Introduction
Immune thrombocytopenia (ITP), formerly idiopathic thrombocytopenic purpura, is a condition arising from immunoglobulin G (IgG) autoantibodies sensitizing circulating platelets, manifesting as a low platelet count, purpura, and hemorrhagic episodes. The diagnosis is typically made by excluding known thrombocytopenia causes. ITP can affect both children and adults. The American Society of Hematology (ASH) defines ITP as a generalized purpuric rash accompanied by a platelet count less than 100,000/μL and normal white blood cell (WBC) count and hemoglobin level.[1] Leukocyte count and hemoglobin abnormalities are not characteristic of ITP and should prompt additional diagnostic testing when detected. (American Society of Hematology)
Primary ITP develops without an underlying cause and must be distinguished from secondary ITP. Conditions that may trigger secondary ITP include drug reactions, autoimmune illnesses like systemic lupus erythematosus (SLE), malignancies like chronic lymphocytic leukemia (CLL), and infections like HIV. Primary ITP may be further categorized based on the timing and persistence of symptoms. Newly diagnosed ITP refers to the condition from the time of diagnosis to 3 months afterward. Persistent ITP arises when symptoms continue 3 to 12 months following the initial diagnosis. Chronic ITP indicates ongoing symptoms beyond 12 months from the initial diagnosis until resolution or further management. Refractory ITP includes cases that do not resolve with splenectomy.
Severe ITP, when platelet counts are below 20,000/μL, warrants medical treatment. Immunosuppressive therapy is the cornerstone of management.[2]
Platelet Physiology
Platelets are essential for hemostasis. These blood cells originate from megakaryocytes in the bone marrow through a process called "thrombopoiesis." Under normal conditions, platelets circulating in the blood range from 150,000 to 350,000 per μL and have a mean life span of 7 to 10 days. Thrombopoietin (TPO), a hormone primarily produced in the liver, kidneys, and bone marrow, regulates platelet production by stimulating megakaryocyte proliferation and differentiation. Factors such as infection, inflammation, and hemorrhage can induce platelet proliferation to maintain normal platelet counts.
Platelets possess distinctive cellular characteristics, including a discoid shape, a lack of nuclei, and abundant granules containing clotting factors and inflammatory mediators. Glycoprotein IIb/IIIa (GP IIb/IIIa) is a key receptor on platelet membranes essential for aggregation, facilitating blood clot formation. This receptor's activation and subsequent fibrinogen binding are crucial steps in hemostasis. The spleen plays a crucial role in platelet homeostasis by removing aged or damaged platelets from circulation.[3][4][5]
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
Infection and immune changes are the 2 most common ITP-inciting events. Viral illnesses often precede infection-associated ITP. Antibodies develop against viral antigens, which exhibit a form of molecular mimicry that leads to cross-reactions with normal platelet antigens. The most common viral infections implicated in ITP include HIV, hepatitis C, cytomegalovirus, and varicella zoster.
ITP cases associated with immune alterations occur due to autoimmune conditions causing loss of peripheral tolerance and promoting the development of autoantibodies. Autoimmune conditions commonly triggering ITP include antiphospholipid syndrome, SLE, Evans syndrome, hematopoietic cell transplantation, common variable immunodeficiency, and autoimmune lymphoproliferative syndrome.[6]
Various medications are known to cause ITP, including the following:
- Abciximab
- β-lactam antibiotics
- Carbamazepine
- Eptifibatide
- Gold compounds
- Heparin
- Phenytoin
- Linezolid
- Measles, mumps, and rubella vaccine
- Piperacillin
- Quinine
- Sulfonamides
- Vancomycin
- Tirofiban
- Rifampin
- Trimethoprim-sulfamethoxazole [7][8]
Malignancies that can cause ITP include CLL, adenocarcinoma, and lymphoma. Endocrine disorders like hypothyroidism and Addison disease may also trigger ITP symptoms. Autoantibody production and failure of self-tolerance are proposed to be the main drivers of ITP development in all these cases.[9]
Epidemiology
The annual incidence of pediatric ITP is estimated to be between 1 and 6.4 cases per 100,000 people. Researchers suggest this figure should be higher because it is based on reported symptomatic hospitalized cases, not total ITP cases. Pediatric ITP can present at any age, but peak incidences occur at ages 2 to 5 and adolescence. The condition is more common among male than female children from infancy to childhood. The opposite is true among adolescents and young adults (ie, 18 to 45 years). Predominance among women in these age groups is attributed to estrogen’s effects, which can promote autoimmunity.[10]
ITP may be acute or chronic. The acute form is more frequent in childhood, often prefaced by a viral infection. Most children have a self-limiting course, typically spontaneously recovering within 3 months. The chronic form more often affects adults, and the inciting factor is usually unclear. However, aging-related immune decline, comorbidities, and drugs are thought to contribute to the condition's development in adults.[11] Approximately 25% of children in the United States develop persistent or chronic ITP, where thrombocytopenia-associated bleeding can be life-threatening.[12]
Seasonal fluctuations have been noted among children with ITP, with the condition’s incidence rising in the spring and early summer when viral infections are common. ITP’s annual incidence in adults is estimated to be 1 to 6 cases per 100,000 people. However, the condition is more of a chronic disease in adults, so the prevalence is approximately 12 per 100,000 cases. Peak incidence in adults occurs around 60 years old. However, incidence increases with age. ITP’s incidence beyond age 60 is nearly similar between male and female adults.[13][14]
Pathophysiology
ITP is mediated by autoantibodies, typically IgG. Autoantibodies directed against platelet membrane proteins, specifically GP IIb/IIIa, Ib/IIa, and VI, develop after exposure to an inciting agent. Antibody-coated platelets are cleared faster than usual by tissue macrophages, specifically in the spleen, shortening platelet half-life and resulting in thrombocytopenia (see Image. Thrombocytopenia).[15] An alternative mechanism involving T-cell-mediated cytotoxicity, particularly attacking megakaryocytes in the bone marrow, has also been proposed, though not fully elucidated.[16]
Histopathology
Studies spanning several decades have investigated bone marrow examinations in ITP, with conflicting findings regarding megakaryocyte numbers and morphology. While some researchers report an increased megakaryocyte population and abnormal appearance, others find these variables normal. Overall, bone marrow examinations are often inconclusive and unreliable for diagnosing typical ITP cases. However, in rare subsets of severe ITP, unique features such as an increased megakaryocyte number may be observed.[17]
History and Physical
History
Some patients with ITP are asymptomatic. When symptomatic, mucocutaneous bleeding is the most common manifestation, ranging from mild petechiae and epistaxis to severe and potentially life-threatening gastrointestinal, intracranial, or urinary tract hemorrhage. Bruising or a petechial rash may be reported. Risk factors for major bleeding include a platelet count of less than 20,000/μL, advanced age, and prior minor bleeding, with a higher incidence in the months following acute ITP diagnosis. Patients may also experience fatigue, though this symptom usually improves with treatment and increased platelet counts.[18]
Possible secondary causes must be investigated. For example, about 60% of children diagnosed with ITP have a preceding viral illness within the past month.[19] Studies have also shown a small increased ITP risk within 6 weeks after vaccination with the measles, mumps, and rubella vaccine. ITP has also been reported after varicella, hepatitis A, and tetanus-diphtheria-acellular pertussis vaccinations in older children.[20]
Patients should be asked about systemic symptoms like fever, weight loss, anorexia, night sweats, or bone pain and the severity and character of bleeding symptoms. Female patients may report menstrual changes. The diet must also be explored, as some beverages and foods like tonic water and “jello shots” contain quinine or other substances that can trigger thrombocytopenia. A personal bleeding history, family history of bleeding or platelet disorders, medication changes, and the presence of certain comorbidities may provide clues to the diagnosis.
Physical Exam
All patients warrant a complete physical examination with special attention to the vital signs, skin and mucosal surfaces, head and neck, abdomen, and neurologic status. The nasal passages, buccal and gingival surfaces, and gastrointestinal and genitourinary tracts are likely bleeding sources in ITP. However, conjunctival and retinal hemorrhages are rarely seen.[21]
Most children are well-appearing, with normal vital signs. The classic petechial rash, which does not blanch when pressure is applied, is usually noted. About two-thirds of adults have bleeding, ranging in severity from a petechial rash to moderate mucosal bleeding and, rarely, hemorrhage.[22] Tachycardia and hypotension may be signs of acute hemorrhage or sepsis. Fever, tachycardia, and lymphadenopathy in a pale patient may indicate severe infection or a hematologic malignancy. Hepatosplenomegaly may be evident in patients with severe ITP. Neurologic deficits may manifest in the presence of intracranial hemorrhage (ICH).
Evaluation
ITP is a diagnosis of exclusion. Diagnostic testing can help determine the cause of thrombocytopenia or rule out other disorders with similar presentations.
Initial Evaluation
Initial laboratory evaluation for children and adults should include a complete blood count (CBC) with differential WBC count, reticulocyte count, and peripheral blood smear (PBS). The WBC count, hemoglobin concentration, red cell indices, and leukocyte differential are usually normal in patients with ITP. However, the platelet count is characteristically less than 100,000/μL. These results may change, depending on the inciting factor or the presence of comorbidities.
For example, patients with a recent infection may exhibit abnormal WBC counts. Bacterial infection usually presents with neutrophil-predominant leukocytosis. Viral infections often manifest with leukopenia, lymphocyte predominance, or both.
CBC with PBS should reveal reduced platelet levels with normal or increased size. White and red blood cells typically appear normal. The reticulocyte count is usually within normal limits unless the patient develops significant acute blood loss. Individuals with chronic bleeding usually have microcytic anemia.[23] WBC precursor cells (ie, blasts) in the circulation may indicate leukemia or lymphoma as the cause of thrombocytopenia.[24] Hemolytic anemia typically manifests with polychromasia, reticulocytosis, or spherocytosis. Schistocytosis usually signifies microangiopathic hemolytic anemia.[25][26]
Blood typing is usually performed if the patient needs a blood transfusion. Meanwhile, direct and indirect antiplatelet autoantibody tests have high specificity but low sensitivity for ITP. Antiplatelet antibody testing is not recommended when evaluating ITP as it does not correlate with clinical outcomes.[27] Immunoglobulin level determination should be considered if an underlying immunodeficiency is suspected.
Further Evaluation
Bone marrow biopsy is no longer performed for typical ITP presentations in both adults and children. Bone marrow aspiration and biopsy are indicated if clinical or laboratory findings suggest malignancy or bone marrow failure. Such findings include lymph node enlargement, splenomegaly, neutropenia, leukocytosis, atypical lymphocytosis, or anemia in the presence of bone pain, fevers, or unintentional weight loss. Unresponsiveness to immunosuppressants also warrants bone marrow analysis.
Prothrombin time and activated partial thromboplastin time may be obtained in individuals with moderate or severe thrombocytopenia, planned invasive procedures, and concern for overt bleeding. Individuals with recurrent epigastric pain suggestive of peptic ulcer must be tested for Helicobacter pylori infection, which is a possible cause of ITP.
Patients requiring splenectomy for ITP treatment or any other surgery may be screened for hypo- or hyperthyroidism before the procedure, as these conditions increase the risk of perioperative complications. Vitamin B12 and folate levels should be measured in patients with highly restrictive diets, neurologic or psychiatric conditions, and findings of hypersegmented neutrophils on a complete blood profile.
About 12.8% of individuals with SLE are initially diagnosed with primary ITP, and 17.5% of patients with ITP have a positive antinuclear antibody (ANA) test. Thus, patients presenting with ITP symptoms and meeting the SLE criteria require additional testing.[28] ANA has a high sensitivity but low specificity for SLE. Meanwhile, anti-double-stranded DNA and anti-Smith antibodies are poorly sensitive but highly specific for SLE. High serum levels of these antibodies suggest SLE.[29] Other tests that may also be considered in the workup for other autoimmune conditions include other autoantibodies like anticardiolipin, complements C3 and C4, cryoglobulins, and immunoglobulins.
Treatment / Management
Initial Management
The ASH 2019 ITP guidelines recommend adult and pediatric algorithms based on the need for treatment and inpatient management. The following variables must first be determined:
- Presence and type of bleeding
- Platelet count
- Bleeding risk and social factors
- Any previous treatments
ASH guidelines recommend corticosteroids over observation in adults with newly diagnosed ITP and a platelet count of less than 30,000/μL if they are asymptomatic or have minor mucocutaneous bleeding. Meanwhile, for adults with newly diagnosed ITP and a platelet count of at least 30,000/μL who are asymptomatic or have minor mucocutaneous bleeding, ASH recommends against corticosteroids and suggests management with observation instead. Factors like platelet count nearing the threshold, additional comorbidities, anticoagulant or antiplatelet use, upcoming procedures, and age (especially older than 60) may warrant consideration for corticosteroid treatment despite ASH recommendations for observation.
For adults with newly diagnosed ITP and a platelet count of less than 20,000/μL who have no symptoms or have minor mucocutaneous bleeding, ASH suggests hospital admission instead of outpatient management. Conversely, outpatient management is suggested over hospital admission for adults with an established ITP diagnosis and a platelet count of less than 20,000/μL who have no symptoms or have minor mucocutaneous bleeding. These recommendations are conditional and depend on individual needs for hospital admission across different platelet count ranges. Patients not admitted to the hospital should receive education and expedited follow-up with a hematologist.
Outpatient management is recommended instead of hospital admission for adults with a platelet count of at least 20,000/μL who are asymptomatic or have minor mucocutaneous bleeding unless other considerations are present, such as social concerns, diagnostic uncertainty, and comorbidities predisposing to bleeding.
For children newly diagnosed with ITP with a platelet count of less than 20,000/μL and have no or mild bleeding (ie, limited to skin manifestations), ASH recommends outpatient management over hospital admission. However, this recommendation is conditional as factors like diagnostic uncertainty, social concerns, distance from the hospital, and uncertainty regarding follow-up may warrant hospital admission despite the platelet count and bleeding severity. Similarly, outpatient management is suggested over hospital admission for children with newly diagnosed ITP, a platelet count of at least 20,000/μL, and no or mild bleeding, with similar considerations for the need for hospital admission as in the previous recommendation.
ASH suggests observation instead of corticosteroids in children with newly diagnosed ITP and no or minor bleeding. ASH also recommends observation over intravenous immunoglobulin (IVIG) and anti-D immunoglobulin for these children. These recommendations underscore the preference for observation as the initial management approach in children with newly diagnosed ITP who have no or minor bleeding to balance the potential risks and benefits of treatment interventions.
Inpatient management strategies are determined based on bleeding severity, platelet count, and other factors like comorbidities, previous treatments, and social circumstances. These strategies are explained below.
Critical bleeding
Critical bleeding indicators include pericardial, intracranial, intraocular, intraspinal, retroperitoneal, intramuscular with compartment syndrome, or any other bleeding episodes leading to hemodynamic instability. The platelet count is typically less than 20,000/μL and often less than 10,000/μL. However, critical bleeding may also occur at higher platelet counts, eg, between 20,000 and 50,000/μL, which require similar treatment. Other reasons for bleeding should also be investigated and treated because thrombocytopenia is an unlikely cause of critical bleeding. Patients are often treated at the intensive care unit (ICU).
Treatment is with immediate platelet transfusion, IVIG, and glucocorticoids. Platelet transfusion is typically administered as 1 apheresis unit or 4 to 6 pooled platelet units. This intervention is the quickest method to elevate the platelet count in critical bleeding. However, platelet count increases following transfusion are generally short-lived, often lasting less than an hour, necessitating repeated transfusions or additional systemic therapies.
IVIG may be administered concurrently with platelet transfusion to enhance its efficacy. IVIG is typically administered at a dose of 1 g/kg as a single dose, which may be repeated the next day if the platelet count remains below 50,000/μL. Glucocorticoids are commonly used with IVIG, with typical regimens including either methylprednisolone 1 g intravenously (IV) once daily for 3 days or dexamethasone 40 mg IV once daily for 4 days. Hemostatic therapies such as tranexamic acid may also be administered.[30]
Severe bleeding
This condition refers to bleeding that does not meet the definition of critical bleeding but leads to a hemoglobin reduction by 2 g/dL. Severe bleeding requires urgent IVIG and glucocorticoid administration. Like critical bleeding, the platelet count in these cases is typically less than 20,000/μL and often less than 10,000/μL. Patients with this condition are also treated in the ICU.
Combining IVIG and glucocorticoids is often considered to have an augmentative effect in ITP management. IVIG is typically administered as a single dose of 1 g/kg, with the option of repetition on the following day if the platelet count remains below 50,000/μL. Glucocorticoids are commonly prescribed in 1 of 2 regimens: methylprednisolone 1 g IV once daily for 3 days or dexamethasone 40 mg IV once daily for 4 days. Hemostatic medications like tranexamic acid may also be given.
Minor bleeding
Minor bleeding does not meet the criteria for critical and severe bleeding. The general management of minor bleeding and severe thrombocytopenia with or without bleeding is similar. As previously mentioned, the decision to treat or observe these patients depends on platelet count and factors like comorbidities and social circumstances. Patients with minor bleeding with a platelet count of less than 50,000/μL and individuals with platelet counts below 20,000/μL should be treated. Conversely, patients with platelet counts of at least 30,000/μL and no bleeding symptoms often require close observation only.[31] (B3)
The ASH 2019 ITP guidelines advise observing children with no or mild bleeding symptoms (eg, bruising or petechiae) regardless of their platelet count. Regular monitoring of bleeding and platelet levels by a pediatric hematologist is recommended. Around 50% to 70% of children typically recover within 3 to 6 months without intervention. The guidelines recommend prednisone 2 to 4 mg/kg/day (maximum 120 mg daily) for 5 to 7 days for children with non-life-threatening mucosal bleeding or reduced quality of life.
The ASH guideline panel recommends either prednisone 0.5-2.0 mg/kg per day or dexamethasone 40 mg daily for 4 days as initial corticosteroid therapy for adults newly diagnosed with ITP. However, the panel cautions against using prednisone for an extended duration exceeding 6 weeks, including both treatment and tapering periods.
IVIG or anti-D immunoglobulin may be considered if corticosteroids are contraindicated or unsuitable. Anti-D, an immunoglobulin against the Rh blood group system’s D antigen, is sometimes used as an alternative in patients with RhD-positive blood group. However, it comes with a US Food and Drug Administration (FDA) boxed warning for hemolytic transfusion reactions. Thus, many clinicians are hesitant to use anti-D immunoglobulin.[32](B3)
Second-Line Therapies
The ASH 2019 guidelines recommend that children with non–life-threatening mucosal bleeding or reduced health-related quality of life unresponsive to first-line therapies or with chronic ITP should be considered for a trial of TPO receptor agonists (TPO-RAs, ie, eltrombopag or romiplostim). I the child does not respond to a TPO-RA, rituximab is recommended as the next therapy of choice.
In adults with ITP, the ASH guideline panel recommends second-line therapies if they are dependent on corticosteroids, unresponsive to corticosteroids for at least 3 months, or considered to have chronic ITP. Second-line treatments for adults include TPO-RAs (ie, eltrombopag, romiplostim, or avatrombopag), rituximab, and splenectomy. The choice of second-line interventions is tailored to the individual based on factors such as ITP chronicity, hospitalizations, comorbidities, and patient preferences. TPO-RAs are preferred for patients seeking a long-term response and avoiding surgery. Rituximab is preferred for individuals unwilling to take medication for an extended period and also wish to avoid surgery.
TPO-RAs are initiated at a low initial dose and uptitrated as necessary.[33] Romiplostim doses typically range from 1 to 10 mcg/kg but usually start at 2 to 3 mcg/kg.[34] The initial response to romiplostim therapy is typically observed within 7 to 14 days. Eltrombopag is initially administered at 50 mg daily, reduced to 25 mg once daily in individuals of East-Asian ancestry or with moderate to severe liver insufficiency.[35] The initial response to eltrombopag therapy is typically seen within 7 to 14 days. Absorption of eltrombopag may be reduced by concomitant intake of calcium-rich foods. Supplements containing polyvalent cations should be administered at least 4 hours before or 2 hours after eltrombopag intake. Avatrombopag is initiated at a dose of 20 mg once daily and adjusted to maintain platelet levels above 50,000/μL. The initial response to avatrombopag therapy is usually observed within 3 to 5 days. Avatrombopag is less hepatotoxic and does not interact with dairy products.(A1)
Rituximab is typically administered at a dose of 375 mg/m IV once a week for 4 consecutive weeks. This drug is contraindicated in patients with hepatitis B infection, as it risks progression to fulminant hepatitis, liver failure, and mortality.[36] Progressive multifocal leukoencephalopathy has also been reported following rituximab treatment for ITP.[37] The initial response to the therapy usually occurs within 7 to 56 days. Immunizations are administered before rituximab treatment to ensure efficacy, as the drug may interfere with the patient’s immune response.(B3)
Splenectomy is typically reserved for children with severe thrombocytopenia and significant hemorrhagic symptoms that require multiple pharmacological interventions. However, the procedure should be delayed as long as possible due to the potential for spontaneous remission and the risk of asplenic sepsis. The current recommendation is to wait at least 12 months from initial diagnosis and until the child is older than 5 before considering splenectomy. In adults, splenectomy should be considered if a long-term response is desired. However, the procedure should be delayed until a year after ITP diagnosis due to the potential for remission within that time frame.
In 2018, the FDA approved the spleen tyrosine kinase inhibitor fostamatinib as an option for ITP refractory to second-line treatments.[38][39] The 2019 ASH guidelines make no recommendations regarding the use of azathioprine, cyclophosphamide, cyclosporine A, danazol, dapsone, mycophenolate mofetil, and the vinca alkaloids vincristine and vinblastine in ITP. (A1)
Platelets should be monitored a week after a dose or drug change and every month once the patient’s condition stabilizes while on the same therapy. Platelets should also be checked whenever a mucosal bleed or petechial rashes occur.
Emerging Treatments
Efgartigimod and rozanolixizumab are compounds undergoing testing for use in myasthenia gravis and ITP. These agents are antibody fragments targeting the neonatal Fc-receptor (FcRn), thus inhibiting IgG recycling. Efgartigimod and rozanolixizumab decrease IgG’s half-life, reducing it to normal or subpathogenic levels. Investigators have found response rates of 38.5% with efgartigimod and more than 50% with rozanolixizumab.[40][41](B3)
Sutimlimab is a humanized monoclonal antibody against the complement pathway inhibitor C1s. This agent decreases complement-dependent cytotoxicity in ITP, thus decreasing platelet destruction. Studies have revealed responses of 33% to 42%, many within days, though overall sample sizes were small.[42](B3)
Rilzabrutinib is an orally administered Bruton Tyrosine kinase inhibitor (BTKI) that has demonstrated efficacy in treating ITP in clinical trials. This agent inhibits Fcγ signal transduction, inhibiting autoantibody production and platelet phagocytosis. Studies show a response rate of about 40%, with minimal toxicity and a time to respond of about a week.[43]
Differential Diagnosis
The differential diagnosis for thrombocytopenia includes but is not limited to the following:
- Leukemia, specifically acute lymphocytic leukemia, can present similarly to primary ITP. However, patients with this condition have clinical and laboratory findings not found in primary ITP, including systemic symptoms (ie, fever, joint pain, or weight loss), hepatosplenomegaly, lymphadenopathy, leukocytosis, and significant anemia (hemoglobin < 10 g/dL).
- An active infection, including HIV, Epstein-Barr virus, cytomegalovirus, and hepatitis C, can induce immune thrombocytopenia.[44]
- Autoimmune hemolytic anemia has corresponding clinical and laboratory findings, including anemia, jaundice, elevated reticulocyte count, spherocytes, and polychromasia. Primary ITP does not have these manifestations.
- Systemic autoimmune disease, particularly SLE and autoimmune lymphoproliferative syndrome, can present initially as thrombocytopenia. Further clinical and laboratory investigation can differentiate these conditions from primary ITP.
- Some immunodeficiency syndromes, specifically common variable immunodeficiency, present with thrombocytopenia following recurrent infections.[45] Wiskott-Aldrich syndrome should be a consideration in a male patient with eczema, small platelet size, bleeding out of proportion to the platelet count, family history, and a lack of response to ITP treatments. DiGeorge syndrome should be considered in people with hypocalcemia, cardiac anomalies, and thrombocytopenia.
- Certain drugs, specifically heparin, quinidine, phenytoin, sulfonamides, valproate, and vancomycin, can cause thrombocytopenia. Withdrawal of the inciting agent can improve platelet levels.
- Bone marrow failure, as seen in aplastic anemia, may manifest with thrombocytopenia. Various drugs, alcohol, infections, and toxins may suppress bone marrow activity.
- Hemolytic uremic syndrome presents with hemolytic anemia, thrombocytopenia, acute kidney injury, and a recent history of gastrointestinal infection.
- Thrombotic thrombocytopenia presents with severe microangiopathic hemolytic anemia, thrombocytopenia, and neurologic symptoms (eg, confusion, somnolence, or headache).
- Disseminated intravascular coagulation presents with thrombocytopenia in the context of hemorrhage and thrombosis with end-organ damage due to sepsis, trauma, or malignancy.
- Posttransfusion purpura is a transfusion-associated reaction occurring about a week after the transfusion. The condition is an amnestic response to platelet antigens, destroying native and transfused platelets.[46] Posttransfusion purpura may occur in multiparous women due to alloimmunization against platelet antigens.[47] This disorder is treated with IVIG.
- Neonatal immune thrombocytopenia arises from the transplacental passage of maternal antiplatelet antibodies.[48]
- Sepsis causes thrombocytopenia by various mechanisms, such as complement activation, histone release, and coagulation activation.
- Megaloblastic anemia and thrombocytopenia may cooccur in vitamin B12 deficiency. This disease can mimic thrombotic thrombocytopenia.[49]
- Refractory anemias, preleukemia, and hematopoietic dysplasia typically have cellular dysmorphism evident on bone marrow exams and often present as lineage cytopenias.[50]
- Hypersplenism presents with thrombocytopenia. The condition is a possible liver cirrhosis complication.[51]
A thorough clinical and diagnostic investigation can differentiate these conditions from primary ITP.
Prognosis
Most children recover from ITP within 3 to 6 months from initial presentation, regardless of treatment. Studies show that approximately 10% to 20% percent of children with acute ITP progress to chronic ITP.[52] Risk factors associated with an increased risk of developing chronic ITP include older age at diagnosis, less severe thrombocytopenia at initial diagnosis, gradual symptom onset, absence of preceding infection or vaccination before ITP diagnosis, and absence of mucosal bleeding at presentation.[53] Spontaneous remission occurs in approximately 50% of cases of children with chronic ITP, often within the first 2 years after diagnosis but can extend up to 5 years from initial diagnosis.[54]
Children younger than 10 with chronic ITP are more likely to undergo remission than older children.[55] ITP mortality in children is rare and is mainly due to bleeding complications, specifically ICH. Most mortality and morbidity in pediatric chronic ITP result from complications of long-term immunosuppressive treatment, mainly infections.[56]
Most adults reach a stable platelet count with 1 or more therapies. Spontaneous remission occurs in about 10% of adults, most often within the first 6 months.[57] About one- to two-thirds of individuals who do not undergo spontaneous remission reach a stable platelet count with first-line treatments. The remainder of adults with ITP have refractory disease, requiring additional second-line therapies or splenectomy. Reported cases include instances where initial ITP was induced via an autoimmune pathway, leading to subsequent SLE or CLL development in adults. However, the overall risk of developing SLE or CLL after ITP remains uncertain.[58]
ITP mortality in adults is only slightly higher than in the age-matched population and is mainly due to bleeding complications, similar to children. Most patients with ITP are more likely to die from non-ITP-related conditions than from ITP or treatment complications.
Complications
The likelihood of developing ITP-associated complications correlates with bleeding risk, especially when the platelet count is less than 20,000/μL. Most people with ITP have bruising and petechiae. Some individuals with ITP may experience mucosal bleeding, such as epistaxis or gum bleeding. Severe ITP may present with gastrointestinal tract bleeding, hematuria, or menorrhagia.
ITP's most feared complication is ICH. The ICH risk in children with newly diagnosed ITP is about 0.5% and is slightly higher in children with chronic ITP, though still less than 1%. Most ICH cases occur at platelet levels less than 10,000/μL. Concerning ICH symptoms in both children and adults include headache, persistent vomiting, altered mental status, seizures, focal neurological findings, and recent head trauma.
Urgent evaluation, including neuroimaging and treatment, is imperative should a patient have ICH. Risk factors for this condition include extremely low platelet counts (< 10,000/μL), head trauma, antiplatelet medication use, and severe bleeding. Epistaxis lasting 5 to 15 minutes, gastrointestinal tract bleeding, or any other severe mucosal bleeding requiring hospitalization or blood transfusion constitutes severe bleeding.
Deterrence and Patient Education
Patients and their families must be educated about the diagnosis of ITP, associated bleeding risks, proper treatment, and the importance of therapeutic compliance. Children with ITP should be restricted from activities predisposing to traumatic bleeding when their platelet count is less than 30,000/μL. Such activities include but are not limited to contact and collision sports, eg, football, boxing, lacrosse, and hockey, or any other activities with significant head injury risks, such as baseball, soccer, skiing, and gymnastics. Both children and adults should avoid antiplatelet medications, eg, nonsteroidal anti-inflammatory drugs like aspirin and ibuprofen. Patients should also avoid anticoagulants like heparin, enoxaparin, and warfarin if their platelet count is less than 20,000/μL.[59] Patients and their caregivers must be counseled on proper treatment and when to seek medical care.
Pearls and Other Issues
The most important points to remember when evaluating and managing ITP include the following:
- The definition of thrombocytopenia is having a blood platelet count of less than 100,000/μL. The condition may not be symptomatic until the platelet count falls below 10,000 to 20,000/μL.
- ITP may be primary or secondary. Primary ITP may be idiopathic or autoimmunity-related, while secondary ITP arises due to an inciting factor, such as infection, drugs, and malignancy. Primary ITP manifests with isolated thrombocytopenia.
- ITP is a diagnosis of exclusion. Diagnostic testing helps determine secondary causes or rule out clinical mimics. CBC with differential WBC count, PBS, and reticulocyte count is recommended as the initial laboratory test. Blood typing may be performed for patients requiring transfusion. Antiplatelet autoantibody tests are often not useful when managing ITP. Other tests, such as bone marrow analysis, vitamin B12 level, and antibody titers, may be obtained if clinically warranted.
- ITP is categorized as either newly diagnosed, persistent, chronic, or refractory, depending on the symptoms' timing and persistence. Children are more likely to present with acute ITP, while chronic disease is more frequent in adults.
- Many patients with ITP are asymptomatic. The decision to treat and hospitalize a patient must be based on the severity of bleeding (if present), platelet count, bleeding risk, social factors, and response to previous therapies.
- Corticosteroids are the first-line therapy in adults and children with minor bleeding. Combining corticosteroids with IVIG has a synergistic effect, making the intervention useful in managing severe-to-critical bleeding. Platelet transfusion and tranexamic acid are also recommended in severe presentations.
- Second-line therapies for chronic ITP include TPO-RAs, rituximab, and splenectomy. Fostamatinib is a recently approved medication that may be given to patients unresponsive to second-line therapies.
- The decision to perform a splenectomy must be postponed until at least a year after diagnosis, as ITP can resolve spontaneously within that time frame.
Patient and family education must cover disease diagnosis, course, prognosis, and treatment. Therapeutic compliance, regular follow-ups, and avoidance of bleeding risk factors must be emphasized when counseling patients and caregivers about symptom prevention.
Enhancing Healthcare Team Outcomes
Clinically significant ITP affects both pediatric and adult populations. While primary care providers may initially identify the condition, collaboration with an interprofessional team comprising hematologists, pathologists, pharmacists, and nurses specialized in ITP management is essential for effective diagnosis and treatment. Care plans often involve patient education on ITP diagnosis, associated bleeding risks, and appropriate treatment options.
Primary care providers should refer patients with complications to a hematologist. Pathologists aid in diagnosis by interpreting laboratory findings. Pharmacists should review any prior medications, coordinate treatment plans with prescribers, ensure medication reconciliation, and communicate potential issues with the team. Nurses facilitate communication between specialists, assist in patient counseling, and oversee care coordination.
Accurate, comprehensive, and updated medical records serve as vital communication tools within the care team. Additionally, patient and caregiver education is crucial to mitigate bleeding risks associated with certain activities and medications. This collaborative, interprofessional approach ensures optimal patient care.
Media
(Click Image to Enlarge)
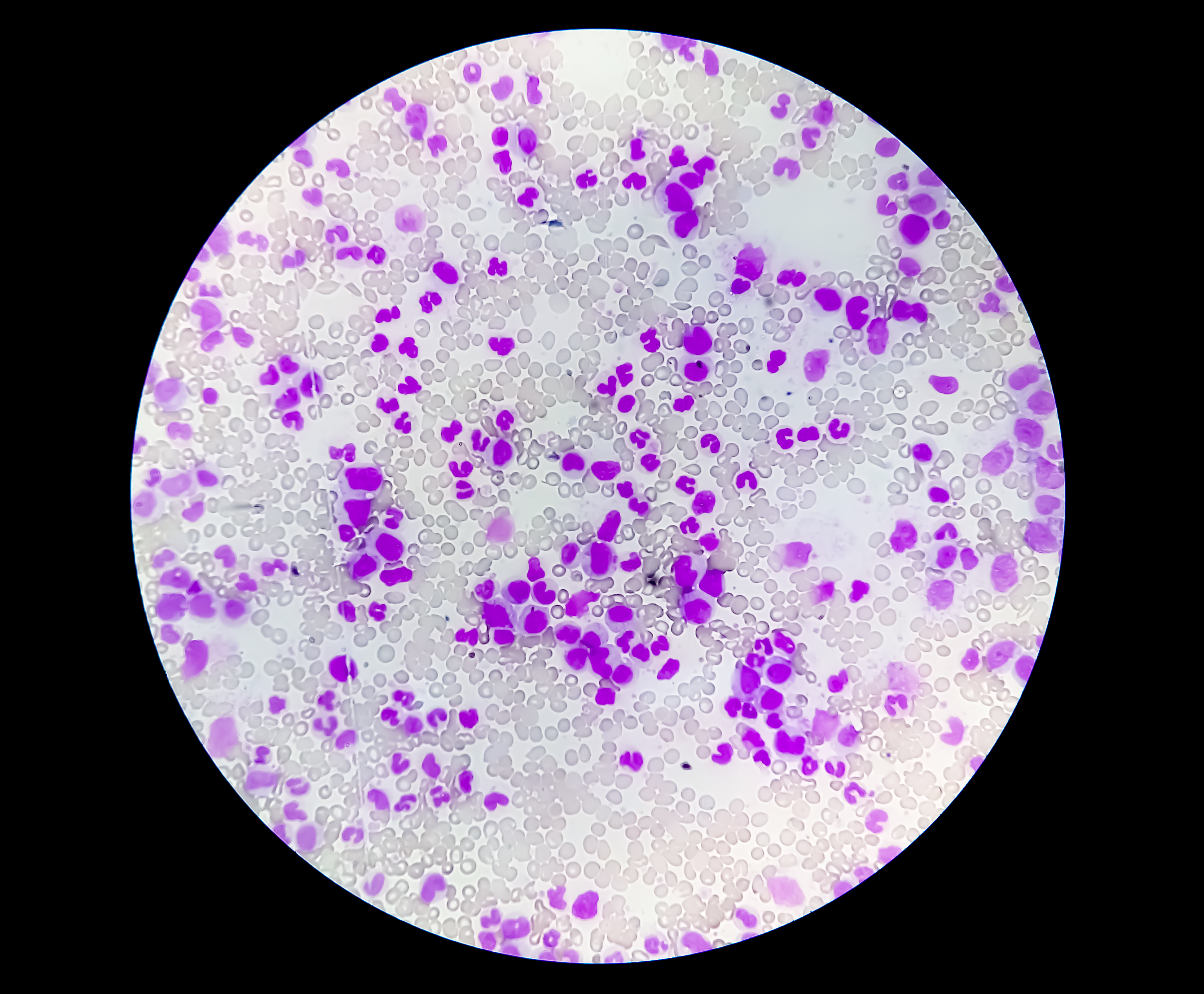
Thrombocytopenia. This image shows normal red and white blood cell counts and a reduced platelet number.
StatPearls
References
Rodeghiero F, Stasi R, Gernsheimer T, Michel M, Provan D, Arnold DM, Bussel JB, Cines DB, Chong BH, Cooper N, Godeau B, Lechner K, Mazzucconi MG, McMillan R, Sanz MA, Imbach P, Blanchette V, Kühne T, Ruggeri M, George JN. Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: report from an international working group. Blood. 2009 Mar 12:113(11):2386-93. doi: 10.1182/blood-2008-07-162503. Epub 2008 Nov 12 [PubMed PMID: 19005182]
Zitek T, Weber L, Pinzon D, Warren N. Assessment and Management of Immune Thrombocytopenia (ITP) in the Emergency Department: Current Perspectives. Open access emergency medicine : OAEM. 2022:14():25-34. doi: 10.2147/OAEM.S331675. Epub 2022 Jan 29 [PubMed PMID: 35125895]
Level 3 (low-level) evidenceWilliams O, Sergent SR. Histology, Platelets. StatPearls. 2025 Jan:(): [PubMed PMID: 32491732]
Cunin P, Nigrovic PA. Megakaryocytes as immune cells. Journal of leukocyte biology. 2019 Jun:105(6):1111-1121. doi: 10.1002/JLB.MR0718-261RR. Epub 2019 Jan 15 [PubMed PMID: 30645026]
Jeon GW. Pathophysiology, classification, and complications of common asymptomatic thrombocytosis in newborn infants. Clinical and experimental pediatrics. 2022 Apr:65(4):182-187. doi: 10.3345/cep.2021.00864. Epub 2021 Oct 18 [PubMed PMID: 34665959]
Cines DB, Bussel JB, Liebman HA, Luning Prak ET. The ITP syndrome: pathogenic and clinical diversity. Blood. 2009 Jun 25:113(26):6511-21. doi: 10.1182/blood-2009-01-129155. Epub 2009 Apr 24 [PubMed PMID: 19395674]
Nusrat S, Borogovac A, George JN, Curtis BR, Reese JA. Drug (vaccine)-induced thrombocytopenia 2021: Diversity of pathogenesis and clinical features. American journal of hematology. 2022 Apr:97(4):E162-E165. doi: 10.1002/ajh.26482. Epub 2022 Feb 15 [PubMed PMID: 35092624]
Mitta A, Curtis BR, Reese JA, George JN. Drug-induced thrombocytopenia: 2019 Update of clinical and laboratory data. American journal of hematology. 2019 Mar:94(3):E76-E78. doi: 10.1002/ajh.25379. Epub 2018 Dec 27 [PubMed PMID: 30549322]
Provan D, Semple JW. Recent advances in the mechanisms and treatment of immune thrombocytopenia. EBioMedicine. 2022 Feb:76():103820. doi: 10.1016/j.ebiom.2022.103820. Epub 2022 Jan 21 [PubMed PMID: 35074629]
Level 3 (low-level) evidenceFrederiksen H, Schmidt K. The incidence of idiopathic thrombocytopenic purpura in adults increases with age. Blood. 1999 Aug 1:94(3):909-13 [PubMed PMID: 10419881]
Schifferli A, Moulis G, Godeau B, Leblanc T, Aladjidi N, Michel M, Leverger G, Elalfy M, Grainger J, Chitlur M, Heiri A, Holzhauer S, Le Gavrian G, Imbach P, Kühne T. Adolescents and young adults with newly diagnosed primary immune thrombocytopenia. Haematologica. 2023 Oct 1:108(10):2783-2793. doi: 10.3324/haematol.2022.282524. Epub 2023 Oct 1 [PubMed PMID: 37051753]
Shaw J, Kilpatrick K, Eisen M, Tarantino M. The incidence and clinical burden of immune thrombocytopenia in pediatric patients in the United States. Platelets. 2020:31(3):307-314. doi: 10.1080/09537104.2019.1635687. Epub 2019 Jul 4 [PubMed PMID: 31271328]
Schoonen WM, Kucera G, Coalson J, Li L, Rutstein M, Mowat F, Fryzek J, Kaye JA. Epidemiology of immune thrombocytopenic purpura in the General Practice Research Database. British journal of haematology. 2009 Apr:145(2):235-44. doi: 10.1111/j.1365-2141.2009.07615.x. Epub 2009 Feb 24 [PubMed PMID: 19245432]
Terrell DR, Beebe LA, Neas BR, Vesely SK, Segal JB, George JN. Prevalence of primary immune thrombocytopenia in Oklahoma. American journal of hematology. 2012 Sep:87(9):848-52. doi: 10.1002/ajh.23262. Epub 2012 Jun 5 [PubMed PMID: 22674643]
Zufferey A, Kapur R, Semple JW. Pathogenesis and Therapeutic Mechanisms in Immune Thrombocytopenia (ITP). Journal of clinical medicine. 2017 Feb 9:6(2):. doi: 10.3390/jcm6020016. Epub 2017 Feb 9 [PubMed PMID: 28208757]
Cines DB, Blanchette VS. Immune thrombocytopenic purpura. The New England journal of medicine. 2002 Mar 28:346(13):995-1008 [PubMed PMID: 11919310]
Mahabir VK, Ross C, Popovic S, Sur ML, Bourgeois J, Lim W, George JN, Wang G, Cook RJ, Toltl LJ, Nazi I, Kelton JG, Arnold DM. A blinded study of bone marrow examinations in patients with primary immune thrombocytopenia. European journal of haematology. 2013 Feb:90(2):121-6. doi: 10.1111/ejh.12041. Epub 2012 Dec 13 [PubMed PMID: 23140198]
Kohli R, Chaturvedi S. Epidemiology and Clinical Manifestations of Immune Thrombocytopenia. Hamostaseologie. 2019 Aug:39(3):238-249. doi: 10.1055/s-0039-1683416. Epub 2019 Mar 13 [PubMed PMID: 30868551]
Kühne T, Buchanan GR, Zimmerman S, Michaels LA, Kohan R, Berchtold W, Imbach P, Intercontinental Childhood ITP Study Group, Intercontinental Childhood ITP Study Group. A prospective comparative study of 2540 infants and children with newly diagnosed idiopathic thrombocytopenic purpura (ITP) from the Intercontinental Childhood ITP Study Group. The Journal of pediatrics. 2003 Nov:143(5):605-8 [PubMed PMID: 14615730]
Level 2 (mid-level) evidenceO'Leary ST, Glanz JM, McClure DL, Akhtar A, Daley MF, Nakasato C, Baxter R, Davis RL, Izurieta HS, Lieu TA, Ball R. The risk of immune thrombocytopenic purpura after vaccination in children and adolescents. Pediatrics. 2012 Feb:129(2):248-55. doi: 10.1542/peds.2011-1111. Epub 2012 Jan 9 [PubMed PMID: 22232308]
Level 2 (mid-level) evidenceKühne T, Berchtold W, Michaels LA, Wu R, Donato H, Espina B, Tamary H, Rodeghiero F, Chitlur M, Rischewski J, Imbach P, Intercontinental Cooperative ITP Study Group. Newly diagnosed immune thrombocytopenia in children and adults: a comparative prospective observational registry of the Intercontinental Cooperative Immune Thrombocytopenia Study Group. Haematologica. 2011 Dec:96(12):1831-7. doi: 10.3324/haematol.2011.050799. Epub 2011 Aug 31 [PubMed PMID: 21880634]
Level 2 (mid-level) evidenceNeunert C, Noroozi N, Norman G, Buchanan GR, Goy J, Nazi I, Kelton JG, Arnold DM. Severe bleeding events in adults and children with primary immune thrombocytopenia: a systematic review. Journal of thrombosis and haemostasis : JTH. 2015 Mar:13(3):457-64. doi: 10.1111/jth.12813. Epub 2015 Jan 14 [PubMed PMID: 25495497]
Level 1 (high-level) evidence. Neunert C, Terrell DR, Arnold DM, et al. American Society of Hematology 2019 guidelines for immune thrombocytopenia. Blood Adv. 2019;3(23):3829-3866. Blood advances. 2020 Jan 28:4(2):252. doi: 10.1182/bloodadvances.2019001380. Epub [PubMed PMID: 31945156]
Level 3 (low-level) evidenceKaseb H, Tariq MA, Gupta G. Lymphoblastic Lymphoma. StatPearls. 2025 Jan:(): [PubMed PMID: 30725922]
Bandaru SS, Killeen RB, Gupta V. Poikilocytosis. StatPearls. 2025 Jan:(): [PubMed PMID: 32965812]
Alkhunein A, Albraikan A, Alayed M, Althaqafi W, Alharbi M. Case report: Microangiopathic hemolytic anemia and thrombocytopenia in a child with Brucella infection. Frontiers in pediatrics. 2023:11():1139622. doi: 10.3389/fped.2023.1139622. Epub 2023 Jun 15 [PubMed PMID: 37397138]
Level 3 (low-level) evidenceVrbensky JR, Moore JE, Arnold DM, Smith JW, Kelton JG, Nazy I. The sensitivity and specificity of platelet autoantibody testing in immune thrombocytopenia: a systematic review and meta-analysis of a diagnostic test. Journal of thrombosis and haemostasis : JTH. 2019 May:17(5):787-794. doi: 10.1111/jth.14419. Epub 2019 Mar 20 [PubMed PMID: 30801909]
Level 1 (high-level) evidencePamuk ON, Ali SM, Hasni S. Development of systemic lupus erythematosus in patients with immune thrombocytopenic purpura: A systematic meta-analysis. Autoimmunity reviews. 2023 Apr:22(4):103297. doi: 10.1016/j.autrev.2023.103297. Epub 2023 Feb 11 [PubMed PMID: 36781038]
Level 1 (high-level) evidenceJustiz Vaillant AA, Goyal A, Varacallo MA. Systemic Lupus Erythematosus. StatPearls. 2025 Jan:(): [PubMed PMID: 30571026]
Baumann MA, Menitove JE, Aster RH, Anderson T. Urgent treatment of idiopathic thrombocytopenic purpura with single-dose gammaglobulin infusion followed by platelet transfusion. Annals of internal medicine. 1986 Jun:104(6):808-9 [PubMed PMID: 2422997]
Neunert C, Terrell DR, Arnold DM, Buchanan G, Cines DB, Cooper N, Cuker A, Despotovic JM, George JN, Grace RF, Kühne T, Kuter DJ, Lim W, McCrae KR, Pruitt B, Shimanek H, Vesely SK. American Society of Hematology 2019 guidelines for immune thrombocytopenia. Blood advances. 2019 Dec 10:3(23):3829-3866. doi: 10.1182/bloodadvances.2019000966. Epub [PubMed PMID: 31794604]
Level 3 (low-level) evidenceTarantino MD, Bussel JB, Cines DB, McCrae KR, Gernsheimer T, Liebman HA, Wong WY, Kulkarni R, Grabowski E, McMillan R. A closer look at intravascular hemolysis (IVH) following intravenous anti-D for immune thrombocytopenic purpura (ITP). Blood. 2007 Jun 15:109(12):5527; author reply 5528 [PubMed PMID: 17554071]
Level 3 (low-level) evidenceGeorge JN, Terrell DR. Novel thrombopoietic agents: a new era for management of patients with thrombocytopenia. Haematologica. 2008 Oct:93(10):1445-9. doi: 10.3324/haematol.13725. Epub [PubMed PMID: 18827261]
Kuter DJ, Bussel JB, Newland A, Baker RI, Lyons RM, Wasser J, Viallard JF, Macik G, Rummel M, Nie K, Jun S. Long-term treatment with romiplostim in patients with chronic immune thrombocytopenia: safety and efficacy. British journal of haematology. 2013 May:161(3):411-23. doi: 10.1111/bjh.12260. Epub 2013 Feb 22 [PubMed PMID: 23432528]
Level 1 (high-level) evidenceTomiyama Y, Miyakawa Y, Okamoto S, Katsutani S, Kimura A, Okoshi Y, Ninomiya H, Kosugi H, Nomura S, Ozaki K, Ikeda Y, Hattori T, Katsura K, Kanakura Y. A lower starting dose of eltrombopag is efficacious in Japanese patients with previously treated chronic immune thrombocytopenia. Journal of thrombosis and haemostasis : JTH. 2012 May:10(5):799-806. doi: 10.1111/j.1538-7836.2012.04695.x. Epub [PubMed PMID: 22409309]
Level 1 (high-level) evidenceHanif N, Anwer F. Rituximab. StatPearls. 2025 Jan:(): [PubMed PMID: 33232044]
Carson KR, Evens AM, Richey EA, Habermann TM, Focosi D, Seymour JF, Laubach J, Bawn SD, Gordon LI, Winter JN, Furman RR, Vose JM, Zelenetz AD, Mamtani R, Raisch DW, Dorshimer GW, Rosen ST, Muro K, Gottardi-Littell NR, Talley RL, Sartor O, Green D, Major EO, Bennett CL. Progressive multifocal leukoencephalopathy after rituximab therapy in HIV-negative patients: a report of 57 cases from the Research on Adverse Drug Events and Reports project. Blood. 2009 May 14:113(20):4834-40. doi: 10.1182/blood-2008-10-186999. Epub 2009 Mar 5 [PubMed PMID: 19264918]
Level 3 (low-level) evidencePodolanczuk A, Lazarus AH, Crow AR, Grossbard E, Bussel JB. Of mice and men: an open-label pilot study for treatment of immune thrombocytopenic purpura by an inhibitor of Syk. Blood. 2009 Apr 2:113(14):3154-60. doi: 10.1182/blood-2008-07-166439. Epub 2008 Dec 18 [PubMed PMID: 19096013]
Level 3 (low-level) evidenceBussel J, Arnold DM, Grossbard E, Mayer J, Treliński J, Homenda W, Hellmann A, Windyga J, Sivcheva L, Khalafallah AA, Zaja F, Cooper N, Markovtsov V, Zayed H, Duliege AM. Fostamatinib for the treatment of adult persistent and chronic immune thrombocytopenia: Results of two phase 3, randomized, placebo-controlled trials. American journal of hematology. 2018 Jul:93(7):921-930. doi: 10.1002/ajh.25125. Epub 2018 May 15 [PubMed PMID: 29696684]
Level 1 (high-level) evidenceBroome C. Efgartigimod alfa for the treatment of primary immune thrombocytopenia. Therapeutic advances in hematology. 2023:14():20406207231172831. doi: 10.1177/20406207231172831. Epub 2023 May 10 [PubMed PMID: 37188068]
Level 3 (low-level) evidenceRobak T, Kaźmierczak M, Jarque I, Musteata V, Treliński J, Cooper N, Kiessling P, Massow U, Woltering F, Snipes R, Ke J, Langdon G, Bussel JB, Jolles S. Phase 2 multiple-dose study of an FcRn inhibitor, rozanolixizumab, in patients with primary immune thrombocytopenia. Blood advances. 2020 Sep 8:4(17):4136-4146. doi: 10.1182/bloodadvances.2020002003. Epub [PubMed PMID: 32886753]
Level 3 (low-level) evidenceBroome CM, Röth A, Kuter DJ, Scully M, Smith R, Wang J, Reuter C, Hobbs W, Daak A. Safety and efficacy of classical complement pathway inhibition with sutimlimab in chronic immune thrombocytopenia. Blood advances. 2023 Mar 28:7(6):987-996. doi: 10.1182/bloodadvances.2021006864. Epub [PubMed PMID: 35973190]
Level 3 (low-level) evidenceKuter DJ, Efraim M, Mayer J, Trněný M, McDonald V, Bird R, Regenbogen T, Garg M, Kaplan Z, Tzvetkov N, Choi PY, Jansen AJG, Kostal M, Baker R, Gumulec J, Lee EJ, Cunningham I, Goncalves I, Warner M, Boccia R, Gernsheimer T, Ghanima W, Bandman O, Burns R, Neale A, Thomas D, Arora P, Zheng B, Cooper N. Rilzabrutinib, an Oral BTK Inhibitor, in Immune Thrombocytopenia. The New England journal of medicine. 2022 Apr 14:386(15):1421-1431. doi: 10.1056/NEJMoa2110297. Epub [PubMed PMID: 35417637]
DiMaggio D, Anderson A, Bussel JB. Cytomegalovirus can make immune thrombocytopenic purpura refractory. British journal of haematology. 2009 Jun:146(1):104-12. doi: 10.1111/j.1365-2141.2009.07714.x. Epub 2009 May 9 [PubMed PMID: 19438507]
Level 3 (low-level) evidenceWang J, Cunningham-Rundles C. Treatment and outcome of autoimmune hematologic disease in common variable immunodeficiency (CVID). Journal of autoimmunity. 2005 Aug:25(1):57-62 [PubMed PMID: 15994061]
Level 2 (mid-level) evidenceHawkins J, Aster RH, Curtis BR. Post-Transfusion Purpura: Current Perspectives. Journal of blood medicine. 2019:10():405-415. doi: 10.2147/JBM.S189176. Epub 2019 Dec 9 [PubMed PMID: 31849555]
Level 3 (low-level) evidenceRafei H, Yunus R, Nassereddine S. Post-Transfusion Purpura: A Case Report of an Underdiagnosed Phenomenon. Cureus. 2017 May 1:9(5):e1207. doi: 10.7759/cureus.1207. Epub 2017 May 1 [PubMed PMID: 28580204]
Level 3 (low-level) evidenceBatton E, Leibel SL. Immune-Mediated Neonatal Thrombocytopenia. NeoReviews. 2022 Jul 1:23(7):e462-e471. doi: 10.1542/neo.23-7-e462. Epub [PubMed PMID: 35773506]
Torrez M, Chabot-Richards D, Babu D, Lockhart E, Foucar K. How I investigate acquired megaloblastic anemia. International journal of laboratory hematology. 2022 Apr:44(2):236-247. doi: 10.1111/ijlh.13789. Epub 2022 Jan 3 [PubMed PMID: 34981651]
Dotson JL, Lebowicz Y. Myelodysplastic Syndrome. StatPearls. 2025 Jan:(): [PubMed PMID: 30480932]
Yoshida H, Shimizu T, Yoshioka M, Matsushita A, Kawano Y, Ueda J, Kawashima M, Taniai N, Mamada Y. The Role of the Spleen in Portal Hypertension. Journal of Nippon Medical School = Nippon Ika Daigaku zasshi. 2023:90(1):20-25. doi: 10.1272/jnms.JNMS.2023_90-104. Epub [PubMed PMID: 36908126]
Imbach P, Kühne T, Müller D, Berchtold W, Zimmerman S, Elalfy M, Buchanan GR. Childhood ITP: 12 months follow-up data from the prospective registry I of the Intercontinental Childhood ITP Study Group (ICIS). Pediatric blood & cancer. 2006 Mar:46(3):351-6 [PubMed PMID: 16086422]
Bennett CM, Neunert C, Grace RF, Buchanan G, Imbach P, Vesely SK, Kuhne T. Predictors of remission in children with newly diagnosed immune thrombocytopenia: Data from the Intercontinental Cooperative ITP Study Group Registry II participants. Pediatric blood & cancer. 2018 Jan:65(1):. doi: 10.1002/pbc.26736. Epub 2017 Aug 9 [PubMed PMID: 28792679]
Neunert CE, Buchanan GR, Imbach P, Bolton-Maggs PH, Bennett CM, Neufeld E, Vesely SK, Adix L, Blanchette VS, Kühne T, Intercontinental Cooperative ITP Study Group Registry II Participants. Bleeding manifestations and management of children with persistent and chronic immune thrombocytopenia: data from the Intercontinental Cooperative ITP Study Group (ICIS). Blood. 2013 May 30:121(22):4457-62. doi: 10.1182/blood-2012-12-466375. Epub 2013 Apr 2 [PubMed PMID: 23550040]
Level 2 (mid-level) evidenceBritish Committee for Standards in Haematology General Haematology Task Force. Guidelines for the investigation and management of idiopathic thrombocytopenic purpura in adults, children and in pregnancy. British journal of haematology. 2003 Feb:120(4):574-96 [PubMed PMID: 12588344]
Level 1 (high-level) evidenceTreutiger I, Rajantie J, Zeller B, Henter JI, Elinder G, Rosthøj S, NOPHO ITP Study Group. Does treatment of newly diagnosed idiopathic thrombocytopenic purpura reduce morbidity? Archives of disease in childhood. 2007 Aug:92(8):704-7 [PubMed PMID: 17460024]
Neylon AJ, Saunders PW, Howard MR, Proctor SJ, Taylor PR, Northern Region Haematology Group. Clinically significant newly presenting autoimmune thrombocytopenic purpura in adults: a prospective study of a population-based cohort of 245 patients. British journal of haematology. 2003 Sep:122(6):966-74 [PubMed PMID: 12956768]
Portielje JE, Westendorp RG, Kluin-Nelemans HC, Brand A. Morbidity and mortality in adults with idiopathic thrombocytopenic purpura. Blood. 2001 May 1:97(9):2549-54 [PubMed PMID: 11313240]
Level 2 (mid-level) evidenceGhanima W, Godeau B, Cines DB, Bussel JB. How I treat immune thrombocytopenia: the choice between splenectomy or a medical therapy as a second-line treatment. Blood. 2012 Aug 2:120(5):960-9. doi: 10.1182/blood-2011-12-309153. Epub 2012 Jun 26 [PubMed PMID: 22740443]
Level 3 (low-level) evidence