Introduction
Immunoglobulin A (IgA), one of the five primary immunoglobulins, plays a pivotal role in mucosal homeostasis in the gastrointestinal, respiratory, and genitourinary tracts, functioning as the dominant antibody of immunity in this role.[1] It is the second most abundant immunoglobulin type found in the body and, consequently, has a crucial role in protection against antigens.[2] IgA production is greater than all other immunoglobulin subtypes, necessary for the many roles it plays systemically.
IgA gets produced by class switching of Ig, which is regulated by various processes. The binding of CD40-CD40L and secretion of other cytokines IL-4, IL-5, IL-6, IL-10, and IL-21 promote maturation of Th2 cells, which promote class switching to different Ig subtypes. Retinoic acid, a metabolite of vitamin A, synergistically acts with IL-5 and IL-6 to induce IgA secretion as well.[3]
Molecular Level
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Molecular Level
IgA has two subtypes, IgA1 and IgA2, which play different roles in the body. The IgA1 hinge portion has multiple O-linked glycans and two N-linked glycosylation sites on each heavy chain. IgA2 has a shortened hinge region with four N-linked glycosylation sites on each heavy chain and no O-linked glycans. Due to the longer hinge region of IgA1, it is more susceptible to cleavage from bacterial proteases. IgA1 makes up around 80% of total IgA and is found on mucosal surfaces, while IgA2 is primarily in the colon. IgA gets cleared from the circulation via the asialoglycoprotein receptor.[1]
Function
Secretory IgA (SIgA) is released into the lumen and is the first-line barrier to pathogens and other irritants. It binds to antigens and prevents their access to the submucosa and circulation.[4] SIgA binds to many receptors such as the IgA transmembrane receptor (FcαRI), transferrin receptor, and Galectin-1, which regulates intestinal immunity.[5] However, Fc-alpha-RI is the primary receptor responsible for IgA mucosal immunity. It gets expressed on myeloid cells such as eosinophils, neutrophils, macrophages, and Kupffer cells. Monomeric IgA has the potential to bind to the extracellular-1 (EC1) domain of the Fc-alpha-RI with high avidity. As IgA immune complexes or IgA-bound pathogens cross-link Fc-alpha-RI, a series of physiologic responses become activated, including phagocytosis, antibody-dependent cellular cytotoxicity, the release of cytokines, the release of other mediators of inflammation, and antigen presentation. Cross-linking with Fc-alpha-RI also releases leukotriene B4 by neutrophils, which is a potent chemoattractant and results in increased migration through a positive feedback loop. Through these mechanisms, any irritants or pathogens in the mucosa are well-contained.[1]
In addition to immune protection from antigens, IgA also has essential anti-inflammatory functions primarily mediated through Fc-alpha-RI, dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin (DC-SIGN), and specific ICAM-3 grabbing nonintegrin-related 1 (SIGNR1) receptors. Secretory IgA has a sugar-dependent interaction with SIGNR1, which induces immune tolerance via regulatory T cells on dendritic cells. DC-SIGN also interacts with sub-epithelial dendritic cells to take up secretory IgA immune complexes in the lamina propria. Furthermore, monomeric IgA interacts with Fc-alpha-RI to phosphorylate Fc receptor gamma-identical activatory motifs (FcRy-ITAM), which results in the recruitment of tyrosine phosphatase SHP-1, causing cellular inhibition through the formation of inhibisomes.[2] As Fc-alpha-RI and IgA play a major role in inflammation and immunity, an inhibitor of Fc-alpha-RI may serve as a potential treatment in allergies or other immune complex conditions that researchers are currently testing in mice models.[1]
Mechanism
IgA is secreted by plasma cells throughout the body into either internal fluids or external secretions. It remains in the monomeric form in internal fluids such as plasma and cerebrospinal fluid and primarily gets released as a dimer held together by a joining (J) chain in external secretions where it plays its role in mucosal immunity. Plasma cells in the mucosal subepithelium secrete monomeric IgA, which then complexes by covalent bondage through J chains to form dimeric IgA. This dimeric IgA binds to the polymeric Ig receptor (pIgR) at the basolateral surface of intestinal epithelial cells to enter the cell via transcytosis.[6] It is shuffled across the cell, and the polymeric Ig receptor still bound to IgA is cleaved at the apical surface to form the secretory component. Thus, pIgR transports dimeric IgA to the mucosal lumen where it gets released as secretory IgA (SIgA), which comprises dimeric IgA, the J chain, and the secretory component deriving from PIgR.[1] This secretory component functions to protect IgA antibodies from proteolytic degradation and prevent their attachment to the epithelial surface in the lumen, better allowing it to interact with pathogens. This SIgA functions to prevent adhesion and penetration of antigens, neutralization of viruses, and opsonization of antigens.
Testing
IgA levels are easily quantified in clinical laboratories by automated turbidimetric and nephelometric methods.
Clinical Significance
Selective IgA deficiency is the most prevalent immunodeficiency in humans and affects 1 in 700 individuals, most of the White race. It characteristically demonstrates an isolated decrease in IgA levels below 7 mg/dl with generally normal levels of IgG and Ig M in patients over four years of age.[7] It is due to an inability of IgA bearing B lymphocytes to switch into IgA secreting plasma cells. While around half of individuals are asymptomatic or have mild symptoms, those with more severe IgA deficiency have repeated bouts of bacterial, enteroviral, or protozoal infections of the gastrointestinal and respiratory tracts due to the lack of mucosal immunity. The most prevalent illnesses associated with IgA deficiency are recurrent sino-pulmonary infections.[7]
Pulmonary infections most commonly result from extracellular encapsulated bacteria and can result in recurrent infections as well as long-term respiratory epithelial damage. While there is an increase in IgM in an attempt to compensate for the missing IgA, it cannot functionally replace IgA completely, especially in the respiratory tract. However, IgM can compensate for IgA more holistically in the gastrointestinal tract leading to fewer gastrointestinal disorders and infections. Despite this, there remains a correlation between IgA deficiency and other disorders such as celiac disease, ulcerative colitis, Crohn disease, and pernicious anemia. There also remains a high chance of contracting giardiasis due to a deficiency of IgA, which has a strong role in preventing this infection. Celiac disease remains the most common disorder concurrently found with IgA deficiency, with an almost 10 to 15% increased chance of being IgA deficient. As Celiac disease is due to an autoimmune reaction of IgA and IgG antibodies directed towards proteins, testing for anti-IgA antibodies can result in a false negative. Thus IgG antibody levels and other tests are required to confirm the diagnosis.
IgA plays an active anti-inflammatory role in the body, leading to allergic diseases and autoimmune conditions when deficient. A decrease in secretory IgA results in a weakened mucosal barrier and allows more allergens to enter into the bloodstream. IgA deficiency can result in the production of IgG anti-IgA antibodies, which can cause a significant risk for anaphylaxis when transfused with blood products that contain IgA. Also, there is a higher incidence of autoimmune diseases such as rheumatoid arthritis, SLE, celiac disease, and thyroiditis in IgA deficient individuals due to unknown mechanisms.[8]
IgA nephropathy, also known as Berger disease, is a type of glomerulonephritis of the kidney. It is the most frequent cause of glomerulonephritis worldwide and has a multitude of clinical manifestations.[9] The hypothesis is that a deficiency in O–linked glycosylation of IgA1 results in immune complexes that trigger the glomerular injury. This condition results from deposits of IgA in the mesangium, a region of the glomerulus that is surrounded by capillaries. On immunofluorescence, a granular pattern is visible due to IgA immune complex deposition. It often presents during childhood as hematuria with red blood cell cells and casts. An upper respiratory tract infection can precede it. IgA levels are increased in both serum and urine but are of no prognostic significance. The presentation can vary according to the age group affected. These individuals have mild proteinuria, and up to 50% may progress to renal failure depending on other confounding factors such as hypertension, decreased glomerular filtration rate, and proteinuria.[9]
IgA Vasculitis (IgAV), also referred to as Henoch-Schonlein purpura, is the most common childhood vasculitis that affects small vessels and is more common in Asian populations. Researchers have advanced impaired IgA1 glycosylation as the possible mechanism of pathogenesis. It results from the deposition of IgA and complement protein C3 in the vessel walls that cause inflammation and leukocytoclastic vasculitis, resulting in erythrocytes extravasating the blood vessels resulting in purpura in the dermis. The peak age of presentation is 4 to 6 years.[10] This condition is associated with a tetrad of symptoms: palpable purpura classically in the lower limbs and buttocks, joint pain, gastrointestinal pain, and renal involvement. The predominant deposition of IgA in the glomeruli results in its renal manifestations. The typical primary presentation is purpura with joint pain that eventually progresses to involve the gastrointestinal tract and kidneys. A feared complication is intussusception, which constitutes a surgical emergency and can present with colicky abdominal pain with bleeding. Usually, it has a very good prognosis.[11]
IgA myeloma is the second most commonly encountered subtype of multiple myeloma, characterized by a plasma cell malignancy that produces IgA. It typically presents with a tetrad of features, including hypercalcemia, renal impairment, and back pain due to osteolytic bone lesions and anemia. Bone marrow biopsy reveals greater than 10 percent plasma cells. Bone destruction is a hallmark of this condition and results in lytic bone lesions primarily in the skull and spine, as well as other regions of the body. The lesions can increase the likelihood of fractures and manifest as severe bone pain. This condition is diagnosable via serum protein electrophoresis, which reveals an M-protein that is then confirmed by immunofixation electrophoresis to be IgA with either kappa or lambda light chains.[12]
Linear immunoglobulin A bullous dermatosis (LABD) is a rare, autoimmune disease that affects the subepidermis. It appears to be caused by IgA antibodies to collagen XVII with concomitant neutrophil accumulation. In this condition, there is a linear deposition of IgA against antigens in the basal membrane zone, which ultimately leads to subepidermal cleavage. LABD results in blisters on a reddish base in the perineum and perioral areas. The skin symptoms can be painful and can lead to more chronic gingivitis, ulcers, and conjunctivitis. It typically presents around six months of age and may spontaneously undergo remission within two years or remain until puberty.[13]
Media
(Click Image to Enlarge)
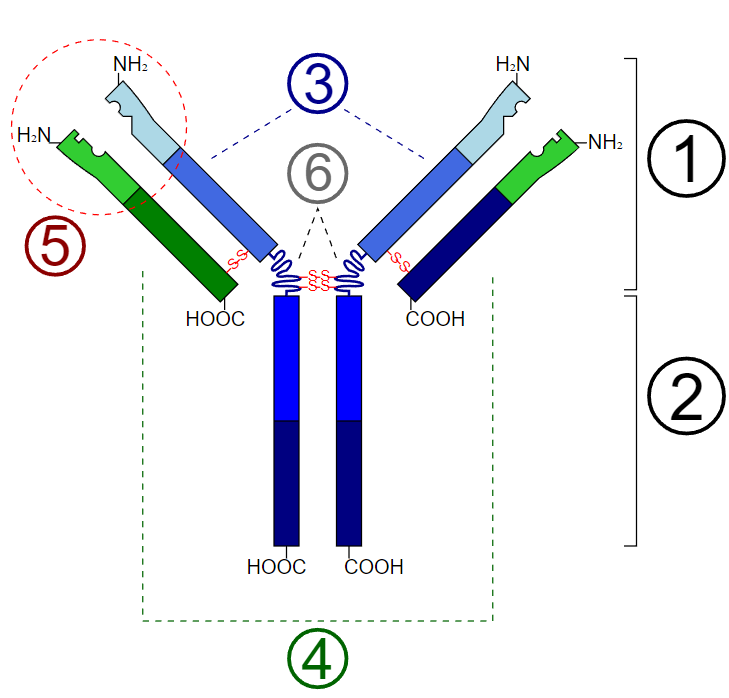
Basic Unit of Immunoglobulin (Antibody). 1. Fab region; 2. Fc region; 3. Heavy chains (consist of VH, CH1, hinge, CH2, and CH3 regions from the N-terminus); 4. Light chains (consist of VL and CL regions from the N-terminus); 5. Antigen binding site; 6. Hinge regions.
Y Tambe, Public Domain, via Wikimedia Commons
References
Breedveld A, van Egmond M. IgA and FcαRI: Pathological Roles and Therapeutic Opportunities. Frontiers in immunology. 2019:10():553. doi: 10.3389/fimmu.2019.00553. Epub 2019 Mar 22 [PubMed PMID: 30984170]
Mkaddem SB, Christou I, Rossato E, Berthelot L, Lehuen A, Monteiro RC. IgA, IgA receptors, and their anti-inflammatory properties. Current topics in microbiology and immunology. 2014:382():221-35. doi: 10.1007/978-3-319-07911-0_10. Epub [PubMed PMID: 25116102]
Level 3 (low-level) evidenceBoyaka PN. Inducing Mucosal IgA: A Challenge for Vaccine Adjuvants and Delivery Systems. Journal of immunology (Baltimore, Md. : 1950). 2017 Jul 1:199(1):9-16. doi: 10.4049/jimmunol.1601775. Epub [PubMed PMID: 28630108]
Corthésy B. Role of secretory immunoglobulin A and secretory component in the protection of mucosal surfaces. Future microbiology. 2010 May:5(5):817-29. doi: 10.2217/fmb.10.39. Epub [PubMed PMID: 20441552]
Corthésy B, Spertini F. Secretory immunoglobulin A: from mucosal protection to vaccine development. Biological chemistry. 1999 Nov:380(11):1251-62 [PubMed PMID: 10614817]
Level 3 (low-level) evidenceGommerman JL, Rojas OL, Fritz JH. Re-thinking the functions of IgA(+) plasma cells. Gut microbes. 2014:5(5):652-62. doi: 10.4161/19490976.2014.969977. Epub [PubMed PMID: 25483334]
Level 3 (low-level) evidenceVo Ngoc DT, Krist L, van Overveld FJ, Rijkers GT. The long and winding road to IgA deficiency: causes and consequences. Expert review of clinical immunology. 2017 Apr:13(4):371-382. doi: 10.1080/1744666X.2017.1248410. Epub 2016 Oct 25 [PubMed PMID: 27776452]
Yazdani R, Azizi G, Abolhassani H, Aghamohammadi A. Selective IgA Deficiency: Epidemiology, Pathogenesis, Clinical Phenotype, Diagnosis, Prognosis and Management. Scandinavian journal of immunology. 2017 Jan:85(1):3-12. doi: 10.1111/sji.12499. Epub [PubMed PMID: 27763681]
Rodrigues JC, Haas M, Reich HN. IgA Nephropathy. Clinical journal of the American Society of Nephrology : CJASN. 2017 Apr 3:12(4):677-686. doi: 10.2215/CJN.07420716. Epub 2017 Feb 3 [PubMed PMID: 28159829]
Oni L, Sampath S. Childhood IgA Vasculitis (Henoch Schonlein Purpura)-Advances and Knowledge Gaps. Frontiers in pediatrics. 2019:7():257. doi: 10.3389/fped.2019.00257. Epub 2019 Jun 27 [PubMed PMID: 31316952]
Level 3 (low-level) evidenceHetland LE, Susrud KS, Lindahl KH, Bygum A. Henoch-Schönlein Purpura: A Literature Review. Acta dermato-venereologica. 2017 Nov 15:97(10):1160-1166. doi: 10.2340/00015555-2733. Epub [PubMed PMID: 28654132]
Giuliani N, Rizzoli V, Roodman GD. Multiple myeloma bone disease: Pathophysiology of osteoblast inhibition. Blood. 2006 Dec 15:108(13):3992-6 [PubMed PMID: 16917004]
Fernandes KA, Galvis KH, Gomes AC, Nogueira OM, Felix PA, Vargas TJ. Linear IgA and IgG bullous dermatosis. Anais brasileiros de dermatologia. 2016 Sep-Oct:91(5 suppl 1):32-34. doi: 10.1590/abd1806-4841.20164630. Epub [PubMed PMID: 28300887]