarely requires reinitiation of phototherapy.[97] PT is considered safe, but recent evidence suggests a possible association with long-term sequelae, including a small risk of epilepsy. However, no studies have proven causation.[98] A few studies also have reported a possible association between solid organ tumors and nonlymphocytic leukemias and children treated with phototherapy.[99][100] Adverse effects of PT include rashes, dehydration, hypocalcemia, retinal damage, hemolysis due to oxidative damage, delay in patent ductus arteriosis closure in preterm infants, and allergic reactions.[101] Bronze baby syndrome is a self-limited condition associated with elevated levels of conjugated bilirubin that rarely occurs with PT, resulting in irregular, bronze-gray pigmentation of the skin, mucous membranes, and urine. The phenomenon's mechanism is unclear but appears to be related to the accumulation of bilirubin and biliverdin photoisomers. Bronze baby syndrome usually resolves within a few days of discontinuing phototherapy; however, the prognosis depends upon the underlying cause of the conjugated hyperbilirubinemia.[102][103](B2)
Exchange Transfusion
In 1947, exchange transfusion (ET) was the first successful treatment for jaundice.[104] Now, ET is the second-line treatment for severe unconjugated hyperbilirubinemia since phototherapy was developed in the 1950s.[105][106] Indications for this therapy include neonatal failure to respond to PT or a TSB level at the exchange transfusion threshold. The threshold to initiate exchange transfusion is calculated based on several factors, including the TSB level and rate of rise, neonatal age (ie, hours or days since birth), and risk factors for neurologic complications.[34] ET rapidly removes bilirubin and hemolysis-causing antibodies from the infant's circulation. A double-volume exchange blood transfusion (160-180 ml/kg) is performed, replacing aliquots of the neonate's blood with crossed-matched donor blood. Since most of the total body bilirubin is extravascular, the TSB level immediately following ET is approximately 60% of the pre-exchange level, but that later increases to 70% to 80% of the pretreatment level as a result of equilibrium. During ET, the neonate's vital signs should be monitored closely. Following the procedure, TSB, CBC, serum calcium, glucose, and electrolytes should be rechecked due to potential complications, including electrolyte abnormalities (eg, hypocalcemia and hyperkalemia), cardiac arrhythmias, thrombocytopenia, blood-borne infections, portal vein thrombosis, graft versus host disease, and necrotizing enterocolitis (NEC).[107][108] Phototherapy should resume after exchange transfusion until the bilirubin reaches a level where PT can be safely discontinued. (B2)
Intravenous Immunoglobulin
Intravenous immunoglobulin (IVIG) is used when immune-mediated hemolysis is the cause of unconjugated hyperbilirubinemia, which prevents RBC hemolysis by coating Fc receptors on RBCs.The AAP recommends IVIG infusion in immune-mediated hemolysis if TSB remains within 2 to 3 mg/dL of the exchange threshold despite intensive phototherapy.[109][110] However, the evidence that IVIG reduces the need for ET is unclear. Nonetheless, IVIG is often used in clinical practice to manage severe unconjugated hyperbilirubinemia.(A1)
Treatment of Conjugated Hyperbilirubinemia
Treatment of conjugated hyperbilirubinemia is tailored to the specific etiology of the jaundice. To achieve the best outcomes, patients diagnosed with biliary atresia require a Kasai operation (hepatic portoenterostomy) within the first 2 months of life to prevent irreversible liver damage.[41] The surgery involves the removal of the atretic biliary ducts and fibrous plate and anastomosis of the jejunum with the remaining ducts by a Roux-en-Y procedure to provide an alternative pathway for biliary drainage.[111] Infectious causes of cholestasis are treated with specific antimicrobial agents, whereas treatment with cholic acid and chenodeoxycholic acid is often curative for many BASDs. Patients with GALD appear to respond well to IVIG and double-volume exchange transfusion. Patients with conditions of cholestasis (eg, GALD and biliary atresia) and severe hepatic damage may require liver transplantation, which is curative but technically challenging in infants.[60] Parenteral nutrition-induced cholestasis is managed with cyclic PN, reducing the duration of exposure and initiating enteral feeds as early as possible. The manganese and copper content of PN should be monitored closely to minimize liver injury.(B2)
Differential Diagnosis
The differential diagnoses of jaundice are extensive. (Refer to the Etiologies section for more information on the causes of jaundice). The onset of neonatal jaundice should prompt a timely evaluation to determine if the underlying etiology is a self-limited physiologic condition or a disease requiring treatment. Any neonate with yellowish skin and sclerae should be clinically diagnosed with jaundice. High carotene levels may cause yellowish skin that mimics jaundice in older infants and children who are ingesting diets of breast milk or foods high in beta-carotene.[112][38] However, with carotenemia, the sclera or mucosa are not involved, and neonates are not born with this benign condition. Visual assessment of the degree of jaundice is an unreliable screening tool for clinically significant hyperbilirubinemia. All newborns should undergo an appropriate evaluation for possible jaundice before hospital discharge.[113][114]
Staging
Acute Bilirubin Encephalopathy
The manifestations of bilirubin encephalopathy in neonates with severe unconjugated hyperbilirubinemia depend on when the symptoms appear. The level at which unconjugated bilirubin becomes neurotoxic is unclear, and kernicterus has been reported in infants without markedly elevated bilirubin levels on autopsy. There are 3 phases of acute bilirubin encephalopathy, including:
- Phase 1: The symptoms of phase 1 appear during the first 1 to 2 days of illness and are notable for poor feeding, lethargy, hypotonia, irritability, or frank seizures.
- Phase 2: If infants continue to deteriorate, they progress to phase 2, characterized by increased extensor muscle tone, exhibiting opisthotonus and retrocollis. This typically occurs during the middle of the first week of illness.
- Phase 3: After the first week, muscle rigidity, stupor or coma, apnea, or seizures may occur.
Chronic Blirubin Encephalopathy
This permanent and disabling neurologic condition, also known as kernicterus, is present in 2 forms, depending on the timing of symptoms.
- In the first year: These patients present with hypotonia, exaggerated deep tendon reflexes, obligatory tonic neck reflexes, and delayed motor milestones.
- Beyond the first year: Patients exhibit movement disorders, most commonly choreo-athetoid cerebral palsy, dental enamel hypoplasia, upward gaze abnormality, and sensorineural hearing loss.[75]
Prognosis
With treatment, the prognosis for most cases of unconjugated hyperbilirubinemia is excellent. In patients with delayed or inadequate treatment, bilirubin encephalopathy may ensue. The burden of bilirubin encephalopathy is significantly higher in developing and resource-limited nations.[71] Reports suggest a resurgence of kernicterus in countries where this complication had virtually disappeared, primarily attributed to the early hospital discharge of newborns with inadequate follow-up. Patients with Crigler-Najjar type I carry a poor prognosis and require liver transplantation for a definitive cure. In the absence of liver transplantation, bilirubin encephalopathy is common.
The prognosis for conjugated hyperbilirubinemia depends on the etiology. The prognosis of patients with biliary atresia is significantly improved by early diagnosis and surgery within 60 days of life. Similarly, patients with bile acid synthesis disorder (BASD) have excellent outcomes and respond well to medical treatment. Historically, the outlook for gestational alloimmune liver disease (GALD) was poor, with up to 80% mortality without liver transplantation. However, with the advent of IVIG use and double volume exchange transfusion, the prognosis has significantly improved in recent years.[115] The outlook for most other causes of cholestasis is often unfavorable, and many patients will require multidisciplinary interventions.
Complications
Newborns with severe hyperbilirubinemia are at risk for bilirubin-induced neurologic dysfunction (BIND). Bilirubin binds to the globus pallidus, hippocampus, cerebellum, and subthalamic nuclear bodies, causing neurotoxicity.[116] Acute bilirubin encephalopathy (ABE) is characterized by lethargy, hypotonia, and decreased sucking. At this stage, the condition is reversible. However, if ABE progresses, patients can develop irreversible chronic bilirubin encephalopathy or kernicterus. ABE manifests as choreo-athetoid cerebral palsy, seizures, arching, posturing, gaze abnormalities, dental enamel defects, and sensorineural hearing loss. Infants with neonatal cholestasis are also at risk of developing liver failure, cirrhosis, and even hepatocellular carcinoma in rare cases. Long-standing cholestasis may also lead to failure to thrive and fat-soluble vitamin deficiencies.
Consultations
A pediatric or neonatal clinician can manage most patients with unconjugated hyperbilirubinemia. However, patients suspected of having genetic causes of hyperbilirubinemia may need consultation and follow-up with a pediatric gastroenterologist, hematologist, and medical geneticist.
However, patients with neonatal cholestasis should be referred to a pediatric gastroenterologist without delay. Most will need a comprehensive investigation, and once the etiology of cholestasis is identified, more consultations may be warranted. Infants diagnosed with biliary atresia also need a referral to an experienced pediatric gastrointestinal surgeon for corrective surgery. Likewise, patients with inborn errors of metabolism require a consultation with a metabolic specialist, a medical geneticist, and a pediatric dietician experienced in treating metabolic disorders. When BIND occurs, neurologic consultation and close follow-up are essential.
Deterrence and Patient Education
In 2022, the AAP published updated clinical guidelines for managing hyperbilirubinemia in the newborn. Prevention of hyperbilirubinemia begins in pregnancy. Pregnant women should be screened with Rh typing and treated appropriately. If the maternal antibody screen is positive or unknown, neonates should undergo blood tests to determine blood type and DAT status. All newborns should be assessed for risk factors increasing the risk of hyperbilirubinemia and frequently monitored to prevent the occurrence of significant jaundice.[34]
Risk factors for significant hyperbilirubinemia include the following:
- Gestational age <38 weeks
- Jaundice at <24 hours of life
- Predischarge TSB or TcB close to the phototherapy threshold
- Evidence of hemolysis or rapidly rising TSB or TcB
- Phototherapy before hospital discharge
- Family history of a sibling or parent who required phototherapy or exchange transfusion
- Family history of RBC disorders, including G6PD deficiency
- Exclusive breastfeeding with suboptimal intake
- Scalp hematoma or significant bruising
- Down syndrome
- Large for gestational age infant of a mother with diabetes [34]
Risk factors for hyperbilirubinemia neurotoxicity that lower the threshold for phototherapy and escalation of care include:
- Gestational age <38 weeks
- Albumen <3.0 g/dL
- Hemolytic conditions
- Sepsis
- Significant clinical instability from any cause [34]
Because suboptimal feeding plays a role in developing hyperbilirubinemia, all breastfeeding mothers should receive support to promote adequate feeding. Breastfeeding within the first hour of life is recommended, followed by on-demand nursing at least 8 times daily. Furthermore, staff should visually assess all hospitalized neonates for jaundice at least every 12 hours. The AAP guidelines recommend universal bilirubin screening before discharge with TSB or TcB.[34] Most patients with unconjugated hyperbilirubinemia have an excellent prognosis, and families should be reassured and educated about the evaluation and indicated treatment when necessary. Neonates with jaundice from etiologies with poor prognoses often require multidisciplinary consultations, and parents should be adequately counseled and supported.
Enhancing Healthcare Team Outcomes
Neonatal jaundice is a common condition with multiple etiologies. Most cases are self-limiting with an excellent prognosis. However, bilirubin encephalopathy is an uncommon but devastating complication of severe hyperbilirubinemia. Healthcare professionals caring for newborns must be aware of this. Nurses and parents are usually the first to notice and report jaundice or difficulties breastfeeding. While many conditions causing jaundice are not diagnosed immediately, timely education about risk factors for significant hyperbilirubinemia and neurotoxicity is critical. Necessary treatment can begin while a diagnostic evaluation is in progress. Before discharge from the hospital, parents should be educated by nurses, nurse practitioners, pediatricians, obstetricians, and family practice clinicians to monitor for jaundice at home and know when to seek follow-up medical care.
A simple two-color icterometer is a simple tool that helps caregivers identify jaundice at home and may result in earlier medical intervention when indicated.[117] Nurses can train mothers to examine the skin and eyes of their babies for jaundice, either visually or with a smartphone application. An interprofessional team of clinicians, nurses, laboratory technicians, subspecialists, and nutritionists optimizes outcomes and must understand their role in caring for jaundiced newborns, including current evidence-based guidelines. Nurses are vital in monitoring treatments, educating parents, and keeping the team apprised of significant changes in the patient's condition. Nurses ensure that the American Academy of Pediatrics recommendation for universal bilirubin screening and risk factor assessment is performed to prevent the development of severe hyperbilirubinemia and improve patient outcomes.[8]
Media
(Click Image to Enlarge)
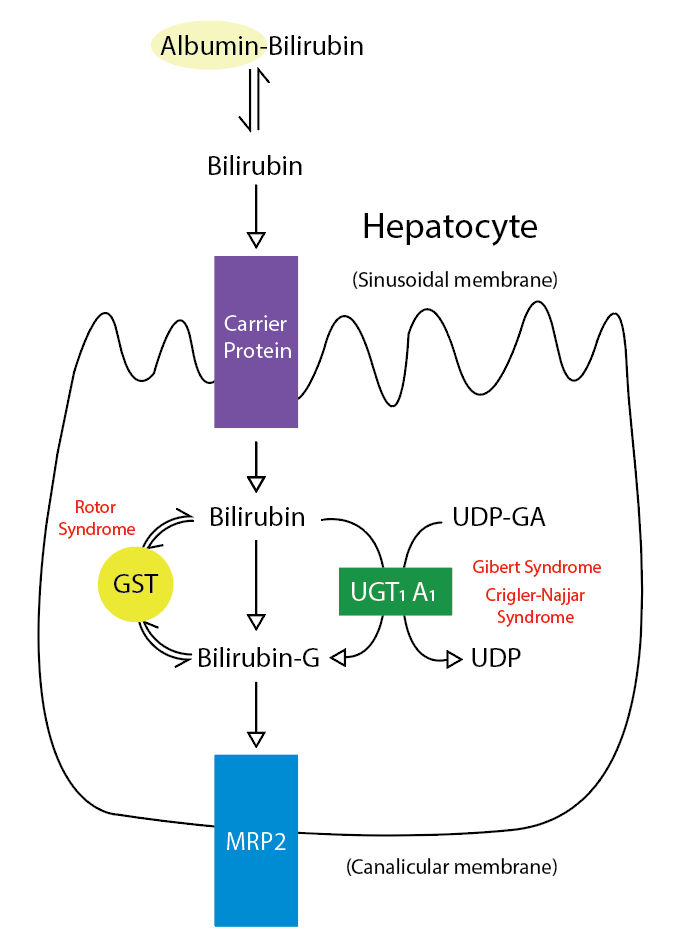
Metabolic Pathway for Bilirubin in the Hepatocyte. Bilirubin-G corresponds to bilirubin glucuronate; the donor is uridine diphosphate glucuronic acid (UDP-GA). This is catalyzed by the enzyme uridine diphosphate-glucuronyltransferase (UGT1A1). Gilbert and Crigler-Najjar syndrome is associated with decreases in UGT1A1 activity. Glutathione-S-transferase (GST) is a carrier protein that assists bilirubin uptake into the cytosol and may be implicated in Rotor syndrome.
Contributed by R Kabir, MD
References
Gale R, Seidman DS, Stevenson DK. Hyperbilirubinemia and early discharge. Journal of perinatology : official journal of the California Perinatal Association. 2001 Jan-Feb:21(1):40-3 [PubMed PMID: 11268867]
Mitra S, Rennie J. Neonatal jaundice: aetiology, diagnosis and treatment. British journal of hospital medicine (London, England : 2005). 2017 Dec 2:78(12):699-704. doi: 10.12968/hmed.2017.78.12.699. Epub [PubMed PMID: 29240507]
Kaplan M, Renbaum P, Levy-Lahad E, Hammerman C, Lahad A, Beutler E. Gilbert syndrome and glucose-6-phosphate dehydrogenase deficiency: a dose-dependent genetic interaction crucial to neonatal hyperbilirubinemia. Proceedings of the National Academy of Sciences of the United States of America. 1997 Oct 28:94(22):12128-32 [PubMed PMID: 9342374]
Level 2 (mid-level) evidenceMoncrieff MW, Dunn J. Phototherapy for hyperbilirubinaemia in very low birthweight infants. Archives of disease in childhood. 1976 Feb:51(2):124-6 [PubMed PMID: 944021]
Bhutani VK, Wong R. Bilirubin-induced neurologic dysfunction (BIND). Seminars in fetal & neonatal medicine. 2015 Feb:20(1):1. doi: 10.1016/j.siny.2014.12.010. Epub 2015 Jan 7 [PubMed PMID: 25577656]
Ip S, Chung M, Kulig J, O'Brien R, Sege R, Glicken S, Maisels MJ, Lau J, American Academy of Pediatrics Subcommittee on Hyperbilirubinemia. An evidence-based review of important issues concerning neonatal hyperbilirubinemia. Pediatrics. 2004 Jul:114(1):e130-53 [PubMed PMID: 15231986]
Level 1 (high-level) evidenceLeung AK, Sauve RS. Breastfeeding and breast milk jaundice. Journal of the Royal Society of Health. 1989 Dec:109(6):213-7 [PubMed PMID: 2513410]
American Academy of Pediatrics Subcommittee on Hyperbilirubinemia. Management of hyperbilirubinemia in the newborn infant 35 or more weeks of gestation. Pediatrics. 2004 Jul:114(1):297-316 [PubMed PMID: 15231951]
Level 1 (high-level) evidenceShahid R, Graba S. Outcome and cost analysis of implementing selective Coombs testing in the newborn nursery. Journal of perinatology : official journal of the California Perinatal Association. 2012 Dec:32(12):966-9. doi: 10.1038/jp.2012.26. Epub 2012 Mar 22 [PubMed PMID: 22441112]
Level 2 (mid-level) evidenceDesjardins L, Blajchman MA, Chintu C, Gent M, Zipursky A. The spectrum of ABO hemolytic disease of the newborn infant. The Journal of pediatrics. 1979 Sep:95(3):447-9 [PubMed PMID: 469673]
Level 2 (mid-level) evidence. ACOG practice bulletin. Prevention of Rh D alloimmunization. Number 4, May 1999 (replaces educational bulletin Number 147, October 1990). Clinical management guidelines for obstetrician-gynecologists. American College of Obstetrics and Gynecology. International journal of gynaecology and obstetrics: the official organ of the International Federation of Gynaecology and Obstetrics. 1999 Jul:66(1):63-70 [PubMed PMID: 10458556]
Level 1 (high-level) evidenceGómez-Manzo S, Marcial-Quino J, Vanoye-Carlo A, Serrano-Posada H, Ortega-Cuellar D, González-Valdez A, Castillo-Rodríguez RA, Hernández-Ochoa B, Sierra-Palacios E, Rodríguez-Bustamante E, Arreguin-Espinosa R. Glucose-6-Phosphate Dehydrogenase: Update and Analysis of New Mutations around the World. International journal of molecular sciences. 2016 Dec 9:17(12): [PubMed PMID: 27941691]
Grace RF, Zanella A, Neufeld EJ, Morton DH, Eber S, Yaish H, Glader B. Erythrocyte pyruvate kinase deficiency: 2015 status report. American journal of hematology. 2015 Sep:90(9):825-30. doi: 10.1002/ajh.24088. Epub 2015 Aug 14 [PubMed PMID: 26087744]
Da Costa L, Galimand J, Fenneteau O, Mohandas N. Hereditary spherocytosis, elliptocytosis, and other red cell membrane disorders. Blood reviews. 2013 Jul:27(4):167-78. doi: 10.1016/j.blre.2013.04.003. Epub 2013 May 9 [PubMed PMID: 23664421]
Perrotta S, Gallagher PG, Mohandas N. Hereditary spherocytosis. Lancet (London, England). 2008 Oct 18:372(9647):1411-26. doi: 10.1016/S0140-6736(08)61588-3. Epub [PubMed PMID: 18940465]
Gallagher PG, Weed SA, Tse WT, Benoit L, Morrow JS, Marchesi SL, Mohandas N, Forget BG. Recurrent fatal hydrops fetalis associated with a nucleotide substitution in the erythrocyte beta-spectrin gene. The Journal of clinical investigation. 1995 Mar:95(3):1174-82 [PubMed PMID: 7883966]
McDonald SJ, Middleton P, Dowswell T, Morris PS. Effect of timing of umbilical cord clamping of term infants on maternal and neonatal outcomes. The Cochrane database of systematic reviews. 2013 Jul 11:2013(7):CD004074. doi: 10.1002/14651858.CD004074.pub3. Epub 2013 Jul 11 [PubMed PMID: 23843134]
Level 1 (high-level) evidenceFogarty M, Osborn DA, Askie L, Seidler AL, Hunter K, Lui K, Simes J, Tarnow-Mordi W. Delayed vs early umbilical cord clamping for preterm infants: a systematic review and meta-analysis. American journal of obstetrics and gynecology. 2018 Jan:218(1):1-18. doi: 10.1016/j.ajog.2017.10.231. Epub 2017 Oct 30 [PubMed PMID: 29097178]
Level 1 (high-level) evidenceFenton C, McNinch NL, Bieda A, Dowling D, Damato E. Clinical Outcomes in Preterm Infants Following Institution of a Delayed Umbilical Cord Clamping Practice Change. Advances in neonatal care : official journal of the National Association of Neonatal Nurses. 2018 Jun:18(3):223-231. doi: 10.1097/ANC.0000000000000492. Epub [PubMed PMID: 29794839]
Level 2 (mid-level) evidenceNakagawa M, Ishida Y, Nagaoki Y, Ohta H, Shimabukuro R, Hirata M, Yamanaka M, Kusakawa I. Correlation between umbilical cord hemoglobin and rate of jaundice requiring phototherapy in healthy newborns. Pediatrics international : official journal of the Japan Pediatric Society. 2015 Aug:57(4):626-8. doi: 10.1111/ped.12583. Epub 2015 Mar 25 [PubMed PMID: 25533043]
Level 2 (mid-level) evidenceBosma PJ, Chowdhury JR, Bakker C, Gantla S, de Boer A, Oostra BA, Lindhout D, Tytgat GN, Jansen PL, Oude Elferink RP. The genetic basis of the reduced expression of bilirubin UDP-glucuronosyltransferase 1 in Gilbert's syndrome. The New England journal of medicine. 1995 Nov 2:333(18):1171-5 [PubMed PMID: 7565971]
Level 2 (mid-level) evidenceAnderson NB, Calkins KL. Neonatal Indirect Hyperbilirubinemia. NeoReviews. 2020 Nov:21(11):e749-e760. doi: 10.1542/neo.21-11-e749. Epub [PubMed PMID: 33139512]
Maruo Y, Nakahara S, Yanagi T, Nomura A, Mimura Y, Matsui K, Sato H, Takeuchi Y. Genotype of UGT1A1 and phenotype correlation between Crigler-Najjar syndrome type II and Gilbert syndrome. Journal of gastroenterology and hepatology. 2016 Feb:31(2):403-8. doi: 10.1111/jgh.13071. Epub [PubMed PMID: 26250421]
Flaherman VJ, Maisels MJ, Academy of Breastfeeding Medicine. ABM Clinical Protocol #22: Guidelines for Management of Jaundice in the Breastfeeding Infant 35 Weeks or More of Gestation-Revised 2017. Breastfeeding medicine : the official journal of the Academy of Breastfeeding Medicine. 2017 Jun:12(5):250-257. doi: 10.1089/bfm.2017.29042.vjf. Epub 2017 Apr 10 [PubMed PMID: 29624434]
Grunebaum E, Amir J, Merlob P, Mimouni M, Varsano I. Breast mild jaundice: natural history, familial incidence and late neurodevelopmental outcome of the infant. European journal of pediatrics. 1991 Feb:150(4):267-70 [PubMed PMID: 2029918]
Preer GL, Philipp BL. Understanding and managing breast milk jaundice. Archives of disease in childhood. Fetal and neonatal edition. 2011 Nov:96(6):F461-6. doi: 10.1136/adc.2010.184416. Epub 2010 Aug 5 [PubMed PMID: 20688866]
Level 3 (low-level) evidenceRubarth LB. Infants of diabetic mothers. Neonatal network : NN. 2013 Nov-Dec:32(6):416-8. doi: 10.1891/0730-0832.32.6.416. Epub [PubMed PMID: 24195802]
Amin SB. Clinical assessment of bilirubin-induced neurotoxicity in premature infants. Seminars in perinatology. 2004 Oct:28(5):340-7 [PubMed PMID: 15686265]
Maisels MJ, Kring E. Risk of sepsis in newborns with severe hyperbilirubinemia. Pediatrics. 1992 Nov:90(5):741-3 [PubMed PMID: 1408547]
Hansen TW. Therapeutic approaches to neonatal jaundice: an international survey. Clinical pediatrics. 1996 Jun:35(6):309-16 [PubMed PMID: 8782955]
Level 3 (low-level) evidenceMaisels MJ, Watchko JF, Bhutani VK, Stevenson DK. An approach to the management of hyperbilirubinemia in the preterm infant less than 35 weeks of gestation. Journal of perinatology : official journal of the California Perinatal Association. 2012 Sep:32(9):660-4. doi: 10.1038/jp.2012.71. Epub 2012 Jun 7 [PubMed PMID: 22678141]
McDonagh AF. Is bilirubin good for you? Clinics in perinatology. 1990 Jun:17(2):359-69 [PubMed PMID: 2196134]
Hegyi T, Goldie E, Hiatt M. The protective role of bilirubin in oxygen-radical diseases of the preterm infant. Journal of perinatology : official journal of the California Perinatal Association. 1994 Jul-Aug:14(4):296-300 [PubMed PMID: 7965225]
Beal JA. American Academy of Pediatrics' Updated Clinical Guidelines for Managing Neonatal Hyperbilirubinemia. MCN. The American journal of maternal child nursing. 2023 Jan-Feb 01:48(1):49. doi: 10.1097/NMC.0000000000000874. Epub [PubMed PMID: 36469895]
Fawaz R, Baumann U, Ekong U, Fischler B, Hadzic N, Mack CL, McLin VA, Molleston JP, Neimark E, Ng VL, Karpen SJ. Guideline for the Evaluation of Cholestatic Jaundice in Infants: Joint Recommendations of the North American Society for Pediatric Gastroenterology, Hepatology, and Nutrition and the European Society for Pediatric Gastroenterology, Hepatology, and Nutrition. Journal of pediatric gastroenterology and nutrition. 2017 Jan:64(1):154-168. doi: 10.1097/MPG.0000000000001334. Epub [PubMed PMID: 27429428]
Plosa EJ, Esbenshade JC, Fuller MP, Weitkamp JH. Cytomegalovirus infection. Pediatrics in review. 2012 Apr:33(4):156-63; quiz 163. doi: 10.1542/pir.33-4-156. Epub [PubMed PMID: 22474112]
Roelofsen H, van der Veere CN, Ottenhoff R, Schoemaker B, Jansen PL, Oude Elferink RP. Decreased bilirubin transport in the perfused liver of endotoxemic rats. Gastroenterology. 1994 Oct:107(4):1075-84 [PubMed PMID: 7926459]
Level 3 (low-level) evidencePan DH, Rivas Y. Jaundice: Newborn to Age 2 Months. Pediatrics in review. 2017 Nov:38(11):499-510. doi: 10.1542/pir.2015-0132. Epub [PubMed PMID: 29093118]
The NS, Honein MA, Caton AR, Moore CA, Siega-Riz AM, Druschel CM, National Birth Defects Prevention Study. Risk factors for isolated biliary atresia, National Birth Defects Prevention Study, 1997-2002. American journal of medical genetics. Part A. 2007 Oct 1:143A(19):2274-84 [PubMed PMID: 17726689]
Level 2 (mid-level) evidenceTakamizawa S, Zaima A, Muraji T, Kanegawa K, Akasaka Y, Satoh S, Nishijima E. Can biliary atresia be diagnosed by ultrasonography alone? Journal of pediatric surgery. 2007 Dec:42(12):2093-6 [PubMed PMID: 18082715]
Level 2 (mid-level) evidenceSerinet MO, Wildhaber BE, Broué P, Lachaux A, Sarles J, Jacquemin E, Gauthier F, Chardot C. Impact of age at Kasai operation on its results in late childhood and adolescence: a rational basis for biliary atresia screening. Pediatrics. 2009 May:123(5):1280-6. doi: 10.1542/peds.2008-1949. Epub [PubMed PMID: 19403492]
Level 2 (mid-level) evidenceBalistreri WF, Bezerra JA. Whatever happened to "neonatal hepatitis"? Clinics in liver disease. 2006 Feb:10(1):27-53, v [PubMed PMID: 16376793]
Soares KC, Arnaoutakis DJ, Kamel I, Rastegar N, Anders R, Maithel S, Pawlik TM. Choledochal cysts: presentation, clinical differentiation, and management. Journal of the American College of Surgeons. 2014 Dec:219(6):1167-80. doi: 10.1016/j.jamcollsurg.2014.04.023. Epub 2014 Jun 27 [PubMed PMID: 25442379]
Amedee-Manesme O, Bernard O, Brunelle F, Hadchouel M, Polonovski C, Baudon JJ, Beguet P, Alagille D. Sclerosing cholangitis with neonatal onset. The Journal of pediatrics. 1987 Aug:111(2):225-9 [PubMed PMID: 3612394]
Ljung R, Ivarsson S, Nilsson P, Solvig J, Wattsgård C, Borulf S. Cholelithiasis during the first year of life: case reports and literature review. Acta paediatrica (Oslo, Norway : 1992). 1992 Jan:81(1):69-72 [PubMed PMID: 1600308]
Level 3 (low-level) evidenceJesina D. Alagille Syndrome: An Overview. Neonatal network : NN. 2017 Nov 1:36(6):343-347. doi: 10.1891/0730-0832.36.6.343. Epub [PubMed PMID: 29185945]
Level 3 (low-level) evidenceEmerick KM, Rand EB, Goldmuntz E, Krantz ID, Spinner NB, Piccoli DA. Features of Alagille syndrome in 92 patients: frequency and relation to prognosis. Hepatology (Baltimore, Md.). 1999 Mar:29(3):822-9 [PubMed PMID: 10051485]
Li L, Wang NL, Gong JY, Wang JS. [Infantile cholestasis caused by CFTR mutation: case report and literature review]. Zhonghua er ke za zhi = Chinese journal of pediatrics. 2016 Nov 2:54(11):851-855. doi: 10.3760/cma.j.issn.0578-1310.2016.11.013. Epub [PubMed PMID: 27806795]
Level 3 (low-level) evidenceTownsend S, Newsome P, Turner AM. Presentation and prognosis of liver disease in alpha-1 antitrypsin deficiency. Expert review of gastroenterology & hepatology. 2018 Aug:12(8):745-747. doi: 10.1080/17474124.2018.1477589. Epub 2018 May 28 [PubMed PMID: 29768056]
Karadag N, Zenciroglu A, Eminoglu FT, Dilli D, Karagol BS, Kundak A, Dursun A, Hakan N, Okumus N. Literature review and outcome of classic galactosemia diagnosed in the neonatal period. Clinical laboratory. 2013:59(9-10):1139-46 [PubMed PMID: 24273939]
Level 2 (mid-level) evidenceChinsky JM, Singh R, Ficicioglu C, van Karnebeek CDM, Grompe M, Mitchell G, Waisbren SE, Gucsavas-Calikoglu M, Wasserstein MP, Coakley K, Scott CR. Diagnosis and treatment of tyrosinemia type I: a US and Canadian consensus group review and recommendations. Genetics in medicine : official journal of the American College of Medical Genetics. 2017 Dec:19(12):. doi: 10.1038/gim.2017.101. Epub 2017 Aug 3 [PubMed PMID: 28771246]
Level 3 (low-level) evidenceJacquemin E. Progressive familial intrahepatic cholestasis. Genetic basis and treatment. Clinics in liver disease. 2000 Nov:4(4):753-63 [PubMed PMID: 11232355]
Davit-Spraul A, Gonzales E, Baussan C, Jacquemin E. Progressive familial intrahepatic cholestasis. Orphanet journal of rare diseases. 2009 Jan 8:4():1. doi: 10.1186/1750-1172-4-1. Epub 2009 Jan 8 [PubMed PMID: 19133130]
Bull LN, Roche E, Song EJ, Pedersen J, Knisely AS, van Der Hagen CB, Eiklid K, Aagenaes O, Freimer NB. Mapping of the locus for cholestasis-lymphedema syndrome (Aagenaes syndrome) to a 6.6-cM interval on chromosome 15q. American journal of human genetics. 2000 Oct:67(4):994-9 [PubMed PMID: 10968776]
Strassburg CP. Hyperbilirubinemia syndromes (Gilbert-Meulengracht, Crigler-Najjar, Dubin-Johnson, and Rotor syndrome). Best practice & research. Clinical gastroenterology. 2010 Oct:24(5):555-71. doi: 10.1016/j.bpg.2010.07.007. Epub [PubMed PMID: 20955959]
Level 3 (low-level) evidenceLauriti G, Zani A, Aufieri R, Cananzi M, Chiesa PL, Eaton S, Pierro A. Incidence, prevention, and treatment of parenteral nutrition-associated cholestasis and intestinal failure-associated liver disease in infants and children: a systematic review. JPEN. Journal of parenteral and enteral nutrition. 2014 Jan:38(1):70-85. doi: 10.1177/0148607113496280. Epub 2013 Jul 26 [PubMed PMID: 23894170]
Level 1 (high-level) evidenceBuchman AL, Iyer K, Fryer J. Parenteral nutrition-associated liver disease and the role for isolated intestine and intestine/liver transplantation. Hepatology (Baltimore, Md.). 2006 Jan:43(1):9-19 [PubMed PMID: 16374841]
Duerksen DR, Van Aerde JE, Chan G, Thomson AB, Jewell LJ, Clandinin MT. Total parenteral nutrition impairs bile flow and alters bile composition in newborn piglet. Digestive diseases and sciences. 1996 Sep:41(9):1864-70 [PubMed PMID: 8794808]
Level 3 (low-level) evidencePan X, Kelly S, Melin-Aldana H, Malladi P, Whitington PF. Novel mechanism of fetal hepatocyte injury in congenital alloimmune hepatitis involves the terminal complement cascade. Hepatology (Baltimore, Md.). 2010 Jun:51(6):2061-8. doi: 10.1002/hep.23581. Epub [PubMed PMID: 20512994]
Feldman AG, Whitington PF. Neonatal hemochromatosis. Journal of clinical and experimental hepatology. 2013 Dec:3(4):313-20. doi: 10.1016/j.jceh.2013.10.004. Epub 2013 Nov 27 [PubMed PMID: 25755519]
Bhutani VK, Stark AR, Lazzeroni LC, Poland R, Gourley GR, Kazmierczak S, Meloy L, Burgos AE, Hall JY, Stevenson DK, Initial Clinical Testing Evaluation and Risk Assessment for Universal Screening for Hyperbilirubinemia Study Group. Predischarge screening for severe neonatal hyperbilirubinemia identifies infants who need phototherapy. The Journal of pediatrics. 2013 Mar:162(3):477-482.e1. doi: 10.1016/j.jpeds.2012.08.022. Epub 2012 Oct 5 [PubMed PMID: 23043681]
Level 2 (mid-level) evidenceBhutani VK. Editorial: building evidence to manage newborn jaundice worldwide. Indian journal of pediatrics. 2012 Feb:79(2):253-5. doi: 10.1007/s12098-011-0631-6. Epub 2011 Dec 20 [PubMed PMID: 22183759]
Level 3 (low-level) evidenceAlkhotani A, Eldin EE, Zaghloul A, Mujahid S. Evaluation of neonatal jaundice in the Makkah region. Scientific reports. 2014 Apr 25:4():4802. doi: 10.1038/srep04802. Epub 2014 Apr 25 [PubMed PMID: 24763104]
Winfield CR, MacFaul R. Clinical study of prolonged jaundice in breast- and bottle-fed babies. Archives of disease in childhood. 1978 Jun:53(6):506-7 [PubMed PMID: 686778]
Sgro M, Campbell D, Shah V. Incidence and causes of severe neonatal hyperbilirubinemia in Canada. CMAJ : Canadian Medical Association journal = journal de l'Association medicale canadienne. 2006 Sep 12:175(6):587-90 [PubMed PMID: 16966660]
Ding G, Zhang S, Yao D, Na Q, Wang H, Li L, Yang L, Huang W, Wang Y, Xu J. An epidemiological survey on neonatal jaundice in China. Chinese medical journal. 2001 Apr:114(4):344-7 [PubMed PMID: 11780450]
Level 2 (mid-level) evidenceBhutani VK, Zipursky A, Blencowe H, Khanna R, Sgro M, Ebbesen F, Bell J, Mori R, Slusher TM, Fahmy N, Paul VK, Du L, Okolo AA, de Almeida MF, Olusanya BO, Kumar P, Cousens S, Lawn JE. Neonatal hyperbilirubinemia and Rhesus disease of the newborn: incidence and impairment estimates for 2010 at regional and global levels. Pediatric research. 2013 Dec:74 Suppl 1(Suppl 1):86-100. doi: 10.1038/pr.2013.208. Epub [PubMed PMID: 24366465]
Level 1 (high-level) evidenceMoore LG, Newberry MA, Freeby GM, Crnic LS. Increased incidence of neonatal hyperbilirubinemia at 3,100 m in Colorado. American journal of diseases of children (1960). 1984 Feb:138(2):157-61 [PubMed PMID: 6695871]
Level 2 (mid-level) evidenceDrew JH, Barrie J, Horacek I, Kitchen WH. Factors influencing jaundice in immigrant Greek infants. Archives of disease in childhood. 1978 Jan:53(1):49-52 [PubMed PMID: 626518]
. Guidelines for detection, management and prevention of hyperbilirubinemia in term and late preterm newborn infants (35 or more weeks' gestation) - Summary. Paediatrics & child health. 2007 May:12(5):401-18 [PubMed PMID: 19030400]
Watchko JF, Tiribelli C. Bilirubin-induced neurologic damage--mechanisms and management approaches. The New England journal of medicine. 2013 Nov 21:369(21):2021-30. doi: 10.1056/NEJMra1308124. Epub [PubMed PMID: 24256380]
Level 3 (low-level) evidenceDick MC, Mowat AP. Hepatitis syndrome in infancy--an epidemiological survey with 10 year follow up. Archives of disease in childhood. 1985 Jun:60(6):512-6 [PubMed PMID: 3874604]
Level 2 (mid-level) evidenceGottesman LE, Del Vecchio MT, Aronoff SC. Etiologies of conjugated hyperbilirubinemia in infancy: a systematic review of 1692 subjects. BMC pediatrics. 2015 Nov 20:15():192. doi: 10.1186/s12887-015-0506-5. Epub 2015 Nov 20 [PubMed PMID: 26589959]
Level 1 (high-level) evidenceD'Alessandro AM, Knechtle SJ, Chin LT, Fernandez LA, Yagci G, Leverson G, Kalayoglu M. Liver transplantation in pediatric patients: twenty years of experience at the University of Wisconsin. Pediatric transplantation. 2007 Sep:11(6):661-70 [PubMed PMID: 17663691]
Dennery PA, Seidman DS, Stevenson DK. Neonatal hyperbilirubinemia. The New England journal of medicine. 2001 Feb 22:344(8):581-90 [PubMed PMID: 11207355]
Poland RL, Odell GB. Physiologic jaundice: the enterohepatic circulation of bilirubin. The New England journal of medicine. 1971 Jan 7:284(1):1-6 [PubMed PMID: 4922346]
Level 1 (high-level) evidenceBrouillard RP. Measurement of red blood cell life-span. JAMA. 1974 Dec 2:230(9):1304-5 [PubMed PMID: 4479604]
Chuniaud L, Dessante M, Chantoux F, Blondeau JP, Francon J, Trivin F. Cytotoxicity of bilirubin for human fibroblasts and rat astrocytes in culture. Effect of the ratio of bilirubin to serum albumin. Clinica chimica acta; international journal of clinical chemistry. 1996 Dec 30:256(2):103-14 [PubMed PMID: 9027422]
Level 3 (low-level) evidenceAmato MM, Kilguss NV, Gelardi NL, Cashore WJ. Dose-effect relationship of bilirubin on striatal synaptosomes in rats. Biology of the neonate. 1994:66(5):288-93 [PubMed PMID: 7873694]
Level 3 (low-level) evidenceHoffman DJ, Zanelli SA, Kubin J, Mishra OP, Delivoria-Papadopoulos M. The in vivo effect of bilirubin on the N-methyl-D-aspartate receptor/ion channel complex in the brains of newborn piglets. Pediatric research. 1996 Dec:40(6):804-8 [PubMed PMID: 8947954]
Level 3 (low-level) evidenceBenchimol EI, Walsh CM, Ling SC. Early diagnosis of neonatal cholestatic jaundice: test at 2 weeks. Canadian family physician Medecin de famille canadien. 2009 Dec:55(12):1184-92 [PubMed PMID: 20008595]
Trauner M, Meier PJ, Boyer JL. Molecular pathogenesis of cholestasis. The New England journal of medicine. 1998 Oct 22:339(17):1217-27 [PubMed PMID: 9780343]
Level 3 (low-level) evidenceChen HL, Wu SH, Hsu SH, Liou BY, Chen HL, Chang MH. Jaundice revisited: recent advances in the diagnosis and treatment of inherited cholestatic liver diseases. Journal of biomedical science. 2018 Oct 26:25(1):75. doi: 10.1186/s12929-018-0475-8. Epub 2018 Oct 26 [PubMed PMID: 30367658]
Level 3 (low-level) evidenceHamza A. Kernicterus. Autopsy & case reports. 2019 Jan-Mar:9(1):e2018057. doi: 10.4322/acr.2018.057. Epub 2019 Jan 14 [PubMed PMID: 30863731]
Level 3 (low-level) evidenceVij M, Rela M. Biliary atresia: pathology, etiology and pathogenesis. Future science OA. 2020 Mar 17:6(5):FSO466. doi: 10.2144/fsoa-2019-0153. Epub 2020 Mar 17 [PubMed PMID: 32518681]
Matthai J, Paul S. Evaluation of cholestatic jaundice in young infants. Indian pediatrics. 2001 Aug:38(8):893-8 [PubMed PMID: 11521001]
Maisels MJ, Bhutani VK, Bogen D, Newman TB, Stark AR, Watchko JF. Hyperbilirubinemia in the newborn infant } or =35 weeks' gestation: an update with clarifications. Pediatrics. 2009 Oct:124(4):1193-8. doi: 10.1542/peds.2009-0329. Epub 2009 Sep 28 [PubMed PMID: 19786452]
Johnson L, Bhutani VK. Guidelines for management of the jaundiced term and near-term infant. Clinics in perinatology. 1998 Sep:25(3):555-74, viii [PubMed PMID: 9779334]
Kemper AR, Newman TB, Slaughter JL, Maisels MJ, Watchko JF, Downs SM, Grout RW, Bundy DG, Stark AR, Bogen DL, Holmes AV, Feldman-Winter LB, Bhutani VK, Brown SR, Maradiaga Panayotti GM, Okechukwu K, Rappo PD, Russell TL. Clinical Practice Guideline Revision: Management of Hyperbilirubinemia in the Newborn Infant 35 or More Weeks of Gestation. Pediatrics. 2022 Sep 1:150(3):. pii: e2022058859. doi: 10.1542/peds.2022-058859. Epub [PubMed PMID: 35927462]
Level 1 (high-level) evidenceWainer S, Rabi Y, Parmar SM, Allegro D, Lyon M. Impact of skin tone on the performance of a transcutaneous jaundice meter. Acta paediatrica (Oslo, Norway : 1992). 2009 Dec:98(12):1909-15. doi: 10.1111/j.1651-2227.2009.01497.x. Epub 2009 Sep 17 [PubMed PMID: 19764923]
Casnocha Lucanova L, Matasova K, Zibolen M, Krcho P. Accuracy of transcutaneous bilirubin measurement in newborns after phototherapy. Journal of perinatology : official journal of the California Perinatal Association. 2016 Oct:36(10):858-61. doi: 10.1038/jp.2016.91. Epub 2016 Jun 9 [PubMed PMID: 27279078]
Hulzebos CV, Dijk PH, van Imhoff DE, Bos AF, Lopriore E, Offringa M, Ruiter SA, van Braeckel KN, Krabbe PF, Quik EH, van Toledo-Eppinga L, Nuytemans DH, van Wassenaer-Leemhuis AG, Benders MJ, Korbeeck-van Hof KK, van Lingen RA, Groot Jebbink LJ, Liem D, Mansvelt P, Buijs J, Govaert P, van Vliet I, Mulder TL, Wolfs C, Fetter WP, Laarman C, BARTrial Study Group. The bilirubin albumin ratio in the management of hyperbilirubinemia in preterm infants to improve neurodevelopmental outcome: a randomized controlled trial--BARTrial. PloS one. 2014:9(6):e99466. doi: 10.1371/journal.pone.0099466. Epub 2014 Jun 13 [PubMed PMID: 24927259]
Level 1 (high-level) evidenceSteinborn M, Seelos KC, Heuck A, von Voss H, Reiser M. MR findings in a patient with Kernicterus. European radiology. 1999:9(9):1913-5 [PubMed PMID: 10602975]
Level 3 (low-level) evidenceMcKiernan PJ, Baker AJ, Kelly DA. The frequency and outcome of biliary atresia in the UK and Ireland. Lancet (London, England). 2000 Jan 1:355(9197):25-9 [PubMed PMID: 10615887]
Level 2 (mid-level) evidenceMorotti RA, Jain D. Pediatric Cholestatic Disorders: Approach to Pathologic Diagnosis. Surgical pathology clinics. 2013 Jun:6(2):205-25. doi: 10.1016/j.path.2013.03.001. Epub 2013 May 4 [PubMed PMID: 26838972]
Bhutani VK, Committee on Fetus and Newborn, American Academy of Pediatrics. Phototherapy to prevent severe neonatal hyperbilirubinemia in the newborn infant 35 or more weeks of gestation. Pediatrics. 2011 Oct:128(4):e1046-52. doi: 10.1542/peds.2011-1494. Epub 2011 Sep 26 [PubMed PMID: 21949150]
Yetman RJ, Parks DK, Huseby V, Mistry K, Garcia J. Rebound bilirubin levels in infants receiving phototherapy. The Journal of pediatrics. 1998 Nov:133(5):705-7 [PubMed PMID: 9821435]
Level 2 (mid-level) evidenceNewman TB, Wu YW, Kuzniewicz MW, Grimes BA, McCulloch CE. Childhood Seizures After Phototherapy. Pediatrics. 2018 Oct:142(4):. pii: e20180648. doi: 10.1542/peds.2018-0648. Epub [PubMed PMID: 30249623]
Newman TB, Wickremasinghe AC, Walsh EM, Grimes BA, McCulloch CE, Kuzniewicz MW. Retrospective Cohort Study of Phototherapy and Childhood Cancer in Northern California. Pediatrics. 2016 Jun:137(6):. pii: e20151354. doi: 10.1542/peds.2015-1354. Epub [PubMed PMID: 27217477]
Level 2 (mid-level) evidenceAuger N, Laverdière C, Ayoub A, Lo E, Luu TM. Neonatal phototherapy and future risk of childhood cancer. International journal of cancer. 2019 Oct 15:145(8):2061-2069. doi: 10.1002/ijc.32158. Epub 2019 Feb 8 [PubMed PMID: 30684392]
Wang J, Guo G, Li A, Cai WQ, Wang X. Challenges of phototherapy for neonatal hyperbilirubinemia (Review). Experimental and therapeutic medicine. 2021 Mar:21(3):231. doi: 10.3892/etm.2021.9662. Epub 2021 Jan 20 [PubMed PMID: 33613704]
Itoh S, Okada H, Kuboi T, Kusaka T. Phototherapy for neonatal hyperbilirubinemia. Pediatrics international : official journal of the Japan Pediatric Society. 2017 Sep:59(9):959-966. doi: 10.1111/ped.13332. Epub 2017 Aug 7 [PubMed PMID: 28563973]
Kar S, Mohankar A, Krishnan A. Bronze baby syndrome. Indian pediatrics. 2013 Jun 8:50(6):624 [PubMed PMID: 23942414]
Level 3 (low-level) evidencePearson CH. Replacement transfusion as a treatment of erythroblastosis fetalis, by Louis K. Diamond, MD, Pediatrics, 1948;2:520-524. Pediatrics. 1998 Jul:102(1 Pt 2):203-5 [PubMed PMID: 9651427]
Hansen TWR, Maisels MJ, Ebbesen F, Vreman HJ, Stevenson DK, Wong RJ, Bhutani VK. Sixty years of phototherapy for neonatal jaundice - from serendipitous observation to standardized treatment and rescue for millions. Journal of perinatology : official journal of the California Perinatal Association. 2020 Feb:40(2):180-193. doi: 10.1038/s41372-019-0439-1. Epub 2019 Aug 16 [PubMed PMID: 31420582]
DIAMOND LK, ALLEN FH Jr, THOMAS WO Jr. Erythroblastosis fetalis. VII. Treatment with exchange transfusion. The New England journal of medicine. 1951 Jan 11:244(2):39-49 [PubMed PMID: 14785788]
Jackson JC. Adverse events associated with exchange transfusion in healthy and ill newborns. Pediatrics. 1997 May:99(5):E7 [PubMed PMID: 9113964]
Patra K, Storfer-Isser A, Siner B, Moore J, Hack M. Adverse events associated with neonatal exchange transfusion in the 1990s. The Journal of pediatrics. 2004 May:144(5):626-31 [PubMed PMID: 15126997]
Level 2 (mid-level) evidenceAlpay F, Sarici SU, Okutan V, Erdem G, Ozcan O, Gökçay E. High-dose intravenous immunoglobulin therapy in neonatal immune haemolytic jaundice. Acta paediatrica (Oslo, Norway : 1992). 1999 Feb:88(2):216-9 [PubMed PMID: 10102158]
Level 1 (high-level) evidenceGottstein R, Cooke RW. Systematic review of intravenous immunoglobulin in haemolytic disease of the newborn. Archives of disease in childhood. Fetal and neonatal edition. 2003 Jan:88(1):F6-10 [PubMed PMID: 12496219]
Level 1 (high-level) evidenceOhi R. Surgery for biliary atresia. Liver. 2001 Jun:21(3):175-82 [PubMed PMID: 11422780]
Karthik SV, Campbell-Davidson D, Isherwood D. Carotenemia in infancy and its association with prevalent feeding practices. Pediatric dermatology. 2006 Nov-Dec:23(6):571-3 [PubMed PMID: 17156001]
Level 2 (mid-level) evidenceRiskin A, Tamir A, Kugelman A, Hemo M, Bader D. Is visual assessment of jaundice reliable as a screening tool to detect significant neonatal hyperbilirubinemia? The Journal of pediatrics. 2008 Jun:152(6):782-7, 787.e1-2. doi: 10.1016/j.jpeds.2007.11.003. Epub 2008 Jan 22 [PubMed PMID: 18492516]
Khurshid F, Rao SP, Sauve C, Gupta S. Universal screening for hyperbilirubinemia in term healthy newborns at discharge: A systematic review and meta-analysis. Journal of global health. 2022 Dec 29:12():12007. doi: 10.7189/jogh.12.12007. Epub 2022 Dec 29 [PubMed PMID: 36579719]
Level 1 (high-level) evidenceRand EB, Karpen SJ, Kelly S, Mack CL, Malatack JJ, Sokol RJ, Whitington PF. Treatment of neonatal hemochromatosis with exchange transfusion and intravenous immunoglobulin. The Journal of pediatrics. 2009 Oct:155(4):566-71. doi: 10.1016/j.jpeds.2009.04.012. Epub 2009 Jun 28 [PubMed PMID: 19560784]
Level 2 (mid-level) evidenceHankø E, Hansen TW, Almaas R, Lindstad J, Rootwelt T. Bilirubin induces apoptosis and necrosis in human NT2-N neurons. Pediatric research. 2005 Feb:57(2):179-84 [PubMed PMID: 15611354]
Olusanya BO, Slusher TM, Imosemi DO, Emokpae AA. Maternal detection of neonatal jaundice during birth hospitalization using a novel two-color icterometer. PloS one. 2017:12(8):e0183882. doi: 10.1371/journal.pone.0183882. Epub 2017 Aug 24 [PubMed PMID: 28837635]