Introduction
Immunoglobulin A (IgA) nephropathy, or IgAN, is one of the leading causes of glomerulonephritis and renal failure.[1][2] This disease is characterized by the deposition of IgA in the glomerular mesangium. Immune-mediated damage to the basement membrane results in hematuria, proteinuria, and renal insufficiency.[3] Berger and Hinglais were the first to describe the disease in 1968, hence it is also known as Berger disease.[4] Pathologically, a spectrum of glomerular lesions may be seen, yet the most frequently encountered change is mesangial proliferation with prominent IgA deposition.
Recent international collaborative efforts have significantly advanced our understanding of the immunopathogenesis of IgAN, leading to important discoveries. Furthermore, the establishment of multicenter networks has facilitated the holistic design and execution of clinical trials, offering crucial insights into immunotherapy for IgAN.
The clinical course typically progresses gradually, yet between 20% and 50% of affected patients develop end-stage renal disease (ESRD) within 20 years of diagnosis. Prevalence varies based on ethnicity, race, geography, and genetics, with Asian patients being more commonly and severely affected.[5][6][7]
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
IgAN results from abnormal immune reactions, resulting in IgA deposits within the glomerulus, increased podocyte permeability, and interstitial fibrosis.[8] Although IgAN is often preceded by an infectious disease, which triggers a dysregulated immune response, it is noteworthy that IgAN itself is not of an infectious etiology. No evidence suggests that specific infectious agents cause IgAN. Instead, the immune system appears to be activated by various clinical and subclinical triggers, along with genetic factors related to IgA glycosylation.
The potential etiologies underlying IgAN include the factors mentioned below.
Familial
- Fewer than 10% of cases are due to familial IgAN.
- At least 18 different gene segments associated with IgAN have been identified.
- Some of these genes include the C1GALT1 (a galactosyl transferase enzyme) and C1GALT1C1 (a molecular chaperone).[9]
Sporadic or Idiopathic IgA Nephropathy
- More than 90% of cases are sporadic.[10]
- No evidence suggests the involvement of a particular infectious agent despite the association between macroscopic hematuria and mucosal inflammation.
- No evidence indicates hypersensitivity to food antigens, except for a small group of patients with celiac disease.
- Abnormal O-glycosylation of the IgA1 hinge region promotes the formation of circulating IgA1-antibody immune complexes, leading to mesangial cell activation and deposition.[10]
Systemic Diseases Causing Secondary IgA Nephropathy
Many systemic conditions are associated with IgA deposits, causing IgAN pathology. Recent reports suggest the pathways leading to glomerular injury are similar between primary and secondary IgAN.[5] Secondary IgAN may arise from various conditions, including liver, gastrointestinal, autoimmune, dermatological, infectious, and drug-related causes.[5][8][11] Notably, it is essential to differentiate primary from secondary IgAN, as secondary forms typically do not require treatment with immunosuppressive agents; instead, the focus is on addressing the underlying cause.[12]
The significance of mucosal-associated lymphoid tissue (MALT) has recently gained prominence in understanding the pathology of various diseases, especially IgAN, as IgA production predominantly occurs in mucosal tissue. The primary role of MALT is to provide defense against environmental toxins and microbes. MALT tissue is predominantly located in the gut, particularly in Peyer's patches and lymphoid follicles, as well as in the tonsils, both of which are implicated in the pathogenesis of IgAN.[13]
Gastrointestinal disorders: Gastrointestinal disorders span a diverse range of conditions, each with distinct implications for IgAN.
- Liver disease: Liver disease is often associated with IgAN through various mechanisms, making it the most common cause of secondary IgAN.[5][12]
- Usually, the liver plays a crucial role in clearing large IgA complexes, but this clearance process is compromised in liver disease, which explains the IgAN association.[14]
- Bacterial translocation from leaky intestines, which is often increased in hepatic disease, can further exacerbate IgAN.
- Cirrohisis is commonly associated with IgAN, and case reports have shown improvement in renal parameters with steroid treatment.[15][16]
- Other rare liver disorders disorders such as hemochromatosis, Wilson disease, and autoimmune hepatitis have also been potentially linked to IgAN.[12][17][18]
- Celiac disease: Celiac disease is strongly correlated with IgAN and shares an autoimmune origin.
- Celiac disease is linked with HLA-DQ2/HLA-DQ8 haplotype.[19]
- The enzyme transglutaminase 2 can activate B cells, leading to the production of IgG and IgA autoantibodies.
- Transglutaminase 2 interacts with transferrin receptor-1, resulting in IgA1 deposition in the renal mesangium.
- For patients diagnosed with IgAN presenting gastrointestinal symptoms, it is reasonable to implement a trial of a gluten-free diet and initiate further diagnostic workup.
- Inflammatory bowel disease: Inflammatory bowel disease poses a risk factor for IgAN.[5]
- Inflammatory bowel disease has 2 forms—Crohn disease and ulcerative colitis, and IgAN is strongly associated with mortality in both conditions.
- A common pathogenesis for both diseases may involve B-cell immunoglobulin oversecretion and CD4+ T cell (helper T cell) dysregulation.[5]
- Abnormal intestinal microbiota: Abnormal intestinal microbiota, comprising commensal gut bacteria, are highly associated with IgAN.[13][20] Studies indicate that patients with IgAN exhibit a reduced diversity of the gut microbiome, notably in the Clostridium, Enterococcus, and Lactobacillus genera, compared to healthy controls.[13] Some researchers have proposed probiotics as a potential therapeutic measure.
Autoimmune disorders: Autoimmune disorders, including Sjögren syndrome, spondyloarthritis, systemic lupus erythematosus, and Behçet disease, have been associated with the development of IgA.[5][9]
Dermatological disorders: Dermatological disorders encompass a range of conditions, some of which are associated with IgAN.
- Psoriasis: Psoriasis is recognized as the predominant skin condition associated with IgAN. Additional dermatological conditions associated with IgAN include dystrophic epidermolysis bullosa, juvenile dermatomyositis, and leukocytic vasculitis.[5]
- Leukocytoclastic vasculitis: Leukocytoclastic vasculitis, a rare type III allergic reaction, leads to immune-complex mediated vasculitis in the dermal small blood vessels, resulting in erythematous macules and palpable purpura, which may progress to ulcers, blood blisters, or necrosis. This condition can manifest renal involvement and be associated with IgA deposits in the kidney, leading to secondary IgAN.[21] Please see StatPearls' companion resource, "Leukocytoclastic Vasculitis," for further information.
Infectious diseases: Clinical symptoms of IgAN, including gross hematuria, are highly associated with preceding viral or bacterial infections, most often an upper respiratory tract infection (URI). Please refer to the Pathophysiology section for further details. Infectious agents induce mucosal injury, resulting in dysregulated IgA production and deposition.
- The co-occurrence of a URI and macroscopic hematuria is so prevalent that it has earned its own designation—synpharyngitic hematuria. Additionally, tonsillar inflammation (attributed to commensal organisms, such as Hemophilus parainfluenza, Prevotella, Fusobacterium, Sphingomonas, and Treponema spp) is considered a contributing factor.[8][22]
- Viral infections caused by COVID-19, HIV, and hepatitis B are also strongly associated with IgAN.[5]
Drug-related IgAN: Drug-induced IgAN involves diagnosing renal injury timing in relation to medication administration and ruling out other potential causes. Tumor necrosis factor-α (TNF-α) inhibitors are notably associated with IgAN due to their immunomodulatory effects. Possible mechanisms include the reaction of IgG antiglycan antibodies against the glycan on TNF-α inhibitor heavy chains or the binding of hypoglycosylated IgA1 to antigens on TNF-α inhibitors. Examples of TNF-α inhibitors include adalimumab and infliximab.[5]
Other medications associated with secondary IgAN include interleukins 12 and 23 inhibitors, immune checkpoint inhibitors, direct oral anticoagulants, warfarin, and thioureylene derivatives used for Graves disease.[5]
Additional Causes
Although uncommon, primary pulmonary diseases, such as sarcoidosis, idiopathic pulmonary fibrosis, or pulmonary hemorrhage, can be associated with IgAN.[23][24] In addition, oncological processes, such as myeloproliferative disease, gastric carcinoma, lung cancers, renal cell carcinomas, and other malignancies, can also be associated with IgA.[25][26][27]
Epidemiology
Although IgAN is the most common cause of glomerulonephritis, its exact prevalence remains uncertain due to many cases being asymptomatic and requiring a renal biopsy for definitive diagnosis. Not all patients undergo a biopsy to confirm the diagnosis, with some opting for conservative management, particularly if they exhibit a benign variant. Approximately 10% of renal biopsies in the United States reveal IgAN, while the prevalence is higher in East Asia (up to 40% of biopsies) and Europe (20% to 30% of biopsies).[4][14][28] A systematic analysis of biopsy-based literature across multiple countries indicates an overall incidence of over 2.5 per 100,000.[14][29]
Routine screening of hematuria in East Asian schoolchildren is believed to contribute to the early detection of microscopic hematuria, thereby contributing to the increased diagnosis of IgAN. However, data from dialysis and biopsy registries indicate a greater burden of IgAN in East and Pacific Asian countries. These findings align with geospatial differences in the prevalence of genetic susceptibility loci observed in global genome-wide association studies.[30][31][32] In addition, evidence also suggests that ESRD is more common among East Asian patients diagnosed with IgAN than non-Asian patients.[33]
In the United States, IgAN appears to affect White populations more than Black individuals disproportionately.[28] The disease commonly manifests in children and young adults, with a peak incidence in the second and third decades of life. A male predominance ratio of about 2.5:1 is present in the United States and Europe, whereas the ratio is closer to 1:1 in East Asia.[9][34]
Pathophysiology
A multi-hit model has been proposed that suggests abnormal immune regulation at the core of IgAN pathogenesis, with susceptibility influenced by various genetic and environmental factors.[35] These "hits" are evident in the IgA moieties detected in biopsies and circulating in IgAN patients. Notably, immune complexes composed of galactose-deficient IgA1 are central in the circulation and glomeruli of individuals with IgAN.
Genetic Factors
The presence of abnormally glycosylated IgA1 is a heritable trait.[36] In a quarter of blood relatives of IgAN patients, galactose-deficient IgA1 levels are elevated, and segregation analysis reveals a likely major dominant gene inheritance with a polygenic background.[37]
Environmental Factors
Glomerular inflammation and mesangial proliferation are thought to occur due to nephritogenic immune complexes. Activation of the renin-angiotensin and complement systems further leads to glomerulosclerosis and tubulointerstitial fibrosis, contributing to impaired renal function. Additionally, risk factors such as smoking and hypertension exacerbate disease progression by inducing microvascular injury.[38] Glmoerulomegaly and maladaptive hyperfiltration injury attributed to obesity may also be implicated in the nonimmunological progression of the disease.[39]
A proposed 4-hit model highlights abnormal mucosal IgA regulation as the central mechanism:[5][10][35]
- Abnormal O-glycosylation of the IgA1 hinge region, leading to increased levels of galactose-deficient IgA1.
- Production of anti-glycan antibodies and IgA1 autoantibodies.
- Formation of IgA1 immune complexes circulating in the bloodstream and depositing in the kidney.
- Activation of mesangial cells, as well as the lectin and alternative complement systems, resulting in the release of cytokines, the deposition of extracellular proteins, and systemic inflammation and fibrosis.
The first "hit" involves a genetically susceptible host predisposed to developing a dysregulated immune response, primarily involving mucosal immunity. IgA comprises 15% of all immunoglobulins and is primarily present in the mucosa and bloodstream. IgA exists in 2 forms—IgA1 and IgA2—and each of these can be monomeric or polymeric.[6] In genetically susceptible patients, a precipitating factor, such as infection, can trigger B cells to produce the abnormal IgA, which is missing a galactose molecule at the hinge region.[7][40] Trivial mucosal infections, chronic exposure to pathogens, and abnormal handling of commensals in the gut have all been hypothesized to trigger the abnormal immune response. The resulting hypoglycosylation enables the aberrant IgA molecules to self-aggregate.[6][7]
Elevated galactose-deficient IgA alone is insufficient to cause IgAN; relatives of IgAN patients have demonstrated elevated levels of abnormally glycosylated IgA1 without developing the disease.[14] The second "hit" occurs when abnormal IgA1 triggers autoantibodies against IgA1 and anti-glycan IgGs.[7][41] Normally, IgA complexes are cleared by the liver, which is why liver disease is highly associated with IgAN.[8]
The third "hit" is the formation of abnormal IgA1 complexes, which circulate in the bloodstream and deposit in the mesangium. This leads to the final "hit" of mesangial cytokine release and complement activation. Cytokine release activates podocytes, causing glomerular permeability and tubular interstitial damage, which leads to interstitial fibrosis, tubular damage, and inflammatory cell infiltration.[6] The inflammatory cell infiltrate especially involves macrophages and dendritic cells.
Activation of complement plays a significant role in the pathogenesis of IgAN, with particular emphasis on the mannose-binding lectin pathway. Polymeric IgA1 can activate this pathway, and components of this lectin pathway, including complement factor H and properdin, are detected within glomerular deposits.[42][43][44] In addition, the alternative complement system is activated, and immune complexes containing C3 (usually in the same distribution as IgA1) are observed in approximately 90% of IgAN kidney biopsy samples on immunofluorescence staining.[7]
These 2 complement activation pathways, the lectin and alternative pathways, are pivotal in causing glomerular basement membrane damage. The damaged basement membrane results in the ultrafiltration of larger molecules and produces hematuria.[7] However, the pathophysiology underlying why some individuals develop asymptomatic hematuria while others progress to rapidly progressive glomerulonephritis, ultimately leading to renal failure, remains poorly understood.[45]
Histopathology
Microscopically, secondary and primary IgAN are indistinguishable unless manifestations of secondary disease are seen.[9] Histologically, IgAN is characterized by the following characteristics (see Image. Histopathological Characteristics of IgA Nephropathy):
- Diffuse proliferation of mesangial cells and matrix.
- Hypercellular or normal glomeruli with diffuse necrotizing crescentic glomerulonephritis.
- Mesangial involvement resembling focal and segmental glomerulosclerosis.
- Immunofluorescence reveals a diffuse granular pattern of IgA deposits in the mesangium.
Light Microscopy
Common findings on light microscopy include focal or diffuse mesangial proliferation and expansion of the extracellular matrix.[46] Morphology reveals intracapillary and extracapillary proliferative lesions. Focal glomerular sclerosis, sometimes indistinguishable from focal segmental glomerulosclerosis, may occur. Interstitial fibrosis, vascular sclerosis, and tubular atrophy are evident in advanced stages. Segmental necrotizing areas with crescent formation may appear in some patients due to extensive capillary disruption.
Electron Microscopy
Electron microscopy reveals mesangial hypercellularity and excess mesangial matrix. An important finding is the presence of mesangial electron-dense deposits of IgA. However, subepithelial and subendothelial deposits of the glomerular capillary wall are seen in a minority of patients, particularly those with the more severe form of the disease.[47]
Immunofluorescence
Immunofluorescence examination reveals mesangial IgA deposits in a diffuse granular pattern, primarily consisting of polymeric IgA of the IgA1 subclass. Additionally, IgG is detected in 43% of patients and IgM in 54%.[35] C3 is commonly present and typically co-deposits with IgA deposits; its deposition correlates with disease severity and is absent in asymptomatic deposits.[9] Other components of the lectin and alternative complement pathways are also frequently found. C1q staining is usually absent, indicating a lack of activation of the classical complement pathway.
C4d, a marker of the lectin complement pathways, imparts a worse prognosis.[7][48] Another potentially quantifiable assessment is the maximal glomerular diameter, which is significantly correlated with loss of glomerular filtration rate (GFR) and proteinuria.[41][49]
Proteinuria
Nephrotic-range proteinuria is more common in children than in adults. In pediatric patients, renal biopsy may show widespread podocyte foot process effacement along with IgA mesangial deposits representing co-occurrence of minimal change disease and IgAN. The mesangial deposits may remain, although foot process effacement and proteinuria often resolve with steroids.[9]
History and Physical
In most patients with IgAN, history and physical examination are typically unremarkable. The primary complaint often revolves around gross hematuria, which can manifest as brown, red, or "coca-cola colored" urine. Acute renal failure may lead to ankle edema, facial puffiness, and hypertension. Other potential symptoms include frothy urine or the presence of a rash. Patients may report a recent history of URIs, such as pharyngitis, preceding the onset of hematuria.[50] Additionally, a history of hematuria associated with intense exercise may be present.[9] Reviewing prior urinalysis reports can provide valuable insights when available.
During a physical examination, it is essential to assess blood pressure and observe for indications of diminished renal function, including edema, ascites, and basal lung crepitations. Hypertension is a common manifestation in IgAN. In addition, IgAN may coexist with cirrhosis, liver diseases, and celiac disease. Therefore, conducting a comprehensive general and abdominal examination is crucial for excluding these clinically significant comorbidities. The range of clinical features of IgAN is broad, ranging from asymptomatic hematuria to rapidly progressive glomerulonephritis. The mode of presentation varies based on age group and histological biopsy patterns. Among the most prevalent clinical phenotypes are asymptomatic hematuria and progressive kidney disease.
Asymptomatic hematuria with mild proteinuria, typically around 0.5 g/d, may be detected during routine screening. A subset of individuals presenting with isolated microscopic hematuria and mild proteinuria may eventually progress to develop substantial proteinuria and hypertension, underscoring the importance of long-term monitoring.[51] Progressive chronic kidney disease (CKD) is frequently observed in various cohorts. Renal survival rates exhibit significant variability depending on factors such as biopsy timing and the presence of lead-time bias. Estimated renal survival at the 10-year mark is thought to be between 57% and 91%.[52]
Synpharyngitic macroscopic hematuria is often the initial presentation of IgAN, with patients seeking medical attention due to the simultaneous occurrence of gross hematuria and pharyngitis or another infection. Recurrent episodes of macroscopic hematuria are also common. While nephrotic-range proteinuria can occur in IgAN, the simultaneous presence of the 2 conditions is rare.[53][54]
Extrarenal manifestations of IgAN are not uncommon or unsurprising, given the systemic nature of the disease. Studies have shown that up to 35% of patients experience gastrointestinal symptoms, including abdominal pain, cramping, and diarrhea.[8]
Less than 5% of patients with IgAN present with acute kidney injury, typically resulting from either the rapid development of crescents or tubular damage or obstruction due to gross hematuria and RBC casts. Although aggressive immunosuppressive treatment is often necessary in crescent formation, damage from large amounts of RBCs is usually reversible with supportive treatment.[9]
Evaluation
The evaluation begins with establishing the diagnosis. The initial investigation involves urine analysis to detect microscopic hematuria. The presence of red cells and red cell casts indicates glomerular injury and potential active disease. Proteinuria is assessed using the protein-to-creatinine ratio in urine or 24-hour urinary protein excretion. Serum creatinine and estimated GFR are measured to quantify renal function.
Confirmation of the diagnosis relies on renal biopsy, which allows for detailed examination of renal histology using light microscopy, electron microscopy, and immunofluorescence. Immunofluorescence, specifically demonstrating the deposit of IgA in the glomerular basement membrane, is the gold standard for diagnosis. Additionally, IgAN should be classified using the Oxford classification to predict prognosis. This Oxford classification system integrates histological, clinical, and biomarker criteria to provide valuable prognostic insights.
The Oxford classification relies on the following criteria:
- Mesangial cellularity
- Endocapillary proliferation
- Segmental glomerulosclerosis
- Tubular atrophy or interstitial fibrosis [55][56]
Various studies have validated these criteria, with tubular atrophy or interstitial fibrosis being the most predictive of poor prognosis.[14][57][58][59] Fibroblast markers, such as FSP1 and URG11, are inversely correlated with GFR and prognosis. Notably, the inclusion of a criterion to assess the presence of crescents remains controversial. Current KDIGO criteria do not support using this as a prognostic indicator; instead, the presence of crescents adds to the severity of the overall clinical presentation.[48][60]
Other possible markers of IgAN include urinary markers assessing components of the complement pathway, such as urinary C4d, serum C3 levels, and other serum markers of alternative and lectin complement pathways.[7] Serum IgA levels lack sensitivity and specificity for IgAN diagnosis. Although galactose-deficient IgA levels are elevated in patients with IgAN, notably, these levels can also be elevated in relatives of IgAN patients without clinical disease. A study suggests that these markers exhibit a sensitivity of 77% and a specificity of 94%.[14][61][62] In addition, IgA and IgG autoantibodies against abnormally glycosylated IgA1 are elevated in patients with IgAN, although the measurement of these levels is not often available outside the research setting.[14][61]
Treatment / Management
The management of IgAN begins with confirming the diagnosis, typically through a renal biopsy, while ruling out secondary causes. Key considerations for formulating the management plan include assessing proteinuria, eGFR, blood pressure, and histological findings. The primary goals of treatment are to induce remission and avert the onset of complications.[63]
First-line agents for managing proteinuria and lowering blood pressure in IgAN patients typically involve angiotensin-converting enzyme (ACE) inhibitors or angiotensin receptor blockers (ARBs). The target blood pressure is typically set at 130/80 mm Hg.[64] If proteinuria is more than 1 g/d, the systolic blood pressure target is less than 125 mm Hg.
Conservative Therapy
Conservative therapy plays a crucial role in managing IgAN by reducing proteinuria and slowing the rate of renal function decline in IgAN, which cannot be overemphasized. In the Supportive Versus Immunosuppressive Therapy of Progressive IgA Nephropathy (STOP-IgAN) study, around one-third of patients achieved satisfactory outcomes without requiring immunosuppressive therapy following a 6-month trial of optimizing conservative measures, including aggressive ACE inhibitors or ARB titration (optimal medical management).[65] In this study, the comparison between immunosuppressive therapy and optimal medical management alone revealed no significant improvement in primary outcomes—death, ESRD, or a decline in GFR exceeding 40%—among those receiving immunosuppressive treatment during the 3-year and 10-year follow-up periods.[66][67][68]
The role of renin-angiotensin system blockade in IgAN is firmly established. However, caution is warranted against dual renin-angiotensin system blockade due to the risk of hyperkalemia.[69] In addition, considering the association between smoking and IgAN progression, advising patients to quit smoking is essential.[38][66][67] Salt intake, lipid management, weight loss, and smoking cessation are also advised. Smoking has been shown to independently increase the risk of IgAN progression, and this effect worsens with GFR decline.[70](A1)
Corticosteroids
Corticosteroids are crucial in reducing proteinuria in IgAN, particularly when levels exceed 1g/d. Steroid administration has especially been shown to decrease proteinuria. Typically, a tapering course of prednisone is prescribed for 2 to 4 months.[71][72][73](A1)
The TESTING trial, which focused on patients with IgAN and proteinuria exceeding 1 g/d, demonstrated that treatment with methylprednisolone for 6 to 9 months significantly slowed disease progression compared to placebo. However, due to an excess of infections and adverse events, the initial dose of methylprednisolone had to be reduced, and cotrimoxazole prophylaxis for opportunistic infections was added.[74][75] Nonetheless, longer-term data suggests that these effects may not be long-lasting.[68][65] (A1)
Novel Agents
In recent years, the approval of several novel medications holds promise for significantly improving outcomes in IgAN.
Sparsentan: Sparsentan is a non-immunosuppressive, novel dual endothelin and angiotensin receptor antagonist, which received accelerated approval from the US Food and Drug Administration (FDA) for the treatment of IgAN in February 2023.[76] The PROTECT trial was a multicenter randomized controlled trial that compared the effects of irbesartan (300 mg) with sparsentan (400 mg) on proteinuria in patients with persistent proteinuria despite maximal ACE inhibitor or ARB therapy. Results showed that the sparsentan group exhibited significantly less proteinuria and a trend toward preserved GFR compared to the irbesartan group.[77][78](A1)
Budesonide: Budesonide, in the form of oral nefecon, is a novel therapy for IgAN. This formulation consists of budesonide packaged in a pH-sensitive capsule, which preferentially releases the active drug at the terminal ileum near Peyer patches—the site of significant IgA-producing lymphoid tissue. This targeted delivery system enables extensive first-pass metabolism, minimizing systemic effects.[68][79] Results from the NEFIGAN and NeflgArd trials have shown significant reductions in proteinuria and preservation of GFR with the use of nefecon. Ongoing data analysis from these trials continues to provide insights into its efficacy.[80][81](A1)
Sodium-glucose cotransporter-2 (SGLT2) inhibitors: SGLT inhibitors demonstrated significant efficacy in slowing the progression of CKD. The DAPA-CKD and EMPA-KIDNEY trials were terminated early due to the evident benefits of SGLT2 inhibitors. Although these trials primarily included patients with all-cause CKD, a significant number of patients from both trials were diagnosed with IgAN. Further studies are needed to evaluate the effects of SGLT2 inhibitors on patients with IgAN.[68][82]
Other Immunosuppressive Agents
Mycophenolate mofetil (MMF): MMF has been studied for its potential to prevent IgAN progression, with studies being equivocal and findings varying across studies, particularly those conducted in Europe and North America. The majority of studies are limited by small sample size.[83] However, some trials conducted in Asia have shown improvement and promising outcomes in terms of reducing proteinuria and preserving GFR with MMF.[84][85] Notably, the MAIN trial, a recent randomized controlled trial in China, showcased significant reductions in proteinuria and preservation of GFR with MMF. This underscores the potential differences in disease progression among East Asian variants of IgAN compared to other regions globally.[84][68](A1)
Rituximab: This drug demonstrated efficacy in various glomerular diseases but has shown less promising results in early studies involving IgAN. A pilot study comparing rituximab to conservative management in patients with proteinuria showed no beneficial effects on proteinuria or renal function.[86](A1)
Iptacopan: Iptacopan binds Factor B in the alternative complement pathway and prevents the formation of C3 convertase (C3bBb). APPLAUSE-IgAN is a phase III study evaluating ipatacopan compared to placebo in patients with biopsy-proven IgAN with a high risk of progression, and preliminary results show a reduction of proteinuria in the iptacopan arm. Upon completion at 24 months, the slope of eGFR decline will also be evaluated.[87][88](A1)
Other medications: Additional medications, such as cyclophosphamide, azathioprine, calcineurin inhibitors, and leflunomide, have been investigated for their potential efficacy in IgAN. These drugs primarily exert their effects on T- and B cells. Although results vary, currently, clear evidence demonstrating a significant benefit from these agents does not exist.[73][89][90][91][92](A1)
Combination Therapy
Combination therapy involving corticosteroids along with another agent is typically considered for cases of progressive IgAN. A randomized prospective trial focusing on high-risk IgAN patients showed improved renal survival in those receiving prednisone along with cyclophosphamide or azathioprine compared to those without immunotherapy.[93] However, results reported in another multicenter trial found no significant difference between patients treated with corticosteroids alone and those receiving combination therapy.[94](A1)
Tonsillectomy
The tonsils are a significant store for MALT, and IgA levels are known to decrease post-tonsillectomy. In addition, a correlation exists between tonsillar dendritic cell concentration and crescent formation in IgAN patients. Multiple retrospective studies have demonstrated the benefits of tonsillectomy in inducing remission or slowing IgAN progression, particularly in Asian populations, notably Japan.[95][96][97][98] Tonsillectomy in conjunction with pulse steroid therapy has also been more recently studied and found to be effective in progressive IgAN.[99][100][101] Timing of a tonsillectomy with pulse steroids immediately before and after may provide augmented results.[102](B3)
However, the efficacy of tonsillectomy in IgAN management has not been consistently demonstrated in randomized controlled trials, leading to its exclusion from standard care guidelines in many regions, particularly outside of Asia.[50][103] Additionally, variations of IgAN observed in Asian populations may respond differently to tonsillectomy compared to non-Asian forms, possibly due to differences in incidence rates and disease aggressiveness.
Renal Transplant
For individuals who progress to ESRD from IgAN, renal transplantation is a viable option. However, the risk of IgAN recurrence in the transplanted kidney persists. A significant study reported a recurrence rate of 23% in transplanted kidneys at 15 years, correlating with a 3.7-fold increase in graft rejection.[104] Despite the recurrence of IgA deposits in the transplanted kidney, progression to recurrent ESRD with the grafted kidney is rare. Utilizing anti-thymocyte globulin and maintaining immunosuppression with corticosteroids can mitigate the recurrence of IgAN.[9] In addition, treatment with ACE inhibitors or ARBs may help delay the progression of recurrent disease in allografts.[105] (B2)
Future Horizons
Recent advancements in understanding the pathways involved in IgAN have spurred the development and evaluation of numerous potential therapeutic agents (see Image. New Drugs Under Evaluation for Treatment of IgA Nephropathy and Image. New Prospective Therapy Targeting the Gut Mucosal Immune System). These medications, currently in the pipeline, span various categories targeting different steps of the disease processes, as mentioned below.
- Endothelin receptor agonists: These are believed to modulate the hemodynamic aspect of IgAN. Among these, Sparsentan is a prominent example, albeit also functioning as an ARB. Moreover, several other drugs within this class are undergoing investigation for their efficacy.
- B-cell inhibitors: B-cell inhibitors are gaining attention for their potential in tackling IgAN, as B cells are responsible for producing deficient IgA1 molecules. Interventions targeting B cells and their functions are currently under investigation, particularly focusing on key regulators such as B-cell activating factor (BAFF) and APRIL (a proliferation-inducing ligand), which influence B-cell modulation and class switching to IgA production. Trials exploring therapies aimed at these factors are currently being investigated.[64][106][107][64]
- Complement activation inhibitors: As previously mentioned, the alternative and lectin complement pathways play a significant role in IgAN deposits, prompting interventions that often target the common pathway, typically at C3, C3a, and C5. Case reports have indicated improvements in patients with IgAN with treatments such as eculizumab, which inhibits C5 cleavage and is utilized in conditions like hemolytic uremic syndrome.[108][109]
- Other investigational agents: Several investigational agents are under scrutiny for their potential in IgAN management, including bortezomib (a proteasome inhibitor), fostamatinib (a tyrosine kinase inhibitor), and nuclear factor erythroid–derived 2-related factor agonists. Additionally, small-interfering RNAs are among the array of novel therapies being explored.[4][9][73] (A1)
Differential Diagnosis
The 2 conditions closely mimicking the symptoms of IgAN are discussed in detail below.
IgA Vasculitis
IgA vasculitis, also known as Henoch-Schönlein purpura, is associated with dysregulated IgA deposits, although these deposits primarily affect the small blood vessels of affected organs. Unlike IgAN, which predominantly affects individuals aged 15 and older, IgA vasculitis primarily affects those aged 15 and younger. Microscopically, patients with IgA vasculitis exhibit more capillary and glomerular injury, while IgAN histology is characterized by mesangial proliferation. Some treatments for both conditions overlap due to their autoimmune involvement. Please see StatPearls' companion resource, "IgA Vasculitis (Henoch-Schönlein Purpura," for further information.
Poststreptococcal Glomerulonephritis
Poststreptococcal glomerulonephritis can also manifest in the context of a URI. A key difference is that while IgAN typically presents concurrently with or shortly after a URI, poststreptococcal glomerulonephritis usually arises 2 to 3 weeks following a URI. Please see StatPearls' companion resource, "Poststreptococcal Glomerulonephritis," for further information.
The following list comprises other potential differentials for IgAN:
- Lupus nephritis (associated with multisystemic presentation)
- Membranoproliferative glomerulonephritis (distinguishable on renal biopsy)
- Thin basement membrane nephropathy (often characterized by microscopic hematuria and a family history of benign microscopic hematuria)
- Alport syndrome (associated with ocular and otic symptoms)
- Malignancies ranging from the kidneys to the urethra (should be ruled out in all patients aged 40 or older)
- Urolithiasis (typically manifests with colicky pain not associated with IgAN; imaging will demonstrate obstruction)
Prognosis
Up to 50% of IgAN patients experience a benign course.[55] Prognosis can be somewhat predictable, particularly based on the Oxford classification outlined in the Pathology section. Any of the MEST criteria (and crescents) serve as poor prognostic indicators. Isolated microscopic hematuria with mild proteinuria suggests a favorable prognosis, indicating a benign and asymptomatic disease course. Additionally, individuals of East Asian origin are significantly associated with an elevated risk of ESRD (see Table below).[33]
Three risk factors highly associated with dialysis or death include:[34]
- Proteinuria of 1 g/d or more
- Sustained hypertension
- Oxford classification MEST-C score (to which some algorithms also include a category for crescents).
Patients with sustained proteinuria of 1 g/d or higher have a 46-fold increased risk of ESRD than those with less than 500 mg/d.[73] About half of the patients with IgAN may obtain complete remission with treatment, whereas the remaining half may show some degree of ongoing disease.[73]
Table. MEST-C Score
| Pathological Characteristics | Explanations | Scoring Criteria |
|
Mesangial hypercellularity (M) |
≥4 Mesangial cells in any mesangial area of a glomerulus |
M0: Mesangial hypercellularity in ≤50% of glomeruli M1: Mesangial hypercellularity in >50% of glomeruli |
|
Endocapillary hypercellularity (E) |
An increased number of cells in the glomerular capillary lumen |
E0: Absence E1: Presence of any glomeruli showing endocapillary hypercellularity |
|
Segmental glomerulosclerosis (S) |
Adhesion or sclerosis that does not involve the entire glomerulus |
S0: Absence S1: Presence of any segmental glomerulosclerosis (podocyte hypertrophy/tip lesions should be noted if present) |
|
Tubular atrophy/interstitial fibrosis (T) |
Percentage of tubular atrophy/interstitial fibrosis of cortical area |
T0: 0-25% T1: 26%-50% T2: >50% |
|
Cellular/fibrocellular crescents (C) |
Extracapillary cell proliferation >2 cell layers thick and <50% matrix |
C0: Absence C1: Presence of cellular/fibrocellular crescents in <25% of glomeruli C2: Presence of cellular/fibrocellular crescents in ≥25% of glomeruli |
A minimum of 8 viable glomeruli is necessary to calculate the MEST-C score accurately. Based on Markowitz G, 2017 (Nat Rev Nephrol. https://doi.org/10.1038/nrneph.2017.67) and Pattrapornpisut P, et al (IgA Nephropathy: Core Curriculum 2021. https://doi.org/10.1053/j.ajkd.2021.01.024).
Complications
Although only a small percentage of patients diagnosed with IgAN progress to ESRD, due to the high prevalence, its high prevalence makes it a frequent cause of ESRD.[54] As the disease progresses, complications of renal failure, such as hypertension, edema, anemia, heart failure, and pulmonary edema, may arise. Common adverse effects and complications of both steroid and steroid-sparing therapy include increased risk of infections, hypertension, fluid retention, weight gain, diabetes mellitus, osteoporosis, and iatrogenic Cushing syndrome.[72] In addition, complications of steroid-sparing agents may include immunosuppression, anaphylaxis, renal injury, and hepatotoxicity.
Postoperative and Rehabilitation Care
A low-antigen diet, which includes restrictions on gluten, meat, and dairy products, is often recommended for individuals with IgAN. Several studies suggest that low-protein diets may slow the deterioration of renal function.[110][111]
Deterrence and Patient Education
Patient education and deterrence strategies are crucial in managing IgAN, especially considering its susceptibility to minor URIs in genetically predisposed individuals.
- A minor URI in IgAN can trigger the disease in genetically susceptible patients.[8]
- Individuals with other autoimmune diseases and a strong family history of IgAN are more likely to have the disease.
- Screening for hematuria is crucial in early detection of the disease, enabling the identification of patients in the asymptomatic stage of the disease progression.
- Patients should receive comprehensive education covering the nature of the disease, the potential progression to ESRD necessitating renal transplantation, the adverse effects associated with steroid therapy, and the critical importance of regular follow-up care.
- The importance of adhering to ACE inhibitors or ARBs and any other prescribed therapies should be reinforced.
- Dietary recommendations include restrictions on salt, protein, and saturated fat intake.
Pearls and Other Issues
A proposed 4-hit model highlights abnormal mucosal IgA regulation as the central mechanism:
- Abnormal O-glycosylation of the IgA1 hinge region, leading to increased levels of galactose-deficient IgA1.
- Production of anti-glycan antibodies and IgA1 autoantibodies.
- Formation of IgA1 immune complexes circulating in the bloodstream and depositing in the kidney.
- Activation of mesangial cells, as well as the lectin and alternative complement systems, resulting in the release of cytokines, the deposition of extracellular proteins, and systemic inflammation and fibrosis.
Other significant issues essential for navigating the diagnosis, treatment, and prognosis of IgAN are mentioned below.
- Distinguishing primary IgAN from secondary IgAN is crucial, as addressing the underlying disease is necessary for managing secondary cases.
- The lectin and alternative complement pathways are highly involved in IgAN pathology.
- IgAN exhibits a higher prevalence and appears to have a more aggressive course among individuals of East Asian origin.
- Asymptomatic microscopic hematuria often indicates a favorable prognosis, while rapidly progressing disease with crescents observed on biopsy suggests a poorer prognosis.
- The Oxford MEST classification system is utilized to stratify the risk of IgAN patients, with some guidelines considering the presence of crescents while others do not.
- When gross hematuria occurs in individuals aged 40 or older, it warrants a comprehensive evaluation to rule out potential underlying kidney or bladder neoplasms.[9]
- Differentiating IgAN from IgA vasculitis and poststreptococcal glomerulonephritis is essential due to variations in treatment approaches for each condition.
- Sparsentan and nefecon are the 2 recently FDA-approved treatments for IgAN.
- Ongoing clinical trials aim to assess therapeutic agents targeting various steps of the IgAN pathways.
Enhancing Healthcare Team Outcomes
The clinical trajectory of IgAN typically spans a protracted course during which an interprofessional healthcare team collaboratively manages patient care. Initial detection often occurs through abnormal urinalysis findings during routine screening by primary care providers. Expedient referral to a nephrologist (or pediatric nephrologist) for comprehensive evaluation is imperative. Coexisting autoimmune diseases may necessitate input from rheumatologists, while systemic manifestations may require the expertise of endocrinologists, gastroenterologists, or other specialists. Moreover, patients should be provided with a "steroid card" for reference.
Pharmacists are critical in educating patients about steroid therapy and facilitating compliance while also maintaining open communication with the clinical team as needed. Laboratory staff must ensure accurate sample collection to prevent false positives for proteinuria. A dietician should design a tailored diet comprising low-salt, low-protein, and low-fat components to support the patient's nutritional needs. Nurses are integral members of the healthcare team, providing comprehensive patient assessment, counseling, and coordination among various specialties involved in the case. In addition, all interprofessional healthcare team members must maintain accurate patient records and promptly communicate any concerns regarding the patient's progress with the rest of the team to ensure optimal care.
Fluid intake should be carefully managed, considering the patient's fluid balance and renal function. In cases where progression to ESRD is anticipated, patients should receive early and comprehensive education about renal replacement therapy options. Social workers are crucial in supporting and guiding patients and their families during this challenging phase. Due to the complexity of IgAN, optimal outcomes are best achieved through the collaborative efforts of an interprofessional healthcare team comprising clinicians, pharmacists, nurses, and allied health professionals dedicated to educating and supporting both the patient and their family.
Media
(Click Image to Enlarge)
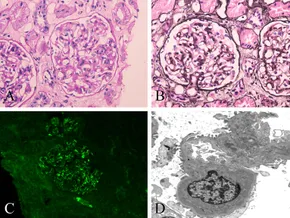
Histopathological Characteristics of IgA Nephropathy. Light microscopy shows a glomerulus with mild-to-moderate mesangial hypercellularity and endocapillary hypercellularity (A and B). Periodic acid-Schiff staining; original magnification ×400 (A). Periodic acid-silver methenamine staining; original magnification ×400 (B). Immunofluorescence image reveals IgA deposits along the glomerular mesangial area; original magnification ×400 (C). Electron microscopy image shows deposits along the mesangium and matrix; original magnification ×5000 (D).
Zhang S, Chen YL, Liu CL, Xie JY, Sun BD, Liu DZ. Case report: IgA nephropathy in a patient with antitranscription intermediary factor-1γ antibody-positive dermatomyositis. Front Immunol. 2022;13:757802. doi: 10.3389/fimmu.2022.757802.
(Click Image to Enlarge)
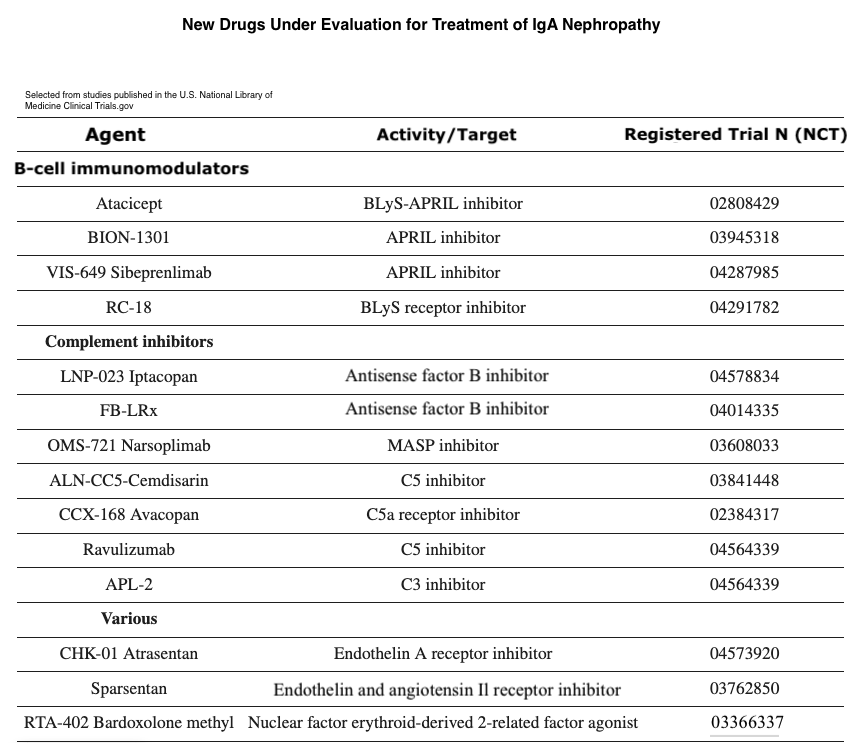
New Drugs Under Evaluation for Treatment of IgA Nephropathy. New drugs are being evaluated for the treatment of IgA nephropathy. These drugs target persistent proteinuria despite months of optimized supportive care. However, it is believed that some of these drugs could be beneficial in managing the most active and progressive cases, including those exhibiting a high presence of crescents.
Trimarchi H, Haas M, Coppo R. Crescents and IgA nephropathy: a delicate marriage. J Clin Med. 2022;11(13):3569. doi: 10.3390/jcm11133569.
(Click Image to Enlarge)
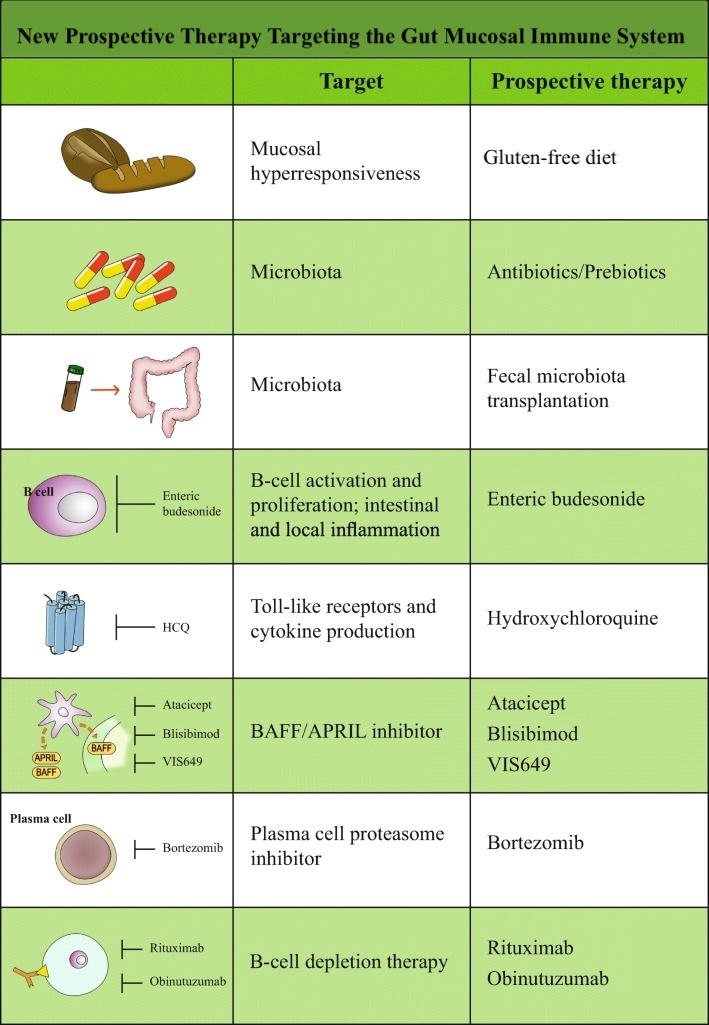
New Prospective Therapy Targeting the Gut Mucosal Immune System. A new prospective therapy targeting the gut mucosal immune system is being developed for IgA nephropathy. Some of these treatments are currently under investigation and available only in research settings.
Gesualdo L, Di Leo V, Coppo R. The mucosal immune system and IgA nephropathy. Semin Immunopathol. 2021;43(5):657-668. doi: 10.1007/s00281-021-00871-y.
References
Pattrapornpisut P, Avila-Casado C, Reich HN. IgA Nephropathy: Core Curriculum 2021. American journal of kidney diseases : the official journal of the National Kidney Foundation. 2021 Sep:78(3):429-441. doi: 10.1053/j.ajkd.2021.01.024. Epub 2021 Jul 9 [PubMed PMID: 34247883]
Working Group of the International IgA Nephropathy Network and the Renal Pathology Society, Coppo R, Troyanov S, Camilla R, Hogg RJ, Cattran DC, Cook HT, Feehally J, Roberts IS, Amore A, Alpers CE, Barratt J, Berthoux F, Bonsib S, Bruijn JA, D'Agati V, D'Amico G, Emancipator SN, Emma F, Ferrario F, Fervenza FC, Florquin S, Fogo AB, Geddes CC, Groene HJ, Haas M, Herzenberg AM, Hill PA, Hsu SI, Jennette JC, Joh K, Julian BA, Kawamura T, Lai FM, Li LS, Li PK, Liu ZH, Mezzano S, Schena FP, Tomino Y, Walker PD, Wang H, Weening JJ, Yoshikawa N, Zhang H. The Oxford IgA nephropathy clinicopathological classification is valid for children as well as adults. Kidney international. 2010 May:77(10):921-7. doi: 10.1038/ki.2010.43. Epub 2010 Mar 3 [PubMed PMID: 20200498]
Shen PC, He LQ, Tang Y, Wang Q, Wang W, Li J. Clinicopathological characteristics and prognostic factors of asymptomatic IgA nephropathy. Journal of investigative medicine : the official publication of the American Federation for Clinical Research. 2010 Mar:58(3):560-5. doi: 10.231/JIM.0b013e3181d20aa1. Epub [PubMed PMID: 20215916]
Rodrigues JC, Haas M, Reich HN. IgA Nephropathy. Clinical journal of the American Society of Nephrology : CJASN. 2017 Apr 3:12(4):677-686. doi: 10.2215/CJN.07420716. Epub 2017 Feb 3 [PubMed PMID: 28159829]
Tota M, Baron V, Musial K, Derrough B, Konieczny A, Krajewska M, Turkmen K, Kusztal M. Secondary IgA Nephropathy and IgA-Associated Nephropathy: A Systematic Review of Case Reports. Journal of clinical medicine. 2023 Apr 6:12(7):. doi: 10.3390/jcm12072726. Epub 2023 Apr 6 [PubMed PMID: 37048809]
Level 1 (high-level) evidenceChang S, Li XK. The Role of Immune Modulation in Pathogenesis of IgA Nephropathy. Frontiers in medicine. 2020:7():92. doi: 10.3389/fmed.2020.00092. Epub 2020 Mar 24 [PubMed PMID: 32266276]
Maillard N, Wyatt RJ, Julian BA, Kiryluk K, Gharavi A, Fremeaux-Bacchi V, Novak J. Current Understanding of the Role of Complement in IgA Nephropathy. Journal of the American Society of Nephrology : JASN. 2015 Jul:26(7):1503-12. doi: 10.1681/ASN.2014101000. Epub 2015 Feb 18 [PubMed PMID: 25694468]
Level 3 (low-level) evidenceRollino C, Vischini G, Coppo R. IgA nephropathy and infections. Journal of nephrology. 2016 Aug:29(4):463-8. doi: 10.1007/s40620-016-0265-x. Epub 2016 Jan 22 [PubMed PMID: 26800970]
Rajasekaran A, Julian BA, Rizk DV. IgA Nephropathy: An Interesting Autoimmune Kidney Disease. The American journal of the medical sciences. 2021 Feb:361(2):176-194. doi: 10.1016/j.amjms.2020.10.003. Epub 2020 Oct 8 [PubMed PMID: 33309134]
Lai KN, Tang SC, Schena FP, Novak J, Tomino Y, Fogo AB, Glassock RJ. IgA nephropathy. Nature reviews. Disease primers. 2016 Feb 11:2():16001. doi: 10.1038/nrdp.2016.1. Epub 2016 Feb 11 [PubMed PMID: 27189177]
Han SH, Kang EW, Kie JH, Yoo TH, Choi KH, Han DS, Kang SW. Spontaneous remission of IgA nephropathy associated with resolution of hepatitis A. American journal of kidney diseases : the official journal of the National Kidney Foundation. 2010 Dec:56(6):1163-7. doi: 10.1053/j.ajkd.2010.08.018. Epub 2010 Oct 8 [PubMed PMID: 20932622]
Level 3 (low-level) evidenceBhandari G, Tiwari V, Gupta A, Gupta P, Bhargava V, Malik M, Gupta A, Bhalla AK, Rana DS. IgA Nephropathy with Wilson's Disease: A Case Report and Literature Review. Indian journal of nephrology. 2021 Sep-Oct:31(5):474-477. doi: 10.4103/ijn.IJN_227_20. Epub 2021 Feb 20 [PubMed PMID: 34880558]
Level 3 (low-level) evidenceGesualdo L, Di Leo V, Coppo R. The mucosal immune system and IgA nephropathy. Seminars in immunopathology. 2021 Oct:43(5):657-668. doi: 10.1007/s00281-021-00871-y. Epub 2021 Oct 12 [PubMed PMID: 34642783]
Canetta PA, Kiryluk K, Appel GB. Glomerular diseases: emerging tests and therapies for IgA nephropathy. Clinical journal of the American Society of Nephrology : CJASN. 2014 Mar:9(3):617-25. doi: 10.2215/CJN.07260713. Epub 2013 Sep 26 [PubMed PMID: 24071652]
Takada D, Sumida K, Sekine A, Hazue R, Yamanouchi M, Suwabe T, Hayami N, Hoshino J, Sawa N, Takaichi K, Fujii T, Ohashi K, Ubara Y. IgA nephropathy featuring massive wire loop-like deposits in two patients with alcoholic cirrhosis. BMC nephrology. 2017 Dec 13:18(1):362. doi: 10.1186/s12882-017-0769-1. Epub 2017 Dec 13 [PubMed PMID: 29237409]
Kaneko T, Arima R, Arakawa Y, Aoki M, Fukuda K, Fukui M, Hirama A, Fujita E, Mii A, Utsumi K, Shimizu A, Iino Y. [Two cases of rapidly progressive nephritic syndrome complicated with alcoholic liver cirrhosis]. Nihon Jinzo Gakkai shi. 2011:53(1):60-7 [PubMed PMID: 21370579]
Level 3 (low-level) evidenceGouet D, Fort E, Roblot P, Maréchaud R, Sudre Y, Touchard G. [Glomerulopathy with mesangial IgA deposits in primary hemochromatosis]. La Revue de medecine interne. 1987 May-Jun:8(3):311-2 [PubMed PMID: 3616239]
Singhal J, Sharma J. IgA nephropathy secondary to liver disease. Pediatric nephrology (Berlin, Germany). 2018 Dec:33(12):2393. doi: 10.1007/s00467-018-4051-y. Epub 2018 Sep 3 [PubMed PMID: 30178238]
Abbad L, Monteiro RC, Berthelot L. Food antigens and Transglutaminase 2 in IgA nephropathy: Molecular links between gut and kidney. Molecular immunology. 2020 May:121():1-6. doi: 10.1016/j.molimm.2020.02.019. Epub 2020 Mar 2 [PubMed PMID: 32135400]
De Angelis M, Montemurno E, Piccolo M, Vannini L, Lauriero G, Maranzano V, Gozzi G, Serrazanetti D, Dalfino G, Gobbetti M, Gesualdo L. Microbiota and metabolome associated with immunoglobulin A nephropathy (IgAN). PloS one. 2014:9(6):e99006. doi: 10.1371/journal.pone.0099006. Epub 2014 Jun 12 [PubMed PMID: 24922509]
Wei LY, Liu C, Zhang YL, Li GL. IgA nephropathy with leucocytoclastic vasculitis. The Journal of international medical research. 2018 Jul:46(7):3009-3014. doi: 10.1177/0300060518775814. Epub 2018 Jun 10 [PubMed PMID: 29888629]
Yamaguchi H, Goto S, Takahashi N, Tsuchida M, Watanabe H, Yamamoto S, Kaneko Y, Higashi K, Mori H, Nakamura Y, Horii A, Kurokawa K, Narita I. Aberrant mucosal immunoreaction to tonsillar microbiota in immunoglobulin A nephropathy. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 2021 Jan 1:36(1):75-86. doi: 10.1093/ndt/gfaa223. Epub [PubMed PMID: 33099625]
Hernández JI, Gómez-Román J, Rodrigo E, Olmos JM, González-Vela C, Ruiz JC, Val JF, Riancho JA. Bronchiolitis obliterans and IgA nephropathy. A new cause of pulmonary-renal syndrome. American journal of respiratory and critical care medicine. 1997 Aug:156(2 Pt 1):665-8 [PubMed PMID: 9279256]
Jelicic I, Mladinov S. Rapidly progressive IgA nephritis and sarcoidosis. Iranian journal of kidney diseases. 2022 Sep:16(5):315-317 [PubMed PMID: 36178867]
Said SM, Leung N, Sethi S, Cornell LD, Fidler ME, Grande JP, Herrmann S, Tefferi A, D'Agati VD, Nasr SH. Myeloproliferative neoplasms cause glomerulopathy. Kidney international. 2011 Oct:80(7):753-9. doi: 10.1038/ki.2011.147. Epub 2011 Jun 8 [PubMed PMID: 21654720]
Wang J, Liu Y, Liu N, Gao M, Yuan H. Paraneoplastic immunoglobulin A nephropathy in a patient with lung adenocarcinoma: A case report and literature review. The Journal of international medical research. 2021 Apr:49(4):300060521996868. doi: 10.1177/0300060521996868. Epub [PubMed PMID: 33926295]
Level 3 (low-level) evidenceMimura I, Tojo A, Kinugasa S, Uozaki H, Fujita T. Renal cell carcinoma in association with IgA nephropathy in the elderly. The American journal of the medical sciences. 2009 Nov:338(5):431-2. doi: 10.1097/MAJ.0b013e3181ae1b12. Epub [PubMed PMID: 19773639]
Kiryluk K, Freedberg DE, Radhakrishnan J, Segall L, Jacobson JS, Mathur M, Mohan S, Neugut AI. Global Incidence of IgA Nephropathy by Race and Ethnicity: A Systematic Review. Kidney360. 2023 Aug 1:4(8):1112-1122. doi: 10.34067/KID.0000000000000165. Epub 2023 May 25 [PubMed PMID: 37227924]
Level 1 (high-level) evidenceMcGrogan A, Franssen CF, de Vries CS. The incidence of primary glomerulonephritis worldwide: a systematic review of the literature. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 2011 Feb:26(2):414-30. doi: 10.1093/ndt/gfq665. Epub 2010 Nov 10 [PubMed PMID: 21068142]
Level 1 (high-level) evidenceUtsunomiya Y, Koda T, Kado T, Okada S, Hayashi A, Kanzaki S, Kasagi T, Hayashibara H, Okasora T. Incidence of pediatric IgA nephropathy. Pediatric nephrology (Berlin, Germany). 2003 Jun:18(6):511-5 [PubMed PMID: 12720079]
Kiryluk K, Li Y, Sanna-Cherchi S, Rohanizadegan M, Suzuki H, Eitner F, Snyder HJ, Choi M, Hou P, Scolari F, Izzi C, Gigante M, Gesualdo L, Savoldi S, Amoroso A, Cusi D, Zamboli P, Julian BA, Novak J, Wyatt RJ, Mucha K, Perola M, Kristiansson K, Viktorin A, Magnusson PK, Thorleifsson G, Thorsteinsdottir U, Stefansson K, Boland A, Metzger M, Thibaudin L, Wanner C, Jager KJ, Goto S, Maixnerova D, Karnib HH, Nagy J, Panzer U, Xie J, Chen N, Tesar V, Narita I, Berthoux F, Floege J, Stengel B, Zhang H, Lifton RP, Gharavi AG. Geographic differences in genetic susceptibility to IgA nephropathy: GWAS replication study and geospatial risk analysis. PLoS genetics. 2012:8(6):e1002765. doi: 10.1371/journal.pgen.1002765. Epub 2012 Jun 21 [PubMed PMID: 22737082]
Level 2 (mid-level) evidenceSchena FP, Nistor I. Epidemiology of IgA Nephropathy: A Global Perspective. Seminars in nephrology. 2018 Sep:38(5):435-442. doi: 10.1016/j.semnephrol.2018.05.013. Epub [PubMed PMID: 30177015]
Level 3 (low-level) evidenceBarbour SJ, Cattran DC, Kim SJ, Levin A, Wald R, Hladunewich MA, Reich HN. Individuals of Pacific Asian origin with IgA nephropathy have an increased risk of progression to end-stage renal disease. Kidney international. 2013 Nov:84(5):1017-24. doi: 10.1038/ki.2013.210. Epub 2013 Jun 5 [PubMed PMID: 23739233]
Level 2 (mid-level) evidenceSoares MF, Roberts IS. IgA nephropathy: an update. Current opinion in nephrology and hypertension. 2017 May:26(3):165-171. doi: 10.1097/MNH.0000000000000312. Epub [PubMed PMID: 28221174]
Level 3 (low-level) evidenceMagistroni R, D'Agati VD, Appel GB, Kiryluk K. New developments in the genetics, pathogenesis, and therapy of IgA nephropathy. Kidney international. 2015 Nov:88(5):974-89. doi: 10.1038/ki.2015.252. Epub 2015 Sep 16 [PubMed PMID: 26376134]
Kiryluk K, Moldoveanu Z, Sanders JT, Eison TM, Suzuki H, Julian BA, Novak J, Gharavi AG, Wyatt RJ. Aberrant glycosylation of IgA1 is inherited in both pediatric IgA nephropathy and Henoch-Schönlein purpura nephritis. Kidney international. 2011 Jul:80(1):79-87. doi: 10.1038/ki.2011.16. Epub 2011 Feb 16 [PubMed PMID: 21326171]
Level 2 (mid-level) evidenceGharavi AG, Moldoveanu Z, Wyatt RJ, Barker CV, Woodford SY, Lifton RP, Mestecky J, Novak J, Julian BA. Aberrant IgA1 glycosylation is inherited in familial and sporadic IgA nephropathy. Journal of the American Society of Nephrology : JASN. 2008 May:19(5):1008-14. doi: 10.1681/ASN.2007091052. Epub 2008 Feb 13 [PubMed PMID: 18272841]
Yamamoto R, Nagasawa Y, Shoji T, Iwatani H, Hamano T, Kawada N, Inoue K, Uehata T, Kaneko T, Okada N, Moriyama T, Horio M, Yamauchi A, Tsubakihara Y, Imai E, Rakugi H, Isaka Y. Cigarette smoking and progression of IgA nephropathy. American journal of kidney diseases : the official journal of the National Kidney Foundation. 2010 Aug:56(2):313-24. doi: 10.1053/j.ajkd.2010.02.351. Epub 2010 May 14 [PubMed PMID: 20471735]
Level 3 (low-level) evidenceKataoka H, Ohara M, Honda K, Mochizuki T, Nitta K. Maximal glomerular diameter as a 10-year prognostic indicator for IgA nephropathy. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 2011 Dec:26(12):3937-43. doi: 10.1093/ndt/gfr139. Epub 2011 Mar 22 [PubMed PMID: 21427079]
Knoppova B, Reily C, Maillard N, Rizk DV, Moldoveanu Z, Mestecky J, Raska M, Renfrow MB, Julian BA, Novak J. The Origin and Activities of IgA1-Containing Immune Complexes in IgA Nephropathy. Frontiers in immunology. 2016:7():117. doi: 10.3389/fimmu.2016.00117. Epub 2016 Apr 12 [PubMed PMID: 27148252]
Kiryluk K, Li Y, Scolari F, Sanna-Cherchi S, Choi M, Verbitsky M, Fasel D, Lata S, Prakash S, Shapiro S, Fischman C, Snyder HJ, Appel G, Izzi C, Viola BF, Dallera N, Del Vecchio L, Barlassina C, Salvi E, Bertinetto FE, Amoroso A, Savoldi S, Rocchietti M, Amore A, Peruzzi L, Coppo R, Salvadori M, Ravani P, Magistroni R, Ghiggeri GM, Caridi G, Bodria M, Lugani F, Allegri L, Delsante M, Maiorana M, Magnano A, Frasca G, Boer E, Boscutti G, Ponticelli C, Mignani R, Marcantoni C, Di Landro D, Santoro D, Pani A, Polci R, Feriozzi S, Chicca S, Galliani M, Gigante M, Gesualdo L, Zamboli P, Battaglia GG, Garozzo M, Maixnerová D, Tesar V, Eitner F, Rauen T, Floege J, Kovacs T, Nagy J, Mucha K, Pączek L, Zaniew M, Mizerska-Wasiak M, Roszkowska-Blaim M, Pawlaczyk K, Gale D, Barratt J, Thibaudin L, Berthoux F, Canaud G, Boland A, Metzger M, Panzer U, Suzuki H, Goto S, Narita I, Caliskan Y, Xie J, Hou P, Chen N, Zhang H, Wyatt RJ, Novak J, Julian BA, Feehally J, Stengel B, Cusi D, Lifton RP, Gharavi AG. Discovery of new risk loci for IgA nephropathy implicates genes involved in immunity against intestinal pathogens. Nature genetics. 2014 Nov:46(11):1187-96. doi: 10.1038/ng.3118. Epub 2014 Oct 12 [PubMed PMID: 25305756]
Level 1 (high-level) evidenceRoos A, Rastaldi MP, Calvaresi N, Oortwijn BD, Schlagwein N, van Gijlswijk-Janssen DJ, Stahl GL, Matsushita M, Fujita T, van Kooten C, Daha MR. Glomerular activation of the lectin pathway of complement in IgA nephropathy is associated with more severe renal disease. Journal of the American Society of Nephrology : JASN. 2006 Jun:17(6):1724-34 [PubMed PMID: 16687629]
Roos A, Bouwman LH, van Gijlswijk-Janssen DJ, Faber-Krol MC, Stahl GL, Daha MR. Human IgA activates the complement system via the mannan-binding lectin pathway. Journal of immunology (Baltimore, Md. : 1950). 2001 Sep 1:167(5):2861-8 [PubMed PMID: 11509633]
Gharavi AG, Kiryluk K, Choi M, Li Y, Hou P, Xie J, Sanna-Cherchi S, Men CJ, Julian BA, Wyatt RJ, Novak J, He JC, Wang H, Lv J, Zhu L, Wang W, Wang Z, Yasuno K, Gunel M, Mane S, Umlauf S, Tikhonova I, Beerman I, Savoldi S, Magistroni R, Ghiggeri GM, Bodria M, Lugani F, Ravani P, Ponticelli C, Allegri L, Boscutti G, Frasca G, Amore A, Peruzzi L, Coppo R, Izzi C, Viola BF, Prati E, Salvadori M, Mignani R, Gesualdo L, Bertinetto F, Mesiano P, Amoroso A, Scolari F, Chen N, Zhang H, Lifton RP. Genome-wide association study identifies susceptibility loci for IgA nephropathy. Nature genetics. 2011 Mar 13:43(4):321-7. doi: 10.1038/ng.787. Epub 2011 Mar 13 [PubMed PMID: 21399633]
Level 2 (mid-level) evidenceSuzuki H, Kiryluk K, Novak J, Moldoveanu Z, Herr AB, Renfrow MB, Wyatt RJ, Scolari F, Mestecky J, Gharavi AG, Julian BA. The pathophysiology of IgA nephropathy. Journal of the American Society of Nephrology : JASN. 2011 Oct:22(10):1795-803. doi: 10.1681/ASN.2011050464. Epub 2011 Sep 23 [PubMed PMID: 21949093]
Hassler JR. IgA nephropathy: A brief review. Seminars in diagnostic pathology. 2020 May:37(3):143-147. doi: 10.1053/j.semdp.2020.03.001. Epub 2020 Mar 16 [PubMed PMID: 32241578]
Kusaba G, Ohsawa I, Ishii M, Inoshita H, Takagi M, Tanifuji C, Takahashi K, Nakamoto J, Yoshida M, Ohi H, Horikoshi S, Kurihara H, Tomino Y. Significance of broad distribution of electron-dense deposits in patients with IgA nephropathy. Medical molecular morphology. 2012 Dec:45(1):29-34. doi: 10.1007/s00795-011-0538-3. Epub 2012 Mar 20 [PubMed PMID: 22431181]
Trimarchi H, Haas M, Coppo R. Crescents and IgA Nephropathy: A Delicate Marriage. Journal of clinical medicine. 2022 Jun 21:11(13):. doi: 10.3390/jcm11133569. Epub 2022 Jun 21 [PubMed PMID: 35806856]
Kataoka H, Ohara M, Shibui K, Sato M, Suzuki T, Amemiya N, Watanabe Y, Honda K, Mochizuki T, Nitta K. Overweight and obesity accelerate the progression of IgA nephropathy: prognostic utility of a combination of BMI and histopathological parameters. Clinical and experimental nephrology. 2012 Oct:16(5):706-12. doi: 10.1007/s10157-012-0613-7. Epub 2012 Feb 17 [PubMed PMID: 22350469]
Appel GB, Waldman M. The IgA nephropathy treatment dilemma. Kidney international. 2006 Jun:69(11):1939-44 [PubMed PMID: 16641925]
Shen P, He L, Huang D. Clinical course and prognostic factors of clinical early IgA nephropathy. The Netherlands journal of medicine. 2008 Jun:66(6):242-7 [PubMed PMID: 18689907]
Level 2 (mid-level) evidenceD'Amico G. Natural history of idiopathic IgA nephropathy and factors predictive of disease outcome. Seminars in nephrology. 2004 May:24(3):179-96 [PubMed PMID: 15156525]
Lai KN, Lai FM, Chan KW, Ho CP, Leung AC, Vallance-Owen J. An overlapping syndrome of IgA nephropathy and lipoid nephrosis. American journal of clinical pathology. 1986 Dec:86(6):716-23 [PubMed PMID: 3538845]
Herlitz LC, Bomback AS, Stokes MB, Radhakrishnan J, D'Agati VD, Markowitz GS. IgA nephropathy with minimal change disease. Clinical journal of the American Society of Nephrology : CJASN. 2014 Jun 6:9(6):1033-9. doi: 10.2215/CJN.11951113. Epub 2014 Apr 10 [PubMed PMID: 24721885]
Level 2 (mid-level) evidenceWalsh M, Sar A, Lee D, Yilmaz S, Benediktsson H, Manns B, Hemmelgarn B. Histopathologic features aid in predicting risk for progression of IgA nephropathy. Clinical journal of the American Society of Nephrology : CJASN. 2010 Mar:5(3):425-30. doi: 10.2215/CJN.06530909. Epub 2010 Jan 14 [PubMed PMID: 20089495]
Level 2 (mid-level) evidenceWorking Group of the International IgA Nephropathy Network and the Renal Pathology Society, Roberts IS, Cook HT, Troyanov S, Alpers CE, Amore A, Barratt J, Berthoux F, Bonsib S, Bruijn JA, Cattran DC, Coppo R, D'Agati V, D'Amico G, Emancipator S, Emma F, Feehally J, Ferrario F, Fervenza FC, Florquin S, Fogo A, Geddes CC, Groene HJ, Haas M, Herzenberg AM, Hill PA, Hogg RJ, Hsu SI, Jennette JC, Joh K, Julian BA, Kawamura T, Lai FM, Li LS, Li PK, Liu ZH, Mackinnon B, Mezzano S, Schena FP, Tomino Y, Walker PD, Wang H, Weening JJ, Yoshikawa N, Zhang H. The Oxford classification of IgA nephropathy: pathology definitions, correlations, and reproducibility. Kidney international. 2009 Sep:76(5):546-56. doi: 10.1038/ki.2009.168. Epub 2009 Jul 1 [PubMed PMID: 19571790]
Herzenberg AM, Fogo AB, Reich HN, Troyanov S, Bavbek N, Massat AE, Hunley TE, Hladunewich MA, Julian BA, Fervenza FC, Cattran DC. Validation of the Oxford classification of IgA nephropathy. Kidney international. 2011 Aug:80(3):310-7. doi: 10.1038/ki.2011.126. Epub 2011 May 4 [PubMed PMID: 21544062]
Level 1 (high-level) evidenceKang SH, Choi SR, Park HS, Lee JY, Sun IO, Hwang HS, Chung BH, Park CW, Yang CW, Kim YS, Choi YJ, Choi BS. The Oxford classification as a predictor of prognosis in patients with IgA nephropathy. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 2012 Jan:27(1):252-8. doi: 10.1093/ndt/gfr295. Epub 2011 May 23 [PubMed PMID: 21606384]
Trimarchi H, Barratt J, Cattran DC, Cook HT, Coppo R, Haas M, Liu ZH, Roberts IS, Yuzawa Y, Zhang H, Feehally J, IgAN Classification Working Group of the International IgA Nephropathy Network and the Renal Pathology Society, Conference Participants. Oxford Classification of IgA nephropathy 2016: an update from the IgA Nephropathy Classification Working Group. Kidney international. 2017 May:91(5):1014-1021. doi: 10.1016/j.kint.2017.02.003. Epub 2017 Mar 22 [PubMed PMID: 28341274]
Rovin BH, Adler SG, Barratt J, Bridoux F, Burdge KA, Chan TM, Cook HT, Fervenza FC, Gibson KL, Glassock RJ, Jayne DRW, Jha V, Liew A, Liu ZH, Mejía-Vilet JM, Nester CM, Radhakrishnan J, Rave EM, Reich HN, Ronco P, Sanders JF, Sethi S, Suzuki Y, Tang SCW, Tesar V, Vivarelli M, Wetzels JFM, Lytvyn L, Craig JC, Tunnicliffe DJ, Howell M, Tonelli MA, Cheung M, Earley A, Floege J. Executive summary of the KDIGO 2021 Guideline for the Management of Glomerular Diseases. Kidney international. 2021 Oct:100(4):753-779. doi: 10.1016/j.kint.2021.05.015. Epub [PubMed PMID: 34556300]
Berthoux F, Suzuki H, Thibaudin L, Yanagawa H, Maillard N, Mariat C, Tomino Y, Julian BA, Novak J. Autoantibodies targeting galactose-deficient IgA1 associate with progression of IgA nephropathy. Journal of the American Society of Nephrology : JASN. 2012 Sep:23(9):1579-87. doi: 10.1681/ASN.2012010053. Epub 2012 Aug 16 [PubMed PMID: 22904352]
Moldoveanu Z, Wyatt RJ, Lee JY, Tomana M, Julian BA, Mestecky J, Huang WQ, Anreddy SR, Hall S, Hastings MC, Lau KK, Cook WJ, Novak J. Patients with IgA nephropathy have increased serum galactose-deficient IgA1 levels. Kidney international. 2007 Jun:71(11):1148-54 [PubMed PMID: 17342176]
Pozzi C. Treatment of IgA nephropathy. Journal of nephrology. 2016 Feb:29(1):21-5. doi: 10.1007/s40620-015-0248-3. Epub 2015 Nov 17 [PubMed PMID: 26577268]
Takahara M, Nagato T, Nozaki Y, Kumai T, Katada A, Hayashi T, Harabuchi Y. A proliferation-inducing ligand (APRIL) induced hyper-production of IgA from tonsillar mononuclear cells in patients with IgA nephropathy. Cellular immunology. 2019 Jul:341():103925. doi: 10.1016/j.cellimm.2019.103925. Epub 2019 May 8 [PubMed PMID: 31088610]
Rauen T, Eitner F, Fitzner C, Sommerer C, Zeier M, Otte B, Panzer U, Peters H, Benck U, Mertens PR, Kuhlmann U, Witzke O, Gross O, Vielhauer V, Mann JF, Hilgers RD, Floege J, STOP-IgAN Investigators. Intensive Supportive Care plus Immunosuppression in IgA Nephropathy. The New England journal of medicine. 2015 Dec 3:373(23):2225-36. doi: 10.1056/NEJMoa1415463. Epub [PubMed PMID: 26630142]
Floege J, Rauen T, Eitner F. Intensive Supportive Care plus Immunosuppression in IgA Nephropathy. The New England journal of medicine. 2016 Mar 10:374(10):992-3. doi: 10.1056/NEJMc1600141. Epub [PubMed PMID: 26962736]
Rauen T, Wied S, Fitzner C, Eitner F, Sommerer C, Zeier M, Otte B, Panzer U, Budde K, Benck U, Mertens PR, Kuhlmann U, Witzke O, Gross O, Vielhauer V, Mann JFE, Hilgers RD, Floege J, STOP-IgAN Investigators. After ten years of follow-up, no difference between supportive care plus immunosuppression and supportive care alone in IgA nephropathy. Kidney international. 2020 Oct:98(4):1044-1052. doi: 10.1016/j.kint.2020.04.046. Epub 2020 May 22 [PubMed PMID: 32450154]
Selvaskandan H, Barratt J, Cheung CK. Novel Treatment Paradigms: Primary IgA Nephropathy. Kidney international reports. 2024 Feb:9(2):203-213. doi: 10.1016/j.ekir.2023.11.026. Epub 2023 Dec 1 [PubMed PMID: 38344739]
Kittiskulnam P, Kanjanabuch T, Tangmanjitjaroen K, Chancharoenthana W, Praditpornsilpa K, Eiam-Ong S. The beneficial effects of weight reduction in overweight patients with chronic proteinuric immunoglobulin a nephropathy: a randomized controlled trial. Journal of renal nutrition : the official journal of the Council on Renal Nutrition of the National Kidney Foundation. 2014 May:24(3):200-7. doi: 10.1053/j.jrn.2014.01.016. Epub [PubMed PMID: 24759301]
Level 1 (high-level) evidenceWang S, Qin A, Pei G, Jiang Z, Dong L, Tan J, Tan L, Tang Y, Qin W. Cigarette smoking may accelerate the progression of IgA nephropathy. BMC nephrology. 2021 Jun 29:22(1):239. doi: 10.1186/s12882-021-02453-4. Epub 2021 Jun 29 [PubMed PMID: 34187402]
Level 2 (mid-level) evidenceGharavi AG, Yan Y, Scolari F, Schena FP, Frasca GM, Ghiggeri GM, Cooper K, Amoroso A, Viola BF, Battini G, Caridi G, Canova C, Farhi A, Subramanian V, Nelson-Williams C, Woodford S, Julian BA, Wyatt RJ, Lifton RP. IgA nephropathy, the most common cause of glomerulonephritis, is linked to 6q22-23. Nature genetics. 2000 Nov:26(3):354-7 [PubMed PMID: 11062479]
Lv J, Xu D, Perkovic V, Ma X, Johnson DW, Woodward M, Levin A, Zhang H, Wang H, TESTING Study Group. Corticosteroid therapy in IgA nephropathy. Journal of the American Society of Nephrology : JASN. 2012 Jun:23(6):1108-16. doi: 10.1681/ASN.2011111112. Epub 2012 Apr 26 [PubMed PMID: 22539830]
Level 1 (high-level) evidenceNatale P, Palmer SC, Ruospo M, Saglimbene VM, Craig JC, Vecchio M, Samuels JA, Molony DA, Schena FP, Strippoli GF. Immunosuppressive agents for treating IgA nephropathy. The Cochrane database of systematic reviews. 2020 Mar 12:3(3):CD003965. doi: 10.1002/14651858.CD003965.pub3. Epub 2020 Mar 12 [PubMed PMID: 32162319]
Level 1 (high-level) evidenceWong MG, Lv J, Hladunewich MA, Jha V, Hooi LS, Monaghan H, Zhao M, Barbour S, Reich HN, Cattran D, Glassock R, Levin A, Jardine MJ, Wheeler DC, Woodward M, Billot L, Chan TM, Liu ZH, Johnson DW, Cass A, Feehally J, Floege J, Remuzzi G, Wu Y, Agarwal R, Zhang H, Perkovic V, TESTING Study Group. The Therapeutic Evaluation of Steroids in IgA Nephropathy Global (TESTING) Study: Trial Design and Baseline Characteristics. American journal of nephrology. 2021:52(10-11):827-836. doi: 10.1159/000519812. Epub 2021 Nov 3 [PubMed PMID: 34731857]
Lv J, Wong MG, Hladunewich MA, Jha V, Hooi LS, Monaghan H, Zhao M, Barbour S, Jardine MJ, Reich HN, Cattran D, Glassock R, Levin A, Wheeler DC, Woodward M, Billot L, Stepien S, Rogers K, Chan TM, Liu ZH, Johnson DW, Cass A, Feehally J, Floege J, Remuzzi G, Wu Y, Agarwal R, Zhang H, Perkovic V, TESTING Study Group. Effect of Oral Methylprednisolone on Decline in Kidney Function or Kidney Failure in Patients With IgA Nephropathy: The TESTING Randomized Clinical Trial. JAMA. 2022 May 17:327(19):1888-1898. doi: 10.1001/jama.2022.5368. Epub [PubMed PMID: 35579642]
Level 1 (high-level) evidenceSyed YY. Sparsentan: First Approval. Drugs. 2023 Apr:83(6):563-568. doi: 10.1007/s40265-023-01864-x. Epub [PubMed PMID: 37022667]
Heerspink HJL, Radhakrishnan J, Alpers CE, Barratt J, Bieler S, Diva U, Inrig J, Komers R, Mercer A, Noronha IL, Rheault MN, Rote W, Rovin B, Trachtman H, Trimarchi H, Wong MG, Perkovic V, PROTECT Investigators. Sparsentan in patients with IgA nephropathy: a prespecified interim analysis from a randomised, double-blind, active-controlled clinical trial. Lancet (London, England). 2023 May 13:401(10388):1584-1594. doi: 10.1016/S0140-6736(23)00569-X. Epub 2023 Apr 1 [PubMed PMID: 37015244]
Level 1 (high-level) evidenceRovin BH, Barratt J, Heerspink HJL, Alpers CE, Bieler S, Chae DW, Diva UA, Floege J, Gesualdo L, Inrig JK, Kohan DE, Komers R, Kooienga LA, Lafayette R, Maes B, Małecki R, Mercer A, Noronha IL, Oh SW, Peh CA, Praga M, Preciado P, Radhakrishnan J, Rheault MN, Rote WE, Tang SCW, Tesar V, Trachtman H, Trimarchi H, Tumlin JA, Wong MG, Perkovic V, DUPRO steering committee and PROTECT Investigators. Efficacy and safety of sparsentan versus irbesartan in patients with IgA nephropathy (PROTECT): 2-year results from a randomised, active-controlled, phase 3 trial. Lancet (London, England). 2023 Dec 2:402(10417):2077-2090. doi: 10.1016/S0140-6736(23)02302-4. Epub 2023 Nov 3 [PubMed PMID: 37931634]
Level 1 (high-level) evidenceSmerud HK, Bárány P, Lindström K, Fernström A, Sandell A, Påhlsson P, Fellström B. New treatment for IgA nephropathy: enteric budesonide targeted to the ileocecal region ameliorates proteinuria. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 2011 Oct:26(10):3237-42. doi: 10.1093/ndt/gfr052. Epub 2011 Mar 4 [PubMed PMID: 21378156]
Barratt J, Lafayette R, Kristensen J, Stone A, Cattran D, Floege J, Tesar V, Trimarchi H, Zhang H, Eren N, Paliege A, Rovin BH, NefIgArd Trial Investigators. Results from part A of the multi-center, double-blind, randomized, placebo-controlled NefIgArd trial, which evaluated targeted-release formulation of budesonide for the treatment of primary immunoglobulin A nephropathy. Kidney international. 2023 Feb:103(2):391-402. doi: 10.1016/j.kint.2022.09.017. Epub 2022 Oct 19 [PubMed PMID: 36270561]
Level 1 (high-level) evidenceFellström BC, Barratt J, Cook H, Coppo R, Feehally J, de Fijter JW, Floege J, Hetzel G, Jardine AG, Locatelli F, Maes BD, Mercer A, Ortiz F, Praga M, Sørensen SS, Tesar V, Del Vecchio L, NEFIGAN Trial Investigators. Targeted-release budesonide versus placebo in patients with IgA nephropathy (NEFIGAN): a double-blind, randomised, placebo-controlled phase 2b trial. Lancet (London, England). 2017 May 27:389(10084):2117-2127. doi: 10.1016/S0140-6736(17)30550-0. Epub 2017 Mar 28 [PubMed PMID: 28363480]
Level 1 (high-level) evidenceBarratt J, Floege J. SGLT-2 inhibition in IgA nephropathy: the new standard of care? Kidney international. 2021 Jul:100(1):24-26. doi: 10.1016/j.kint.2021.04.002. Epub 2021 Apr 17 [PubMed PMID: 33878337]
Beck L, Bomback AS, Choi MJ, Holzman LB, Langford C, Mariani LH, Somers MJ, Trachtman H, Waldman M. KDOQI US commentary on the 2012 KDIGO clinical practice guideline for glomerulonephritis. American journal of kidney diseases : the official journal of the National Kidney Foundation. 2013 Sep:62(3):403-41. doi: 10.1053/j.ajkd.2013.06.002. Epub 2013 Jul 18 [PubMed PMID: 23871408]
Level 2 (mid-level) evidenceChen Y, Li Y, Yang S, Li Y, Liang M. Efficacy and safety of mycophenolate mofetil treatment in IgA nephropathy: a systematic review. BMC nephrology. 2014 Dec 5:15():193. doi: 10.1186/1471-2369-15-193. Epub 2014 Dec 5 [PubMed PMID: 25475967]
Level 1 (high-level) evidenceTian L, Shao X, Xie Y, Wang L, Wang Q, Che X, Ni Z, Mou S. The long-term efficacy and safety of immunosuppressive therapy on the progression of IgA nephropathy: a meta-analysis of controlled clinical trials with more than 5-year follow-up. Expert opinion on pharmacotherapy. 2015 Jun:16(8):1137-47. doi: 10.1517/14656566.2015.1038238. Epub 2015 Apr 20 [PubMed PMID: 25892092]
Level 3 (low-level) evidenceLafayette RA, Canetta PA, Rovin BH, Appel GB, Novak J, Nath KA, Sethi S, Tumlin JA, Mehta K, Hogan M, Erickson S, Julian BA, Leung N, Enders FT, Brown R, Knoppova B, Hall S, Fervenza FC. A Randomized, Controlled Trial of Rituximab in IgA Nephropathy with Proteinuria and Renal Dysfunction. Journal of the American Society of Nephrology : JASN. 2017 Apr:28(4):1306-1313. doi: 10.1681/ASN.2016060640. Epub 2016 Nov 7 [PubMed PMID: 27821627]
Level 1 (high-level) evidenceRizk DV, Rovin BH, Zhang H, Kashihara N, Maes B, Trimarchi H, Perkovic V, Meier M, Kollins D, Papachristofi O, Charney A, Barratt J. Targeting the Alternative Complement Pathway With Iptacopan to Treat IgA Nephropathy: Design and Rationale of the APPLAUSE-IgAN Study. Kidney international reports. 2023 May:8(5):968-979. doi: 10.1016/j.ekir.2023.01.041. Epub 2023 Feb 9 [PubMed PMID: 37180505]
Zhang H, Rizk DV, Perkovic V, Maes B, Kashihara N, Rovin B, Trimarchi H, Sprangers B, Meier M, Kollins D, Papachristofi O, Milojevic J, Junge G, Nidamarthy PK, Charney A, Barratt J. Results of a randomized double-blind placebo-controlled Phase 2 study propose iptacopan as an alternative complement pathway inhibitor for IgA nephropathy. Kidney international. 2024 Jan:105(1):189-199. doi: 10.1016/j.kint.2023.09.027. Epub 2023 Oct 31 [PubMed PMID: 37914086]
Level 1 (high-level) evidenceCoppo R. Treatment of IgA nephropathy: Recent advances and prospects. Nephrologie & therapeutique. 2018 Apr:14 Suppl 1():S13-S21. doi: 10.1016/j.nephro.2018.02.010. Epub [PubMed PMID: 29606258]
Level 3 (low-level) evidenceLai KN, Leung JC, Tang SC. The Treatment of IgA Nephropathy. Kidney diseases (Basel, Switzerland). 2015 May:1(1):19-26. doi: 10.1159/000381508. Epub 2015 Apr 15 [PubMed PMID: 27536661]
Tang SC, Tang AW, Wong SS, Leung JC, Ho YW, Lai KN. Long-term study of mycophenolate mofetil treatment in IgA nephropathy. Kidney international. 2010 Mar:77(6):543-9. doi: 10.1038/ki.2009.499. Epub 2009 Dec 23 [PubMed PMID: 20032964]
Barbour S, Feehally J. An update on the treatment of IgA nephropathy. Current opinion in nephrology and hypertension. 2017 Jul:26(4):319-326. doi: 10.1097/MNH.0000000000000336. Epub [PubMed PMID: 28399021]
Level 3 (low-level) evidenceBallardie FW, Roberts ISD. Controlled prospective trial of prednisolone and cytotoxics in progressive IgA nephropathy. Journal of the American Society of Nephrology : JASN. 2002 Jan:13(1):142-148. doi: 10.1681/ASN.V131142. Epub [PubMed PMID: 11752031]
Level 1 (high-level) evidencePozzi C, Andrulli S, Pani A, Scaini P, Roccatello D, Fogazzi G, Pecchini P, Rustichelli R, Finocchiaro P, Del Vecchio L, Locatelli F. IgA nephropathy with severe chronic renal failure: a randomized controlled trial of corticosteroids and azathioprine. Journal of nephrology. 2013 Jan-Feb:26(1):86-93. doi: 10.5301/jn.5000110. Epub [PubMed PMID: 22460183]
Level 1 (high-level) evidenceMaeda I, Hayashi T, Sato KK, Shibata MO, Hamada M, Kishida M, Kitabayashi C, Morikawa T, Okada N, Okumura M, Konishi M, Konishi Y, Endo G, Imanishi M. Tonsillectomy has beneficial effects on remission and progression of IgA nephropathy independent of steroid therapy. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 2012 Jul:27(7):2806-13. doi: 10.1093/ndt/gfs053. Epub 2012 Apr 5 [PubMed PMID: 22492824]
Kumon S, Moriyama T, Kamiyama T, Karasawa K, Nitta K. The impact of tonsillectomy combined with steroid pulse therapy in patients with advanced IgA nephropathy and impaired renal function. Clinical and experimental nephrology. 2020 Apr:24(4):295-306. doi: 10.1007/s10157-019-01828-0. Epub 2019 Dec 16 [PubMed PMID: 31845064]
Ohya M, Otani H, Minami Y, Yamanaka S, Mima T, Negi S, Yukawa S, Shigematsu T. Tonsillectomy with steroid pulse therapy has more effect on the relapse rate than steroid pulse monotherapy in IgA nephropathy patients. Clinical nephrology. 2013 Jul:80(1):47-52. doi: 10.5414/CN107861. Epub [PubMed PMID: 23557791]
Xie Y, Chen X, Nishi S, Narita I, Gejyo F. Relationship between tonsils and IgA nephropathy as well as indications of tonsillectomy. Kidney international. 2004 Apr:65(4):1135-44 [PubMed PMID: 15086452]
Watanabe-Kusunoki K, Nakazawa D, Yamamoto J, Matsuoka N, Kaneshima N, Nakagaki T, Yamamoto R, Maoka T, Iwasaki S, Tsuji T, Fukasawa Y, Nishimoto N, Nishio S, Atsumi T. Comparison of administration of single- and triple-course steroid pulse therapy combined with tonsillectomy for immunoglobulin A nephropathy. Medicine. 2021 Dec 17:100(50):e27778. doi: 10.1097/MD.0000000000027778. Epub [PubMed PMID: 34918629]
Kamei D, Moriyama T, Takei T, Wakai S, Nitta K. Comparison between consecutive and intermittent steroid pulse therapy combined with tonsillectomy for clinical remission of IgA nephropathy. Clinical and experimental nephrology. 2014 Apr:18(2):320-8. doi: 10.1007/s10157-013-0822-8. Epub 2013 Jun 7 [PubMed PMID: 23744063]
Komatsu H, Fujimoto S. Tonsillectomy combined with steroid pulse therapy induces clinical remission of IgA nephropathy. Advances in oto-rhino-laryngology. 2011:72():57-9. doi: 10.1159/000324606. Epub 2011 Aug 18 [PubMed PMID: 21865690]
Level 3 (low-level) evidenceAdachi M, Sato M, Miyazaki M, Hotta O, Hozawa K, Sato T, Taguma Y, Katori Y. Steroid pulse therapy transiently destroys the discriminative histological structure of tonsils in IgA nephropathy: Tonsillectomy should be performed before or just after steroid pulse therapy. Auris, nasus, larynx. 2018 Dec:45(6):1206-1213. doi: 10.1016/j.anl.2018.04.009. Epub 2018 May 19 [PubMed PMID: 29789195]
Feriozzi S, Polci R. The role of tonsillectomy in IgA nephropathy. Journal of nephrology. 2016 Feb:29(1):13-9. doi: 10.1007/s40620-015-0247-4. Epub 2015 Nov 18 [PubMed PMID: 26582216]
Uffing A, Pérez-Saéz MJ, Jouve T, Bugnazet M, Malvezzi P, Muhsin SA, Lafargue MC, Reindl-Schwaighofer R, Morlock A, Oberbauer R, Buxeda A, Burballa C, Pascual J, von Moos S, Seeger H, La Manna G, Comai G, Bini C, Russo LS, Farouk S, Nissaisorakarn P, Patel H, Agrawal N, Mastroianni-Kirsztajn G, Mansur J, Tedesco-Silva H, Ventura CG, Agena F, David-Neto E, Akalin E, Alani O, Mazzali M, Manfro RC, Bauer AC, Wang AX, Cheng XS, Schold JD, Berger SP, Cravedi P, Riella LV. Recurrence of IgA Nephropathy after Kidney Transplantation in Adults. Clinical journal of the American Society of Nephrology : CJASN. 2021 Aug:16(8):1247-1255. doi: 10.2215/CJN.00910121. Epub [PubMed PMID: 34362788]
Courtney AE, McNamee PT, Nelson WE, Maxwell AP. Does angiotensin blockade influence graft outcome in renal transplant recipients with IgA nephropathy? Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 2006 Dec:21(12):3550-4 [PubMed PMID: 16968729]
Level 2 (mid-level) evidenceMyette JR, Kano T, Suzuki H, Sloan SE, Szretter KJ, Ramakrishnan B, Adari H, Deotale KD, Engler F, Shriver Z, Wollacott AM, Suzuki Y, Pereira BJG. A Proliferation Inducing Ligand (APRIL) targeted antibody is a safe and effective treatment of murine IgA nephropathy. Kidney international. 2019 Jul:96(1):104-116. doi: 10.1016/j.kint.2019.01.031. Epub 2019 Mar 16 [PubMed PMID: 31027890]
Zhai YL, Zhu L, Shi SF, Liu LJ, Lv JC, Zhang H. Increased APRIL Expression Induces IgA1 Aberrant Glycosylation in IgA Nephropathy. Medicine. 2016 Mar:95(11):e3099. doi: 10.1097/MD.0000000000003099. Epub [PubMed PMID: 26986150]
Kaartinen K, Safa A, Kotha S, Ratti G, Meri S. Complement dysregulation in glomerulonephritis. Seminars in immunology. 2019 Oct:45():101331. doi: 10.1016/j.smim.2019.101331. Epub 2019 Nov 9 [PubMed PMID: 31711769]
Rizk DV, Maillard N, Julian BA, Knoppova B, Green TJ, Novak J, Wyatt RJ. The Emerging Role of Complement Proteins as a Target for Therapy of IgA Nephropathy. Frontiers in immunology. 2019:10():504. doi: 10.3389/fimmu.2019.00504. Epub 2019 Mar 19 [PubMed PMID: 30941137]
Luvizotto MJ, Menezes-Silva L, Woronik V, Monteiro RC, Câmara NOS. Gut-kidney axis in IgA nephropathy: Role on mesangial cell metabolism and inflammation. Frontiers in cell and developmental biology. 2022:10():993716. doi: 10.3389/fcell.2022.993716. Epub 2022 Nov 17 [PubMed PMID: 36467425]
Ferri C, Puccini R, Longombardo G, Paleologo G, Migliorini P, Moriconi L, Pasero G, Cioni L. Low-antigen-content diet in the treatment of patients with IgA nephropathy. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 1993:8(11):1193-8 [PubMed PMID: 8302454]