Introduction
Phosphate is one of the most important molecular elements to normal cellular functions within the body.[1] It acts as an integral component of nucleic acids and is used to replicate DNA and RNA.[2] It is an energy source for molecular functions through its role in adenosine triphosphate (ATP).[2][3] It incorporates into the phospholipids of cell membranes. It adds and deletes phosphate groups to or from proteins and functions as an on/off switch to regulate molecular activity.[3] Given its widespread role in nearly every molecular and cellular function, aberrations in serum phosphate levels can be highly impactful.
Hypophosphatemia is defined as an adult serum phosphate level of less than 2.5 mg/dL.[4] The normal serum phosphate level in children is considerably higher, between 4.0 to 7.0 mg/dL for children.[5] Hypophosphatemia is a relatively common laboratory abnormality and is often found incidentally.[6][7][8][9] About 85% of phosphorus is found in bones and teeth, 14% intracellularly, and 1% in the serum/extracellular fluid component. Free phosphate within the body is predominantly intracellular.[10]
Phosphorus Homeostasis
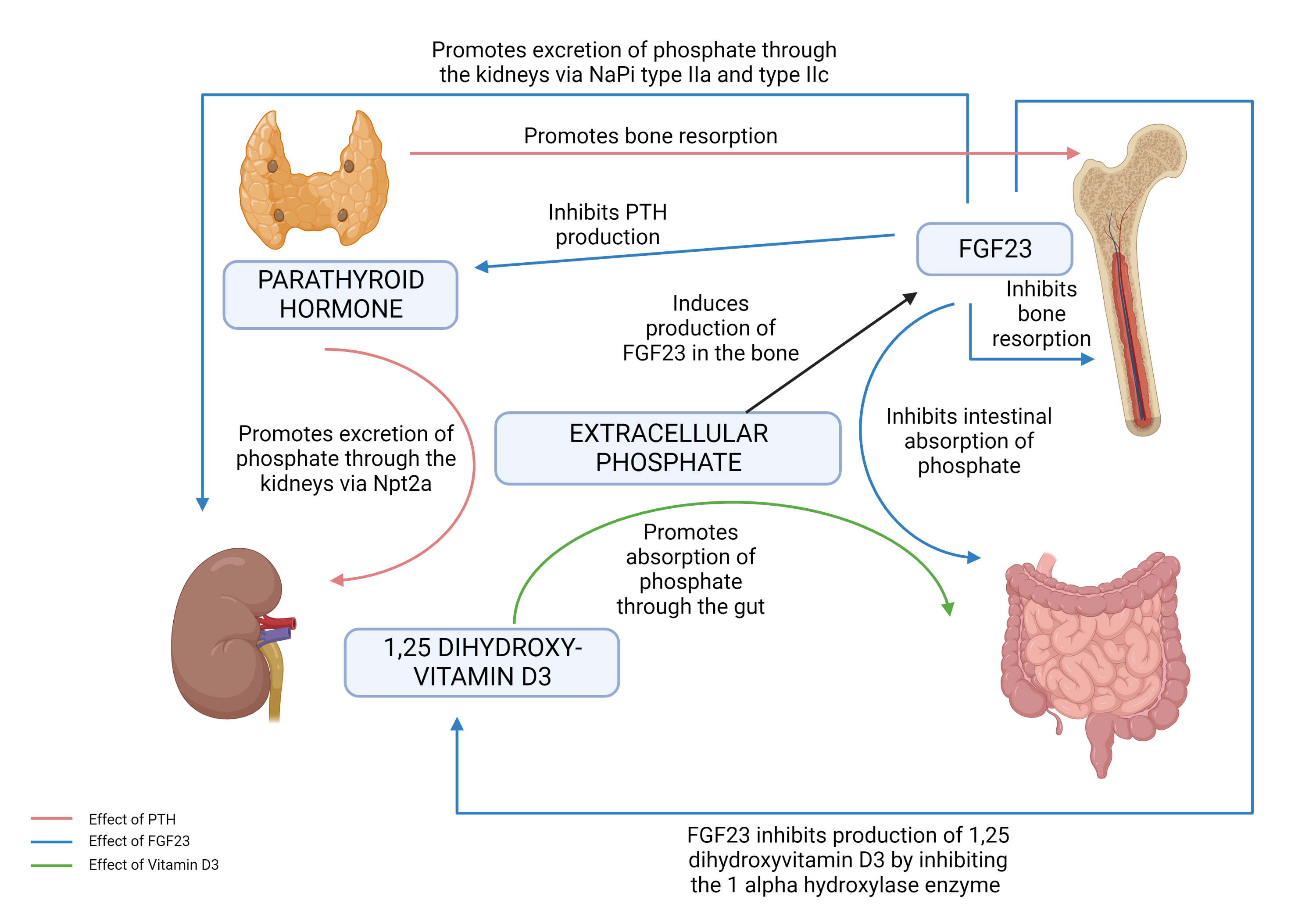
In general, phosphate levels increase through upregulated absorption in the intestines and decrease through renal excretion. Excess is stored in the bones, which act as a buffer to maintain a relatively stable total body content. A typical, nutritious diet provides 1000 to 2000 mg of phosphate daily.[11][12] Of this, 600 mg to 1200 mg is absorbed via the intestines.[13] Phosphate exists primarily in the crystallized extracellular matrix of bones, where it is relatively stable and inert. In the absence of pathology, the homeostasis of bone phosphate is neutral, with resorption and deposition of approximately 3 mg/kg per day.[14] Bone homeostasis of phosphate is regulated primarily by the parathyroid hormone, vitamin D, and sex hormones. The serum phosphate levels are predominantly maintained by absorption via the kidneys, of which 70% to 80% occurs in the proximal convoluted tubule.[4][15] Sodium-dependent phosphate type 2 (NaPi II) cotransporters in the proximal tubule are a primary site of regulation. The three types of type II transporters are type IIa, type IIb, and type IIc. Type IIa and IIc transporters are expressed predominately in the kidney. Type IIb transporters are expressed in the small intestine and control the dietary uptake of phosphate.[15][16][17]
Three important regulatory hormones central to phosphate regulation are:
- Parathyroid hormone (PTH)[12]
- 1,25 dihydroxy vitamin D3 (1,25D)
- FGF23 (Fibroblast growth factor 23)
The role of each of these hormones on phosphate metabolism is complex (Figure 1: Phosphate Homeostasis) and incompletely understood. Phosphate regulation is intricately connected to calcium metabolism through regulation by these three important hormones.
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
Hypophosphatemia most commonly occurs by one of the following three mechanisms (table 1):
- Inadequate intestinal phosphate absorption
- Increased phosphate excretion
- Shift from extracellular phosphate into the intracellular space or into the mineral space of the bone.
| Causes of hypophosphatemia | ||
| Increased phosphate excretion | PTH dependent |
Primary hyperparathyroidism Secondary hyperparathyroidism Vitamin D deficiency |
| FGF23 dependent [18] |
X-linked hypophosphatemic rickets (XLH) Autosomal dominant hypophosphatemic rickets (ADHR) Autosomal recessive hypophosphatemic rickets (ARHR) Tumor-induced osteomalacia (TIO) seen in mesenchymal tumors Fibrous dysplasia (FD)/McCune Albright Syndrome (MAS) Neurofibromatosis (NF) Hypophosphatemic rickets and hyperparathyroidism (HRHPT) |
|
| Intrinsic renal causes |
Fanconi syndrome Acute kidney injury Recovery from acute tubular necrosis Renal transplant Sodium-phosphate cotransporter mutations |
|
| Drugs [19][20][21] |
Diuretics Corticosteroids Carbonic anhydrase inhibitors Bisphosphonates Estrogen Acyclovir Imatinib Denosumab |
|
| Inadequate intestinal phosphate absorption | Decreased intake |
Malnutrition Alcoholism |
| Malabsorption |
Bariatric surgery Crohn’s disease Chronic diarrhea |
|
| Drugs [19] |
Phosphate–binding antacids and binders |
|
| Redistribution | Intracellular shift |
Respiratory alkalosis Refeeding syndrome Insulin therapy for hyperglycemia Intravenous glucose therapy Catecholamines (epinephrine, albuterol, terbutaline, dopamine) Thyrotoxic periodic paralysis |
| Shift into the bone matrix |
Post-parathyroidectomy (Hungry bone syndrome) Use of cinacalcet for hyperparathyroidism [22] |
|
| Other drugs [19] |
Alcohol (induced metabolic acidosis) Phenytoin, phenobarbital (causing vitamin D deficiency or resistance) Intravenous iron Acetaminophen poisoning |
|
| Other causes |
Diabetic ketoacidosis Poorly controlled diabetes Renal replacement therapy Prolonged fasting Partial hepatectomy (affecting Vitamin D-25 hydroxylation) |
PTH: Parathyroid hormone. FGF23: Fibroblast growth factor.
Epidemiology
Hypophosphatemia is typically asymptomatic, so it is difficult to estimate prevalence in the general population. However, 2.2%–3.1% of hospitalized patients and 29%–34% of ICU patients are documented with this condition.[23] It is much more prevalent in alcoholism, diabetic ketoacidosis, or sepsis, with a frequency of up to 80%.[24][25][26] The presence of severe hypophosphatemia is associated with increased mortality by almost four-fold.[27] The morbidity of hypophosphatemia is highly dependent on its etiology and severity.
Pathophysiology
Hypophosphatemia's most common causes are inadequate phosphate absorption, increased phosphate excretion, and a shift from extracellular phosphate into intracellular space.[28][29][30]
Inadequate Intake
Hypophosphatemia secondary to inadequate phosphate intake occurs in the setting of prolonged poor phosphate dietary phosphate sources, intestinal malabsorption, and intestinal binding by exogenous agents. Almost all diet types contain a surplus of phosphate sufficient to maintain needs. Additionally, renal adaptations typically can compensate for short-term deficiency.
Gastrointestinal Loss
Activated vitamin D (1,25D) is crucial to intestinal absorption of both calcium and phosphorus. Conditions associated with decreased 1,25D include chronic kidney disease (CKD), hepatic dysfunction, and hypercalcemia. Intestinal malabsorption may be due to a variety of causes. Notably, chronic diarrhea has been shown to increase phosphate losses. Certain medications, such as antacids containing aluminum, magnesium, and calcium, are known to bind with phosphate, decreasing the available free ion form by creating non-absorbable phosphate salts. Phosphate binders are also used in CKD to decrease the ever-present hyperphosphatemia, but overuse can lead to low phosphorus.
Renal Loss
Increased excretion of phosphate occurs primarily in the renal system. The proximal renal tubule normally reabsorbs about 70% to 80% of filtered phosphate, and the distal tubule reabsorbs about 15% of filtered phosphate.[15] Phosphate reabsorption is stimulated by low serum phosphate concentration, which directly triggers the sodium-phosphate cotransporters of the proximal tubule and increases the expression and formation of new sodium-phosphate cotransporters. Conversely, the parathyroid hormone increases phosphate excretion by inhibiting the activity of sodium-phosphate cotransporters.
FGF23, along with its cofactor Klotho, decreases the expression of sodium-phosphorus cotransporter 2 in the proximal tubule. Osteocytes secrete FGF23 in response to increased phosphorus and 1,25D; FGF23 also reduces 1,25D and PTH levels. FGF23 is highly linked to CKD progression, left ventricular hypertrophy, and cardiovascular disease, and levels are increased prior to detectable abnormalities of serum phosphate or PTH. Therefore, FGF23 may be a more sensitive early test for these conditions than either abnormal phosphorus or PTH.[31][32][33] To a much lesser degree, fibroblast growth factor 7, extracellular matrix phosphoglycoprotein, and secreted frizzled-related protein-4 are also associated with phosphaturia.[34] Classic examples of increased FGF23 activity are X-linked hypophosphatemic rickets (XLH) and tumor-induced osteomalacia (TIO). XLH is an inherited form of rickets associated with mutations in the PHEX gene. TIO is an acquired paraneoplastic condition leading to overproduction of FGF23.[10]
Primary renal phosphate-wasting syndromes also exist related to structural tubular abnormalities causing defects in the sodium-phosphate cotransporter or defects in general proximal tubular reabsorption causing renal wasting of electrolytes and other substances.
- Mutations in the sodium-phosphate cotransporter gene SLC34A3 cause type 2c sodium-phosphate cotransporter failure, and mutations in the SLC34A1 gene cause defective type 2a sodium-phosphate cotransporters.[35]
- The sodium-hydrogen exchanger regulatory factor 1 creates the sodium gradient, which powers most ion reabsorption. Mutations in this lead to losses of many ions and substances, including phosphate.[36]
- Fanconi syndrome is another classic cause of renal loss.[37] It is a generalized impairment in proximal tubular function leading to urinary wasting. Often, this is due to medications, hereditary conditions, or illnesses, such as multiple myeloma (where immunoglobulin light chains induce renal tubular damage) or Wilson disease (associated with copper accumulation in tubules.)[38][39][40]
- Polyuria also will lead to obligate phosphate loss; causes include glucosuria, alcohol, lithium, diuretics, and rapid fluid volume expansion from oral or intravenous fluids.[19]
Hyperparathyroidism
Primary and secondary hyperparathyroidism are common causes of hypophosphatemia in the outpatient setting. Primary hyperparathyroidism is due to an increased production of PTH from a parathyroid adenoma, resulting in hypercalcemia and hypophosphatemia. Secondary hyperparathyroidism results from multiple other causes, including vitamin D deficiency, hypocalcemia, and chronic renal failure. PTH induces renal phosphate loss via the NaPi IIa co-transporter in the proximal tubule.[41] Secondary hyperparathyroidism has additional factors leading to low phosphate levels based on the underlying cause.
Redistribution
Intracellular shifting of phosphate stores may occur in a variety of clinical scenarios. Refeeding syndrome occurs when a patient who has been starved of nutrition (and depleted of total body phosphate) suddenly is replenished with carbohydrates, proteins, and lipids. The body begins to process the newfound foods to produce ATP for energy, and insulin and glucose help drive phosphate intracellularly.[42] Cells uptake all available free phosphate, leading to profound hypophosphatemia. In another redistribution physiology, hungry bone syndrome occurs after the correction of hyperparathyroidism, where osteopenic bones begin to reabsorb and store phosphate and calcium. This leads to increased bone demand for these ions and hypophosphatemia.[43] Acute respiratory alkalosis induces hypophosphatemia via changes in cellular pH. Increased pH stimulates phosphofructokinase, thus stimulating glycolysis to produce ATP, thus consuming phosphate from the cellular space. Serum phosphate is shifted intracellularly to meet this demand. While typically mild, extreme hyperventilation with subsequent PCO2 changes to less than 20 mmHg can lower phosphate concentrations to below 1 mg/dL (or 0.32 mmol/L). This is thought to be a common cause of marked hypophosphatemia in hospitalized patients.[44][45]
End Stage Renal Disease
In patients with renal failure, hypophosphatemia can be seen as a result of dialysis therapy removing phosphate in bulk.[46][47] Patients with renal transplants are also prone to hypophosphatemia due to a combination of hypercalcemia, hypophosphatemia, and tertiary hyperparathyroidism.[48]
History and Physical
Most patients with hypophosphatemia are asymptomatic, and the most common symptom is generalized mild to moderate weakness. The history of presenting illness will sometimes indicate possible hypophosphatemia. Associated conditions include poor nutritional status, intestinal malabsorption, frequent or recurrent bone pain, bone fractures, history or suspicion of multiple myeloma, parenteral nutrition supplementation, diabetic ketoacidosis, and medication use—including chronic glucocorticoids, antacids, cisplatin, or pamidronate.
Clinical symptoms usually appear with moderate or severe hypophosphatemia. Neurologic symptoms include altered mental status, dysarthria, neurological instability including seizures, and focal neurologic findings such as numbness or reflexive weakness. Cardiac and respiratory failure can occur in severe cases. In addition to muscle weakness, muscle pain may signify hypophosphatemia-related rhabdomyolysis.[23]
Evaluation
Hypophosphatemia is diagnosed with a simple serum measurement. It is important to remember that measured serum phosphate represents only 1% of total body phosphate, so small fluctuations can be a portent of serious depletion.
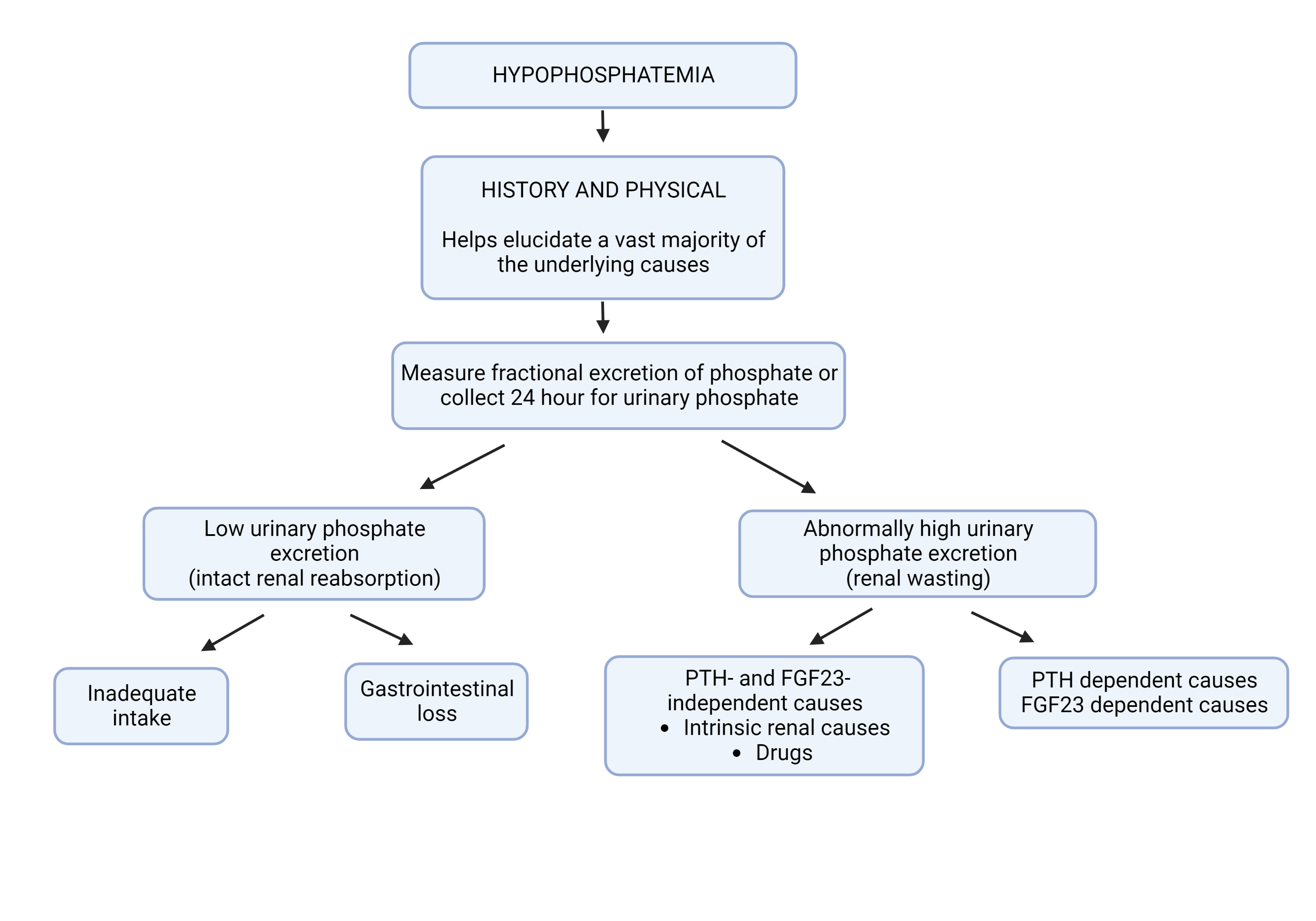
Etiology is typically evident from history. However, if unknown, it is essential to determine renal phosphate excretion. Renal phosphate excretion can be measured either from a 24-hour urine collection or by calculation of the fractional excretion of filtered phosphate (FEPO4) from a random urine specimen. FEPO4 is calculated as follows—where U represents urine values, and P represents plasma values of phosphate (PO4) and creatinine (Cr):
- FEPO4 = (UPO4 x PCr x 100) / (PPO4 x UCr)
A 24-hour urine phosphate excretion of less than 100 mg or FEPO4 less than 5% shows decreased phosphate excretion, indicating hypophosphatemia is from a redistribution within the body or decreased intestinal absorption. A 24-hour urine phosphate excretion greater than 100 mg or FEPO4 greater than 5% indicates renal phosphate wasting. Hypophosphatemia in this scenario is likely due to hyperparathyroidism or vitamin D deficiency (Figure 2: Work-up of unexplained hypophosphatemia).
Treatment / Management
The effects of hypophosphatemia are broad and impact nearly every system. Symptoms of this deficiency become apparent below 1 mg/dL (or 0.32 mmol/L).[49][50] While symptoms are not clinically present in mild/moderate cases, it is essential to address and replace phosphate whenever abnormalities are noted. The appropriate regimen for replacement is determined depending on clinical symptoms.
Treatment of Hypophosphatemia with Oral Replacement Regimens
- Serum phosphorus >1.5 mg/dL: Total dose 1 mmol/kg (maximum 80 mmol) in 3-4 divided doses in 24 hours
- Serum phosphorus <1.5 mg/dL: Total dose 1.3 mmol/kg (maximum 100 mmol) in 3-4 divided doses in 24 hours
Severe, symptomatic cases are appropriate for intravenous phosphate if the serum phosphate is less than 1 mg/dL (or 0.32 mmol/L) and should be changed to oral replacement when the serum phosphate exceeds 2 mg/dL (or 0.48 mmol/L). Intravenous phosphate replacement can also be used in patients who cannot take oral medications or tolerate oral replacement.[51](B3)
Treatment of Hypophosphatemia with Intravenous Replacement Regimens
Commonly used preparations: Sodium phosphate and potassium phosphate, containing equivalent phosphate content of 0.011 g/mL.
- Serum phosphorus <1 mg/dL: 0.6 mmol/kg over 6 hours
- Serum phosphorus 1.0-1.7 mg/dL: 0.4 mmol/kg over 6 hours
- Serum phosphorus 1.7-2.2 mg/dL: 0.2 mmol/kg over 6 hours
With renal dysfunction, the phosphate replacement dose should be reduced by 50%. In patients greater than 130% of their ideal body weight, adjusted body weight should be used to calculate the doses.[51](B3)
Differential Diagnosis
The most common manifestation is generalized weakness. As such, any other electrolyte aberrations should also be suspected, including hypokalemia, hypocalcemia and hypomagnesemia.
Additionally, the following conditions can mimic hypophosphatemia: benzodiazepine toxicity, delirium tremens, Guillain-Barre syndrome, hypothyroidism, hyperparathyroidism, insulin overdose, myopathies, primary muscle disorders, rhabdomyolysis, multiple myeloma, uremic encephalopathy.
Prognosis
A vast majority of the patients with hypophosphatemia have mild to moderate hypophosphatemia and will recover without complications. Even patients with severe hypophosphatemia can avoid complications if this condition is recognized early and the replacement of phosphate is promptly initiated. The presence of severe hypophosphatemia has been shown to be associated with significantly higher mortality rate in critically ill patients with sepsis or septic shock; whether this is due to hypophosphatemia alone or in combination with other factors remains to be determined.[52][53][54]
Complications
Severe hypophosphatemia (<1 mg/dL) can be associated with a wide range of adverse clinical outcomes, especially if prolonged and there is a delay in management. The most significant effects are seen in the bones, but many other systems are also affected.
Bone: Prolonged hypophosphatemia leads to osteopenia, osteoporosis, rickets, or osteomalacia due to decreased bone mineralization and a resultant increase in bone fractures.[4][55] Dental abnormalities such as periodontitis are also seen.[10]
Muscle: Generalized muscular weakness can occur. Intracellular ATP depletion and mitochondrial dysfunction can cause rhabdomyolysis, resulting in renal injury and increased creatinine phosphokinases. This is especially pronounced in chronic hypophosphatemia, such as with alcohol use disorder.[56][57]
Central Nervous System: The central nervous system may manifest with metabolic encephalopathy due to ATP depletion and may include an altered mental state, irritability, paresthesias, numbness, seizures, or coma.[58][59]
Cardiac: Cardiac function is impacted by ATP depletion. The myocytes become less stable, and arrhythmias become more likely.[60][61] Some studies show a significantly increased risk of heart failure, possibly related to lower levels of 2,3 DPG in erythrocytes and depleted ATP causing cardiomyopathy.[10]
Respiratory: Respiratory depression is associated with hypophosphatemia.[62] Decreased diaphragmatic function impacts pulmonary function with subsequent hypoventilation. Ventilator-dependent patients are shown to have longer hospital courses and worse outcomes when hypophosphatemia is present.[62][63][64]
Gastrointestinal: Dysfunction occurs as a result of ATP deficiency, with possible dysphagia, ileus, or constipation.[4]
Hematologic: Depletion of ATP within erythrocytes can impair oxygen-carrying ability and result in hemolytic anemia. In addition, phosphorus is necessary for 2,3-diphosphoglycerate formation, facilitating oxygen release from hemoglobin.[65][66]
Deterrence and Patient Education
Patients should be instructed to keep track of their diet and water intake. A diet rich in phosphate should be encouraged, including dairy products, meat, poultry, and peanuts. They should follow up with the physician as advised or if any of these problems arise:
- Confusion
- Irritable behavior
- Muscle pain
- Nausea, vomiting, or diarrhea
- Constipation lasting longer than 2 days
Pearls and Other Issues
- The primary causes of hypophosphatemia are gastrointestinal malabsorption and increased renal excretion.
- The primary hormones involved are PTH, 1,25D, and FGF23 (with its cofactor Klotho).
- FGF23 is emerging as a significant predictor of cardiovascular mortality and CKD progression.
- FGF23 decreases expression of sodium phosphate cotransporters 2 (NaPi II) and downregulates PTH and 1,25D.
- In contrast, PTH increases expression of 1,25D.
- PTH is primarily regulated by serum calcium levels.
- Intestinal absorption of calcium and phosphorus is mediated by 1,25D.
- Hypophosphatemia can weaken the heart and respiratory muscles, leading to CHF and ventilator dependence.
Enhancing Healthcare Team Outcomes
Because of the diverse presentation of hypophosphatemia, this condition is best managed by an interprofessional team that consists of an internist, endocrinologist, intensivist, radiologist, and nephrologist, as needed. Pharmacists review oral and parenteral therapy, including dosage and drug compatibility. The effects of hypophosphatemia are broad and impact nearly every system. Symptoms of this deficiency become apparent below 1 mg/dL (or 0.32 mmol/L). In general, all symptomatic patients need treatment with phosphate; asymptomatic patients may be observed and monitored.
Providing patient-centered care for individuals with hypophosphatemia requires a collaborative effort among healthcare professionals, including physicians, advanced practice practitioners, nurses, pharmacists, and others. First and foremost, healthcare providers must possess the necessary clinical skills and expertise when diagnosing, evaluating, and treating this condition. This includes proficiency in interpreting laboratory findings and understanding the interplay among serum markers, urine electrolytes, and hormonal regulators. Moreover, a strategic approach involving evidence-based guidelines and individualized care plans tailored to each patient's unique circumstances is vital.
Responsibilities within the interprofessional team should be clearly defined, with each member contributing their specialized knowledge and skills to optimize patient care. Effective interprofessional communication fosters a collaborative environment where information is shared, questions are encouraged, and concerns are addressed promptly.
Lastly, care coordination is pivotal in ensuring seamless and efficient patient care. Physicians, advanced practitioners, nurses, pharmacists, and other healthcare professionals must work together to streamline the patient's journey, from diagnosis through treatment and follow-up. This coordination minimizes errors, reduces delays, and enhances patient safety, ultimately leading to improved outcomes and patient-centered care that prioritizes the well-being and satisfaction of those affected by hypophosphatemia. The outlook for patients depends on the primary condition causing hypophosphatemia. If the cause is benign, then treatment outcomes are excellent.
Media
(Click Image to Enlarge)
Phosphate homeostasis. FGF23: fibroblast growth factor 23. PTH: parathyroid hormone. NaPi: Sodium phosphate co-transporter. Contributed by Dr. Jasleen Kaur. Generated using Biorender.
(Click Image to Enlarge)
Work-up of unexplained hypophostatemia Contributed by Jasleen Kaur, MD. Generated using Biorender.
References
Takeda E, Taketani Y, Sawada N, Sato T, Yamamoto H. The regulation and function of phosphate in the human body. BioFactors (Oxford, England). 2004:21(1-4):345-55 [PubMed PMID: 15630224]
Peacock M. Phosphate Metabolism in Health and Disease. Calcified tissue international. 2021 Jan:108(1):3-15. doi: 10.1007/s00223-020-00686-3. Epub 2020 Apr 7 [PubMed PMID: 32266417]
Bonora M,Patergnani S,Rimessi A,De Marchi E,Suski JM,Bononi A,Giorgi C,Marchi S,Missiroli S,Poletti F,Wieckowski MR,Pinton P, ATP synthesis and storage. Purinergic signalling. 2012 Sep; [PubMed PMID: 22528680]
Amanzadeh J, Reilly RF Jr. Hypophosphatemia: an evidence-based approach to its clinical consequences and management. Nature clinical practice. Nephrology. 2006 Mar:2(3):136-48 [PubMed PMID: 16932412]
Koljonen L, Enlund-Cerullo M, Hauta-Alus H, Holmlund-Suila E, Valkama S, Rosendahl J, Andersson S, Pekkinen M, Mäkitie O. Phosphate Concentrations and Modifying Factors in Healthy Children From 12 to 24 Months of Age. The Journal of clinical endocrinology and metabolism. 2021 Sep 27:106(10):2865-2875. doi: 10.1210/clinem/dgab495. Epub [PubMed PMID: 34214153]
Haider DG, Lindner G, Wolzt M, Ahmad SS, Sauter T, Leichtle AB, Fiedler GM, Fuhrmann V, Exadaktylos AK. Hyperphosphatemia Is an Independent Risk Factor for Mortality in Critically Ill Patients: Results from a Cross-Sectional Study. PloS one. 2015:10(8):e0133426. doi: 10.1371/journal.pone.0133426. Epub 2015 Aug 7 [PubMed PMID: 26252874]
Level 2 (mid-level) evidenceKing AL, Sica DA, Miller G, Pierpaoli S. Severe hypophosphatemia in a general hospital population. Southern medical journal. 1987 Jul:80(7):831-5 [PubMed PMID: 3603104]
Berger MM, Appelberg O, Reintam-Blaser A, Ichai C, Joannes-Boyau O, Casaer M, Schaller SJ, Gunst J, Starkopf J, ESICM-MEN section. Prevalence of hypophosphatemia in the ICU - Results of an international one-day point prevalence survey. Clinical nutrition (Edinburgh, Scotland). 2021 May:40(5):3615-3621. doi: 10.1016/j.clnu.2020.12.017. Epub 2020 Dec 29 [PubMed PMID: 33454128]
Level 3 (low-level) evidenceWang L, Xiao C, Chen L, Zhang X, Kou Q. Impact of hypophosphatemia on outcome of patients in intensive care unit: a retrospective cohort study. BMC anesthesiology. 2019 May 24:19(1):86. doi: 10.1186/s12871-019-0746-2. Epub 2019 May 24 [PubMed PMID: 31122196]
Level 2 (mid-level) evidenceKim KJ, Song JE, Kim JH, Hong N, Kim SG, Lee J, Rhee Y. Elevated morbidity and mortality in patients with chronic idiopathic hypophosphatemia: a nationwide cohort study. Frontiers in endocrinology. 2023:14():1229750. doi: 10.3389/fendo.2023.1229750. Epub 2023 Aug 10 [PubMed PMID: 37635983]
Uribarri J, Calvo MS. Hidden sources of phosphorus in the typical American diet: does it matter in nephrology? Seminars in dialysis. 2003 May-Jun:16(3):186-8 [PubMed PMID: 12753675]
Peacock M. Calcium metabolism in health and disease. Clinical journal of the American Society of Nephrology : CJASN. 2010 Jan:5 Suppl 1():S23-30. doi: 10.2215/CJN.05910809. Epub [PubMed PMID: 20089499]
Level 3 (low-level) evidenceStremke ER, Hill Gallant KM. Intestinal Phosphorus Absorption in Chronic Kidney Disease. Nutrients. 2018 Sep 23:10(10):. doi: 10.3390/nu10101364. Epub 2018 Sep 23 [PubMed PMID: 30249044]
Berndt T, Kumar R. Novel mechanisms in the regulation of phosphorus homeostasis. Physiology (Bethesda, Md.). 2009 Feb:24():17-25. doi: 10.1152/physiol.00034.2008. Epub [PubMed PMID: 19196648]
Level 3 (low-level) evidenceBlaine J, Chonchol M, Levi M. Renal control of calcium, phosphate, and magnesium homeostasis. Clinical journal of the American Society of Nephrology : CJASN. 2015 Jul 7:10(7):1257-72. doi: 10.2215/CJN.09750913. Epub 2014 Oct 6 [PubMed PMID: 25287933]
Level 3 (low-level) evidenceSegawa H, Shiozaki Y, Kaneko I, Miyamoto K. The Role of Sodium-Dependent Phosphate Transporter in Phosphate Homeostasis. Journal of nutritional science and vitaminology. 2015:61 Suppl():S119-21. doi: 10.3177/jnsv.61.S119. Epub [PubMed PMID: 26598821]
Lederer E. Regulation of serum phosphate. The Journal of physiology. 2014 Sep 15:592(18):3985-95. doi: 10.1113/jphysiol.2014.273979. Epub 2014 Jun 27 [PubMed PMID: 24973411]
Level 3 (low-level) evidenceHuang X, Jiang Y, Xia W. FGF23 and Phosphate Wasting Disorders. Bone research. 2013 Jun:1(2):120-32. doi: 10.4248/BR201302002. Epub 2013 Jun 28 [PubMed PMID: 26273497]
Liamis G, Milionis HJ, Elisaf M. Medication-induced hypophosphatemia: a review. QJM : monthly journal of the Association of Physicians. 2010 Jul:103(7):449-59. doi: 10.1093/qjmed/hcq039. Epub 2010 Mar 30 [PubMed PMID: 20356849]
Kaur U, Chakrabarti SS, Gambhir IS. Zoledronate Induced Hypocalcemia and Hypophosphatemia in Osteoporosis: A Cause of Concern. Current drug safety. 2016:11(3):267-9 [PubMed PMID: 27113952]
Clark SL, Nystrom EM. A Case of Severe, Prolonged, Refractory Hypophosphatemia After Zoledronic Acid Administration. Journal of pharmacy practice. 2016 Apr:29(2):172-6. doi: 10.1177/0897190015624050. Epub 2016 Jan 5 [PubMed PMID: 26739479]
Level 3 (low-level) evidenceNowack R, Wachtler P. Hypophosphatemia and hungry bone syndrome in a dialysis patient with secondary hyperparathyroidism treated with cinacalcet--proposal for an improved monitoring. Clinical laboratory. 2006:52(11-12):583-7 [PubMed PMID: 17175888]
Level 3 (low-level) evidenceKalantar-Zadeh K, Ganz T, Trumbo H, Seid MH, Goodnough LT, Levine MA. Parenteral iron therapy and phosphorus homeostasis: A review. American journal of hematology. 2021 May 1:96(5):606-616. doi: 10.1002/ajh.26100. Epub 2021 Feb 9 [PubMed PMID: 33471363]
van der Vaart A, Waanders F, van Beek AP, Vriesendorp TM, Wolffenbutel BHR, van Dijk PR. Incidence and determinants of hypophosphatemia in diabetic ketoacidosis: an observational study. BMJ open diabetes research & care. 2021 Feb:9(1):. doi: 10.1136/bmjdrc-2020-002018. Epub [PubMed PMID: 33597187]
Level 2 (mid-level) evidenceBarak V, Schwartz A, Kalickman I, Nisman B, Gurman G, Shoenfeld Y. Prevalence of hypophosphatemia in sepsis and infection: the role of cytokines. The American journal of medicine. 1998 Jan:104(1):40-7 [PubMed PMID: 9528718]
Gaasbeek A, Meinders AE. Hypophosphatemia: an update on its etiology and treatment. The American journal of medicine. 2005 Oct:118(10):1094-101 [PubMed PMID: 16194637]
Camp MA, Allon M. Severe hypophosphatemia in hospitalized patients. Mineral and electrolyte metabolism. 1990:16(6):365-8 [PubMed PMID: 2089250]
Level 2 (mid-level) evidenceUday S, Sakka S, Davies JH, Randell T, Arya V, Brain C, Tighe M, Allgrove J, Arundel P, Pryce R, Högler W, Shaw NJ. Elemental formula associated hypophosphataemic rickets. Clinical nutrition (Edinburgh, Scotland). 2019 Oct:38(5):2246-2250. doi: 10.1016/j.clnu.2018.09.028. Epub 2018 Sep 28 [PubMed PMID: 30314926]
Christov M, Jüppner H. Phosphate homeostasis disorders. Best practice & research. Clinical endocrinology & metabolism. 2018 Oct:32(5):685-706. doi: 10.1016/j.beem.2018.06.004. Epub 2018 Jun 18 [PubMed PMID: 30449549]
Jacquillet G, Unwin RJ. Physiological regulation of phosphate by vitamin D, parathyroid hormone (PTH) and phosphate (Pi). Pflugers Archiv : European journal of physiology. 2019 Jan:471(1):83-98. doi: 10.1007/s00424-018-2231-z. Epub 2018 Nov 5 [PubMed PMID: 30393837]
Isakova T, Wahl P, Vargas GS, Gutiérrez OM, Scialla J, Xie H, Appleby D, Nessel L, Bellovich K, Chen J, Hamm L, Gadegbeku C, Horwitz E, Townsend RR, Anderson CA, Lash JP, Hsu CY, Leonard MB, Wolf M. Fibroblast growth factor 23 is elevated before parathyroid hormone and phosphate in chronic kidney disease. Kidney international. 2011 Jun:79(12):1370-8. doi: 10.1038/ki.2011.47. Epub 2011 Mar 9 [PubMed PMID: 21389978]
Kurpas A, Supeł K, Idzikowska K, Zielińska M. FGF23: A Review of Its Role in Mineral Metabolism and Renal and Cardiovascular Disease. Disease markers. 2021:2021():8821292. doi: 10.1155/2021/8821292. Epub 2021 May 17 [PubMed PMID: 34055103]
Gutiérrez OM, Mannstadt M, Isakova T, Rauh-Hain JA, Tamez H, Shah A, Smith K, Lee H, Thadhani R, Jüppner H, Wolf M. Fibroblast growth factor 23 and mortality among patients undergoing hemodialysis. The New England journal of medicine. 2008 Aug 7:359(6):584-92. doi: 10.1056/NEJMoa0706130. Epub [PubMed PMID: 18687639]
Level 2 (mid-level) evidencePrasad N, Bhadauria D. Renal phosphate handling: Physiology. Indian journal of endocrinology and metabolism. 2013 Jul:17(4):620-7. doi: 10.4103/2230-8210.113752. Epub [PubMed PMID: 23961477]
Lederer E, Miyamoto K. Clinical consequences of mutations in sodium phosphate cotransporters. Clinical journal of the American Society of Nephrology : CJASN. 2012 Jul:7(7):1179-87. doi: 10.2215/CJN.09090911. Epub 2012 Apr 19 [PubMed PMID: 22516291]
King AJ, Siegel M, He Y, Nie B, Wang J, Koo-McCoy S, Minassian NA, Jafri Q, Pan D, Kohler J, Kumaraswamy P, Kozuka K, Lewis JG, Dragoli D, Rosenbaum DP, O'Neill D, Plain A, Greasley PJ, Jönsson-Rylander AC, Karlsson D, Behrendt M, Strömstedt M, Ryden-Bergsten T, Knöpfel T, Pastor Arroyo EM, Hernando N, Marks J, Donowitz M, Wagner CA, Alexander RT, Caldwell JS. Inhibition of sodium/hydrogen exchanger 3 in the gastrointestinal tract by tenapanor reduces paracellular phosphate permeability. Science translational medicine. 2018 Aug 29:10(456):. doi: 10.1126/scitranslmed.aam6474. Epub [PubMed PMID: 30158152]
Ansermet C, Moor MB, Centeno G, Auberson M, Hu DZ, Baron R, Nikolaeva S, Haenzi B, Katanaeva N, Gautschi I, Katanaev V, Rotman S, Koesters R, Schild L, Pradervand S, Bonny O, Firsov D. Renal Fanconi Syndrome and Hypophosphatemic Rickets in the Absence of Xenotropic and Polytropic Retroviral Receptor in the Nephron. Journal of the American Society of Nephrology : JASN. 2017 Apr:28(4):1073-1078. doi: 10.1681/ASN.2016070726. Epub 2016 Oct 31 [PubMed PMID: 27799484]
Mandry JM, Posner MR, Tucci JR, Eil C. Hyperphosphatemia in multiple myeloma due to a phosphate-binding immunoglobulin. Cancer. 1991 Sep 1:68(5):1092-4 [PubMed PMID: 1913479]
Level 3 (low-level) evidenceGoyal JP, Kumar N, Rao SS, Shah VB. Wilson's Disease Presenting as Resistant Rickets. Gastroenterology research. 2011 Feb:4(1):34-35 [PubMed PMID: 27957011]
Subrahmanyam DK, Vadivelan M, Giridharan S, Balamurugan N. Wilson's disease - A rare cause of renal tubular acidosis with metabolic bone disease. Indian journal of nephrology. 2014 May:24(3):171-4. doi: 10.4103/0971-4065.132017. Epub [PubMed PMID: 25120295]
Level 3 (low-level) evidenceDéliot N, Hernando N, Horst-Liu Z, Gisler SM, Capuano P, Wagner CA, Bacic D, O'Brien S, Biber J, Murer H. Parathyroid hormone treatment induces dissociation of type IIa Na+-P(i) cotransporter-Na+/H+ exchanger regulatory factor-1 complexes. American journal of physiology. Cell physiology. 2005 Jul:289(1):C159-67 [PubMed PMID: 15788483]
Level 3 (low-level) evidenceMehanna HM, Moledina J, Travis J. Refeeding syndrome: what it is, and how to prevent and treat it. BMJ (Clinical research ed.). 2008 Jun 28:336(7659):1495-8. doi: 10.1136/bmj.a301. Epub [PubMed PMID: 18583681]
Witteveen JE, van Thiel S, Romijn JA, Hamdy NA. Hungry bone syndrome: still a challenge in the post-operative management of primary hyperparathyroidism: a systematic review of the literature. European journal of endocrinology. 2013 Mar:168(3):R45-53. doi: 10.1530/EJE-12-0528. Epub 2013 Feb 20 [PubMed PMID: 23152439]
Level 1 (high-level) evidenceHoppe A, Metler M, Berndt TJ, Knox FG, Angielski S. Effect of respiratory alkalosis on renal phosphate excretion. The American journal of physiology. 1982 Nov:243(5):F471-5 [PubMed PMID: 6291407]
Level 3 (low-level) evidencePaleologos M, Stone E, Braude S. Persistent, progressive hypophosphataemia after voluntary hyperventilation. Clinical science (London, England : 1979). 2000 May:98(5):619-25 [PubMed PMID: 10781395]
Tejeda A, Saffarian N, Uday K, Dave M. Hypophosphatemia in end stage renal disease. Nephron. 1996:73(4):674-8 [PubMed PMID: 8856268]
Level 3 (low-level) evidenceHendrix RJ, Hastings MC, Samarin M, Hudson JQ. Predictors of Hypophosphatemia and Outcomes during Continuous Renal Replacement Therapy. Blood purification. 2020:49(6):700-707. doi: 10.1159/000507421. Epub 2020 Apr 22 [PubMed PMID: 32320987]
Torregrosa JV, Ferreira AC, Cucchiari D, Ferreira A. Bone Mineral Disease After Kidney Transplantation. Calcified tissue international. 2021 Apr:108(4):551-560. doi: 10.1007/s00223-021-00837-0. Epub 2021 Mar 25 [PubMed PMID: 33765230]
Halevy J, Bulvik S. Severe hypophosphatemia in hospitalized patients. Archives of internal medicine. 1988 Jan:148(1):153-5 [PubMed PMID: 3122679]
Lentz RD, Brown DM, Kjellstrand CM. Treatment of severe hypophosphatemia. Annals of internal medicine. 1978 Dec:89(6):941-4 [PubMed PMID: 102230]
Felsenfeld AJ, Levine BS. Approach to treatment of hypophosphatemia. American journal of kidney diseases : the official journal of the National Kidney Foundation. 2012 Oct:60(4):655-61. doi: 10.1053/j.ajkd.2012.03.024. Epub 2012 Aug 3 [PubMed PMID: 22863286]
Level 3 (low-level) evidenceAl Harbi SA, Al-Dorzi HM, Al Meshari AM, Tamim H, Abdukahil SAI, Sadat M, Arabi Y. Association between phosphate disturbances and mortality among critically ill patients with sepsis or septic shock. BMC pharmacology & toxicology. 2021 May 28:22(1):30. doi: 10.1186/s40360-021-00487-w. Epub 2021 May 28 [PubMed PMID: 34049590]
Shor R, Halabe A, Rishver S, Tilis Y, Matas Z, Fux A, Boaz M, Weinstein J. Severe hypophosphatemia in sepsis as a mortality predictor. Annals of clinical and laboratory science. 2006 Winter:36(1):67-72 [PubMed PMID: 16501239]
Miller CJ, Doepker BA, Springer AN, Exline MC, Phillips G, Murphy CV. Impact of Serum Phosphate in Mechanically Ventilated Patients With Severe Sepsis and Septic Shock. Journal of intensive care medicine. 2020 May:35(5):485-493. doi: 10.1177/0885066618762753. Epub 2018 Mar 8 [PubMed PMID: 29519205]
Prié D, Huart V, Bakouh N, Planelles G, Dellis O, Gérard B, Hulin P, Benqué-Blanchet F, Silve C, Grandchamp B, Friedlander G. Nephrolithiasis and osteoporosis associated with hypophosphatemia caused by mutations in the type 2a sodium-phosphate cotransporter. The New England journal of medicine. 2002 Sep 26:347(13):983-91 [PubMed PMID: 12324554]
Level 3 (low-level) evidenceFunabiki Y, Tatsukawa H, Ashida K, Matsubara K, Kubota Y, Uwatoko H, Kitamura K. Disturbance of consciousness associated with hypophosphatemia in a chronically alcoholic patient. Internal medicine (Tokyo, Japan). 1998 Nov:37(11):958-61 [PubMed PMID: 9868960]
Level 3 (low-level) evidenceSinghal PC, Kumar A, Desroches L, Gibbons N, Mattana J. Prevalence and predictors of rhabdomyolysis in patients with hypophosphatemia. The American journal of medicine. 1992 May:92(5):458-64 [PubMed PMID: 1580292]
Junge O. [Acute polyneuropathy due to phosphate deficiency during parenteral feeding]. Fortschritte der Medizin. 1979 Feb 22:97(8):335-8 [PubMed PMID: 105978]
Level 3 (low-level) evidenceImel EA, Econs MJ. Approach to the hypophosphatemic patient. The Journal of clinical endocrinology and metabolism. 2012 Mar:97(3):696-706. doi: 10.1210/jc.2011-1319. Epub [PubMed PMID: 22392950]
Level 3 (low-level) evidenceFuller TJ, Nichols WW, Brenner BJ, Peterson JC. Reversible depression in myocardial performance in dogs with experimental phosphorus deficiency. The Journal of clinical investigation. 1978 Dec:62(6):1194-1200 [PubMed PMID: 748374]
Level 3 (low-level) evidenceDavis SV, Olichwier KK, Chakko SC. Reversible depression of myocardial performance in hypophosphatemia. The American journal of the medical sciences. 1988 Mar:295(3):183-7 [PubMed PMID: 3354591]
Gravelyn TR, Brophy N, Siegert C, Peters-Golden M. Hypophosphatemia-associated respiratory muscle weakness in a general inpatient population. The American journal of medicine. 1988 May:84(5):870-6 [PubMed PMID: 3364446]
Zhao Y, Li Z, Shi Y, Cao G, Meng F, Zhu W, Yang GE. Effect of hypophosphatemia on the withdrawal of mechanical ventilation in patients with acute exacerbations of chronic obstructive pulmonary disease. Biomedical reports. 2016 Apr:4(4):413-416 [PubMed PMID: 27073623]
Alsumrain MH, Jawad SA, Imran NB, Riar S, DeBari VA, Adelman M. Association of hypophosphatemia with failure-to-wean from mechanical ventilation. Annals of clinical and laboratory science. 2010 Spring:40(2):144-8 [PubMed PMID: 20421625]
Altuntas Y, Innice M, Basturk T, Seber S, Serin G, Ozturk B. Rhabdomyolysis and severe haemolytic anaemia, hepatic dysfunction and intestinal osteopathy due to hypophosphataemia in a patient after Billroth II gastrectomy. European journal of gastroenterology & hepatology. 2002 May:14(5):555-7 [PubMed PMID: 11984155]
Level 3 (low-level) evidenceMelvin JD, Watts RG. Severe hypophosphatemia: a rare cause of intravascular hemolysis. American journal of hematology. 2002 Mar:69(3):223-4 [PubMed PMID: 11891812]
Level 3 (low-level) evidence