Introduction
Hyperbilirubinemia is a condition defined as elevated serum or plasma bilirubin levels above the reference range of the laboratory, and it is due to disorders of bilirubin metabolism. Depending on the form of bilirubin present in serum, hyperbilirubinemia can be further classified as unconjugated (indirect) or conjugated (direct). Unconjugated hyperbilirubinemia (albumin-bound) usually results from increased production, impaired hepatic uptake, and decreased conjugation of bilirubin.[1][2] In neonates, jaundice typically occurs due to unconjugated hyperbilirubinemia, which is characterized by the increased levels of indirect or unconjugated bilirubin (UCB) in the serum. In newborns, the increased concentration of UCB can cross the blood-brain barrier, and deposit in the basal ganglia or cerebellum causing a bilirubin-induced encephalopathy or kernicterus[3]. Several inherited disorders can also produce unconjugated hyperbilirubinemia, including Gilbert syndrome, Crigler-Najjar syndromes type I and II, and inherited disorders causing hemolytic anemia.[4][5]
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
Bilirubin, a yellow-orange bile pigment, is the catabolic product of heme metabolism. Approximately 85 percent of the heme moiety comes from the hemoglobin degradation of red blood cells, while the remaining derives from the ineffective erythropoiesis and the breakdown of other hemoproteins such as cytochromes, myoglobin, and catalase.[6] The conversion of heme to bilirubin is a two-step reaction, in the first step the microsomal heme oxygenase enzyme of the reticuloendothelial system, converts heme to biliverdin, which in turn is reduced to unconjugated bilirubin (UCB) by a second enzyme biliverdin reductase.[7] The UCB is lipophilic. UCB tightly bound to albumin is transported to the liver. The entry of UCB into the liver has not been elucidated, and the best candidate appears to be a bilirubin transporter. In liver hepatocytes, UCB dissociates from albumin and binds to proteins of the glutathione-S-transferases family that present it for conjugation and prevent it from effluxing from the liver. Next, unconjugated bilirubin gets conjugated with one or two molecules of glucuronic acid by the enzyme uridine diphospho-glucuronate glucuronosyltransferase (UGT1A1), which forms bilirubin monoglucuronide and bilirubin diglucuronide respectively.[7][8] Conjugation increases the solubility of bilirubin in plasma and thereby enhances its elimination from the body. The conjugated bilirubin (CB) is then pumped into bile via an energy-requiring process requiring the multidrug resistance-associated protein 2 (MRP2).[9] This process also reduces the ability of bilirubin to diffuse across the blood-brain barrier. Therefore, unconjugated hyperbilirubinemia can result from dysfunction of any of these conjugation steps. In neonates, inefficient conjugation of bilirubin leads to unconjugated hyperbilirubinemia (physiologic neonatal jaundice). See Figure. Metabolic Pathway for Bilirubin in the Hepatocyte.
Unconjugated hyperbilirubinemia arises in one of the three major pathophysiologic conditions or a combination of them:
- Increased bilirubin production
- Impaired bilirubin uptake
- Impaired bilirubin conjugation
Increased bilirubin production and consequential unconjugated hyperbilirubinemia can result from increased catabolic degradation of hemoglobin and other heme proteins, typically due to accelerated hemolysis, a large hematoma, dyserythropoiesis (e.g., megaloblastic and sideroblastic anemias), or sometimes due to destruction of transfused erythrocytes. In these conditions, patients with normal liver function efficiently conjugate and excrete the excess bilirubin.[2][10] As a result, the serum levels of unconjugated bilirubin remain modest (1 to 4 mg/dL) and rarely exceed 4 mg/dL. Prolonged hemolysis can lead to severe unconjugated hyperbilirubinemia in patients with concurrent hepatic dysfunction.
The impaired hepatic uptake of bilirubin can be the result of decreased bilirubin delivery to the liver and inefficient uptake of bilirubin by hepatocytes, usually resulting from reduced hepatic blood flow (congestive heart failure and portosystemic shunts) and drugs/contrast administration. The unconjugated hyperbilirubinemia induced by several drugs (rifampin, flavaspidic acid, novobiocin, and various cholecystographic contrast agents), generally resolves within 48 hours of drug discontinuation.[11]
Impaired bilirubin conjugation can result from hereditary defects, including Gilbert syndrome and the Crigler-Najjar syndrome type I and II, that cause a decrease or loss of UDP-glucuronosyltransferase (UGT1A1) activity, an enzyme responsible for conjugation of bilirubin with glucuronic acid.[4] Lucy-Driscoll syndrome, also known as maternal serum jaundice, a form of transient familial neonatal unconjugated hyperbilirubinemia, is a rare metabolic disorder caused by a UGT1A1 inhibitor usually present in the maternal serum.[12] Most newborns develop unconjugated hyperbilirubinemia (neonatal jaundice) because of hepatic immaturity and low activity of UGT1A1 during days 2 to 5. Breast milk feeding increases bilirubin levels in infants which results in maternal milk jaundice.[13][14] Lactation failure also results in hyperbilirubinemia due to insufficient caloric intake, which results in decreased bilirubin clearance, and increased enterohepatic circulation.[15] Drugs such as novobiocin, pregnanediol, chloramphenicol, gentamycin, and several HIV protease inhibitors can induce hyperbilirubinemia by inhibiting the UGT1A1 enzyme.[16] In newborns, ABO/Rh incompatibility may lead to hyperbilirubinemia and consequently, neonatal jaundice.[10]
Epidemiology
Approximately 50% of full-term and 80% of preterm infants develop jaundice in the first 2 to 4 days after birth when serum bilirubin levels are greater than or equal to 5mg/dL.[17] Crigler-Najjar syndrome is a very rare disease with less than fifty cases in the United States and approximately 1 case per million births worldwide.[18] In the United States, the prevalence of Gilbert syndrome is relatively higher; around 9% of the population is homozygous for UGT1A1 mutation. In Gilbert syndrome, ethnic groups' specific mutations occur at a different site of the UGT1A1 gene.[19] For example, Gilbert syndrome in the white population is commonly associated with the mutation in the TATAA element of the UGT1A1 promoter region.[20] Neonatal jaundice is common in male infants, while Crigler-Najjar syndrome does not correlate with sex. At the stage of puberty, Gilbert syndrome is more common in males than females, which can be due to the high rate of bilirubin production in males than females.[21] In 0.5 to 2.4% of infants, breast milk jaundice occurs between days 2 and 5 after birth due to increased enterohepatic circulation of bilirubin, peaks at two weeks, and resolves between 3 to 12 weeks.[14]
Pathophysiology
Jaundice is evident when the serum bilirubin level exceeds 3mg/dL. Gilbert syndrome and Crigler-Najjar syndrome type I and II, are hereditary non-hemolytic unconjugated hyperbilirubinemias that result from the mutations in the UGT1A1 gene and impair bilirubin conjugation. In Gilbert syndrome, UGT1A1 enzyme activity is 10 to 30% of normal, which results in mild unconjugated hyperbilirubinemia with bilirubin levels below 5 mg/dL.[22] The decreased activity of UGT1A1 results from the addition of extra thymine-adenine (TA) repeats in the TATAA box region of the UGT1A1 gene promoter.[23] Individuals with Gilbert syndrome have a relatively benign course. Crigler-Najjar types I and II syndromes are inherited autosomal recessive disorders that result from the mutations in any one or more of the five exons of the gene that encodes the UGT1A1 enzyme.[24] In Crigler-Najjar type I (CNS1), UGT1A1 enzyme activity is absent, leading to a severe increase in unconjugated bilirubin levels ranging from 20 to 45 mg/dL. In the neonatal period, infants with Crigler-Najjar type I syndrome usually develop bilirubin encephalopathy (Kernicterus) with seizures, opisthotonos, and die from severe neurologic diseases in the absence of treatment.[10]
Individuals with Crigler-Najjar type II (CNS2) have less than 10% of the normal UGT1A1 activity, resulting in bilirubin levels of 5 to 25 mg/dL. Kernicterus is rare in Crigler-Najjar type II, but patients with increased bilirubin levels (greater than 15 mg/dL) have high chances of its occurrence.[25][26] Kernicterus can occur under stress and with inter-current illnesses. Also, CNS2 therapy with phenobarbital can lower the UCB levels by 25%.[27]
History and Physical
Patients with unconjugated hyperbilirubinemia may present with jaundice or other signs, or the condition may be noticed in the asymptomatic patient by the finding of high levels of bilirubin in the serum during routine blood testing. As far as the differential diagnosis of unconjugated hyperbilirubinemia is involved, it requires a detailed history and physical examination of patients for the evaluation. The assessment of the patient’s clinical history includes; age at the disease manifestation, ethnic origin/race, hereditary disorder, family history of anemia or jaundice, recently used medications, diet history, and history of liver disease.
Approximately 50% of neonates present with physiologic jaundice during the first five days after birth. Nonphysiologic, Lucey-Driscoll syndrome, or maternal serum jaundice in neonates results from maternal serum during the initial four days after delivery.[12] The onset of asymptomatic jaundice usually characterizes the overproduction of bilirubin due to ineffective erythropoiesis (early labeled bilirubin production). Patients with Gilbert syndrome are generally asymptomatic and present with mild or intermittent unconjugated hyperbilirubinemia without any liver disease or hemolysis, and on clinical examination may manifest jaundice. Some nonspecific symptoms, such as fatigue, abdominal cramps, and malaise, are commonly observed in this disease. Infants with Crigler-Najjar type I syndrome present jaundice in the first few days after birth with neurological sequelae if UCB is greater than 20 mg/dL. By the second week of an infant’s life, it rapidly progresses to life-threatening severe jaundice that requires treatment with exchange transfusion, intensive phototherapy, and liver transplantation.[28] However, patients with Crigler-Najjar type II syndrome, are mostly asymptomatic and may rarely present with jaundice and kernicterus. During the physical examination, the color of the skin and sclerae should be examined carefully under light for the evaluation of jaundice. For example, in patients with mild jaundice or hyperbilirubinemia (2 to 3 mg/dL), only scleral icterus can be seen. Also, with hemolytic anemia, there might be anemia and splenomegaly.
Evaluation
The initial assessment of the patient presenting with jaundice depends on whether hyperbilirubinemia is the result of unconjugated or conjugated bilirubin. A urine test positive for bilirubin indicates conjugated hyperbilirubinemia. Conjugated bilirubin is soluble in water; therefore, it can be excreted via urine but not unconjugated bilirubin due to water insolubility. Further, first-line laboratory testing should include measurement of the serum total bilirubin levels with fractionation (direct with indirect obtained by subtraction from total), followed by serum aspartate transaminase (AST), alanine transaminase (ALT), alkaline phosphatase, and γ-glutamyl transpeptidase (GGT) levels, complete blood count (CBC) with an examination of the smear, a reticulocyte count, a direct Coombs test, lactate dehydrogenase (LDH) and haptoglobin levels.[29] Jaundice, with a toxic appearance, fever, or abnormal count of white blood cells indicates sepsis that requires prompt evaluation and adequate treatment. The increased levels of AST and ALT are markers of hepatocyte damage/injury. However, increased levels of alkaline phosphatase and γ-glutamyltransferase suggest cholestasis.
A complete blood count is a beneficial test for the determination of hemolysis, which is characterized by the presence of damaged red blood cells and increased reticulocytes on the smear. In patients, when the serum total bilirubin levels exceed 3.0 mg/dL, jaundice becomes clinically apparent. Hyperbilirubinemia is unconjugated when the conjugated bilirubin level is less than 15% of the TB. While in conjugated hyperbilirubinemia, conjugated bilirubin level is high and more than 20% of the total bilirubin. Generally, patients with carotenemia also present yellow skin but have no elevation in serum bilirubin levels. During the physical examination, carotenemia due to excess carotene intake, e.g., carrots, peaches, etc., can be distinguished from jaundice due to the absence of scleral icterus in carotenemia and with the pigmentation of soles and palms and other areas. Computerized axial tomography (CT) and ultrasonography scanning are useful tools in differentiating cholestasis (intra-hepatic and extra-hepatic) from hepatocellular disease in the assessment of a patient with conjugated hyperbilirubinemia, which is not the focus of the review.
Treatment / Management
Neonatal Jaundice
In most cases of mild neonatal jaundice, no treatment is necessary, and it resolves on its own within a few weeks. In cases of moderate or severe jaundice (TB over 15 mg/dL), phototherapy is a safe option. During phototherapy, the infant’s skin is exposed to light in the blue-green spectrum (460-490 nm) in a way that transforms bilirubin into lumirubin, a water-soluble isomer, and reduces its toxicity by increasing its elimination in both the urine and stool.[30][31] It is crucial to maintain proper hydration and urine output during phototherapy.[17][32] Unfiltered sunlight has ultraviolet light (<400 nm) which increases the risk of sunburn and skin cancers. Hence unfiltered sunlight is not utilized for phototherapy.
In some cases, infants carry antibodies from the mother that can result in the quick breakdown of neonatal red blood cells (due to differences in the blood type), intravenous transfusion of an immunoglobulin (IVIG) that can reduce the levels of antibodies may be the choice of treatment. In a rare instance of severe neonatal jaundice, when other treatments do not work, exchange transfusion of blood may be performed.[33] In this procedure, a small amount of infant’s blood repeatedly gets withdrawn and replaced with donor blood to dilute the levels of bilirubin and maternal antibodies. Total bilirubin decreases to 50-75 percent of the pre-exchange level with double volume exchange transfusion. (B2)
Gilbert Syndrome
The most crucial part for clinicians in the management and care of patients with Gilbert syndrome (GS) is to recognize that it is relatively benign with a good prognosis. The diagnosis of GS requires normal liver function tests, elevated bilirubin levels, and genetic testing to confirm the disorder further. Owing to the benign and inconsequential nature of this syndrome, the use of medications is not justified for its treatment. There exists an increased risk of side effects and toxicity from the use of certain drugs such as acetaminophen and irinotecan that are conjugated by the liver.[5] Phenobarbital has been shown to normalize bilirubin levels.
Crigler-Najjar Type II Syndrome
Patients with Crigler-Najjar syndrome type II respond to phenobarbital therapy. Since CNS2 is milder, phenobarbital is effective in reducing plasma bilirubin levels in these patients by 25 %. In some cases, patients with severe hyperbilirubinemia may need phototherapy or exchange transfusions. However, often, patients affected with type II syndrome may not require any therapy but may need routine monitoring. The goal is to maintain TB below 15 mg/dL.
Crigler-Najjar Type I Syndrome
Unlike CNS2, patients with CNS1 do not respond to phenobarbital therapy due to the absence of the enzyme. Rapid treatment of kernicterus is needed to prevent devastating neurologic sequelae. The emergent treatment for bilirubin encephalopathy relies on repeated plasma exchange transfusions followed by long-term phototherapy.[28] The process of plasma exchange transfusion flushes out bilirubin-saturated albumin and provides free protein, which lures bilirubin from the tissues. To maintain the levels of unconjugated hyperbilirubinemia below the neurotoxic threshold, patients with Crigler-Najjar syndrome type I generally need phototherapy for 10 to 12 hours a day. Phototherapy helps in converting bilirubin to more water-soluble bilirubin isoforms that can get excreted in the urine. The efficacy of phototherapy is dose-dependent and depends on the intensity of light, exposure of the body surface, or reflecting mirrors used. Short-term complications of phototherapy are reduced cardiac output and renal perfusion, hyperthermia, increased cerebral perfusion, oxidative stress, skin rash, breastfeeding interruption, and rarely bronze baby syndrome.[30] The bronze baby syndrome is a rare, reversible dark, grayish-brown skin, serum, and urine discoloration, caused by bronze pigmented photoisomers, identified in infants treated with phototherapy.[34] Pigments are eliminated slowly after phototherapy discontinuation, and resolution is seen within weeks without any sequelae.[35] Some side effects associated with long-term phototherapy are restriction of activity, delay in normal development, diarrhea, reduced weight gain, problems in maintaining body temperature, and risk of seizures (greater in boys).[36](B3)
Oral calcium phosphate may be a valuable adjuvant to phototherapy in Crigler-Najjar type I syndrome, by interrupting the enterohepatic circulation of bilirubin.[37] Treatment with inhibitors of heme oxygenase (tin-mesoporphyrin) helps decrease plasma bilirubin levels but is not recommended for the long term owing to their side effects.[38](A1)
Plasmapheresis is also being employed to lower bilirubin levels in the blood rapidly.[28] In this procedure, unwanted substances (toxins and plasma components) get removed from the blood, and plasma gets replaced with other human plasma before transfusing blood back into the patient.
In patients with Crigler-Najjar syndrome type I, orthotopic liver transplantation may be considered a definite form of therapy and should be performed before the onset of neurologic damage.[39] Transplantation of hepatocytes is being investigated as an alternative to liver transplantation.[40] Gene therapy is another approach for the treatment of CNS1. Significant improvement leading to the life-long cure of the disease can be achieved clinically with gene therapy by replacing a defective gene with a normal one.[41] Presently gene transfer that can reduce the bilirubin levels for several years by partially replacing the gene controlling bilirubin glucuronosyltransferase activity in the patients is being considered for treatment of CNS1. Long-term monitoring is important for patients at risk for a sudden increase in serum bilirubin levels.(B3)
Pharmacologic management
Patients with Crigler-Najjar type II syndrome do not require any treatment and can be managed with phenobarbital. Also, for Gilbert syndrome, no medical therapy is needed, and phenobarbital has been effectively shown to decrease bilirubin production. The common medications used for the treatment of patients with Crigler-Najjar type I syndrome are phenobarbital, calcium (infusions), ursodeoxycholic acid, metalloporphyrins, chlorpromazine, and cholestyramine.
Phenobarbital increases both the conjugation and excretion of bilirubin by stimulating the gene for the UGT1A1 enzyme, which induces the production of conjugated bilirubin and thereby reduces the serum bilirubin levels by 25%. Ursodeoxycholic acid increases the flow of the bile, the elimination of bile into the gastrointestinal tract, thereby reducing the total serum bilirubin levels.[42]
Metalloporphyrins like tin-mesoporphyrin (SnMp) inhibit the activity of the heme oxygenase enzyme, which is a rate-limiting step in the production of bilirubin from heme metabolism.[43][44] They reduce the necessity of phototherapy and the need for continued hospitalization.[45][46][47] Calcium (infusions) like calcium phosphate bind to bilirubin in the gut and increases bilirubin’s fecal excretion. (A1)
Differential Diagnosis
Unconjugated hyperbilirubinemia can be caused by dysregulation in bilirubin metabolism that includes bilirubin production, uptake in hepatocytes, and conjugation. The clinician should distinguish unconjugated hyperbilirubinemia from other conditions with similar clinical manifestations. Hemolytic anemias and resorption of hematoma significantly increase the levels of unconjugated bilirubin. Hemolytic anemias may lead to a mild increase in bilirubin levels with or without clinical symptoms. Hemolytic disorders such as glucose-6-phosphate dehydrogenase (G6PD) deficiency, sickle cell anemia, thalassemia, iso-immune mediated hemolysis, and autoimmune disorders should be a consideration during diagnosis.[48]
Inherited disorders of the enzyme glucuronosyltransferase (UGT1A1) such as Gilbert syndrome and Crigler-Najjar syndromes (CNS1 and CNS2), may impede the complete conjugation of bilirubin and thereby cause varying degrees of unconjugated hyperbilirubinemia, depending on inhibition of enzyme with each disease. In addition to inherited disorders, other conditions such as portosystemic shunts, congestive heart failure, liver damage, dyserythropoiesis, and hyperthyroidism can also contribute to impaired bilirubin conjugation and should merit consideration during diagnosis. Additional considerations in the diagnosis of unconjugated hyperbilirubinemia include drugs interfering with bilirubin uptake or conjugation (rifampicin, probenecid, or other rifamycin antibiotics, ketoconazole, amitryptiline, and protease inhibitors), chronic liver diseases, thyrotoxicosis, infections, and physiologic neonatal jaundice resulting from the immature conjugating ability of the UGT1A1 enzyme.[49][50][51]
Prognosis
The prognosis in neonatal jaundice is excellent if the patient receives the recommended standard of care. In most cases, jaundice gets better within one or two weeks. In some cases, such as breast milk jaundice and maternal serum jaundice, jaundice can persist for several weeks. Thus, the patient’s serum bilirubin levels need to be monitored closely, with adequate adjustments in treatment made accordingly to prevent persistent hyperbilirubinemia and thereby its consequence, kernicterus and bilirubin induced neurologic dysfunction. The prognosis of patients with ineffective erythropoiesis, which causes the overproduction of bilirubin is also excellent and depends on the cause. Patients with Crigler Najjar syndrome type I have a high risk of kernicterus, and thereby present a poor prognosis and need intensive, aggressive management. However, the prognosis of Crigler Najjar syndrome type II patients is more favorable and rarely associated with kernicterus. Gilbert syndrome is a benign condition, and patients with this syndrome have an excellent prognosis. An individual affected with this inherited disorder can live a normal life. Recent studies have shown that a person with Gilbert syndrome has a low risk of cardiovascular diseases, possibly due to an increase in bilirubin ameliorating oxidative stress.[52][53] Long-term survival is identified in patients that had liver transplantation.[54]
Complications
Extreme hyperbilirubinemia (TB of 25 to 30 mg/dL) can cause bilirubin encephalopathy, Kernicterus, which is usually characterized by the deposition of unconjugated bilirubin (yellow stain) in brain cells. Neuronal necrosis/damage occurs in the basal ganglia, hippocampus, hypothalamic nuclei, diencephalon, midbrain responsible for neurohumoral and electrolyte control, brainstem nuclei accounting for oculomotor and auditory function, and in the cerebellum depositing as a bilirubin-phosphatidylcholine precipitate. Vision, hearing, speech, cognition, gait, and language are typically affected.[55][56][57][58] Clinical manifestations include cerebral palsy, deafness, seizures, abnormalities of the gaze, and hypoplasia of the dental enamel.[59]
Deterrence and Patient Education
Patients and their families should receive education regarding jaundice and its consequences. Patients need to understand the role of unconjugated hyperbilirubinemia in the manifestation of jaundice and its severity. Patients require counseling regarding the physical appearance or symptoms of jaundice. In the case of neonatal jaundice, parents will need to know about the cause of physiologic or non-physiologic jaundice and the treatment options available for it. Patients with any change in mental or neural behavior should immediately consult with a physician. Future parents with a family history of inherited Crigler-Najjar syndromes should seek genetic counseling. Education on the autosomal recessive pattern of inheritance must be provided to the patients and families.
Enhancing Healthcare Team Outcomes
The treatment of unconjugated hyperbilirubinemia requires interprofessional teamwork, as it can result from multiple disorders or diseases. As far as the differential diagnoses of unconjugated hyperbilirubinemia are involved, it necessitates good coordination between primary care physicians, gastroenterologists, hematologists, a pediatrician, and other specialists. A detailed history of the patient's medications that causes liver dysfunction is critical for the treatment. The outcome of most patients with unconjugated hyperbilirubinemia is excellent when they receive the treatment according to the accepted guidelines with no or minimal neural and developmental sequelae.[60] Crigler Najjar syndromes, especially Type 1 on the other hand have a high risk of the life-threatening kernicterus that requires lifelong monitoring and treatment.
Media
(Click Image to Enlarge)
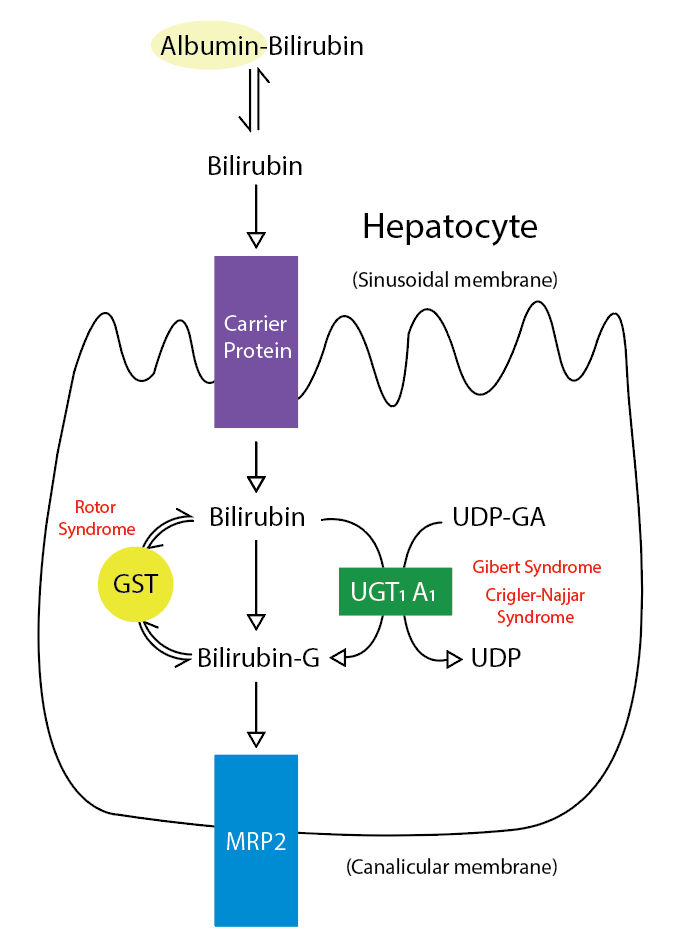
Metabolic Pathway for Bilirubin in the Hepatocyte. Bilirubin-G corresponds to bilirubin glucuronate; the donor is uridine diphosphate glucuronic acid (UDP-GA). This is catalyzed by the enzyme uridine diphosphate-glucuronyltransferase (UGT1A1). Gilbert and Crigler-Najjar syndrome is associated with decreases in UGT1A1 activity. Glutathione-S-transferase (GST) is a carrier protein that assists bilirubin uptake into the cytosol and may be implicated in Rotor syndrome.
Contributed by R Kabir, MD
References
Fargo MV, Grogan SP, Saguil A. Evaluation of Jaundice in Adults. American family physician. 2017 Feb 1:95(3):164-168 [PubMed PMID: 28145671]
Roche SP, Kobos R. Jaundice in the adult patient. American family physician. 2004 Jan 15:69(2):299-304 [PubMed PMID: 14765767]
Memon N, Weinberger BI, Hegyi T, Aleksunes LM. Inherited disorders of bilirubin clearance. Pediatric research. 2016 Mar:79(3):378-86. doi: 10.1038/pr.2015.247. Epub 2015 Nov 23 [PubMed PMID: 26595536]
Radlović N, Hereditary hyperbilirubinemias. Srpski arhiv za celokupno lekarstvo. 2014 Mar-Apr; [PubMed PMID: 24839786]
Ramakrishnan N, Bittar K, Jialal I. Impaired Bilirubin Conjugation. StatPearls. 2024 Jan:(): [PubMed PMID: 29494090]
Franchini M, Targher G, Lippi G. Serum bilirubin levels and cardiovascular disease risk: a Janus Bifrons? Advances in clinical chemistry. 2010:50():47-63 [PubMed PMID: 20521440]
Level 3 (low-level) evidenceVítek L, Schwertner HA. The heme catabolic pathway and its protective effects on oxidative stress-mediated diseases. Advances in clinical chemistry. 2007:43():1-57 [PubMed PMID: 17249379]
Level 3 (low-level) evidenceVítek L,Ostrow JD, Bilirubin chemistry and metabolism; harmful and protective aspects. Current pharmaceutical design. 2009; [PubMed PMID: 19754364]
Level 3 (low-level) evidenceJedlitschky G, Hoffmann U, Kroemer HK. Structure and function of the MRP2 (ABCC2) protein and its role in drug disposition. Expert opinion on drug metabolism & toxicology. 2006 Jun:2(3):351-66 [PubMed PMID: 16863439]
Level 3 (low-level) evidenceDennery PA, Seidman DS, Stevenson DK. Neonatal hyperbilirubinemia. The New England journal of medicine. 2001 Feb 22:344(8):581-90 [PubMed PMID: 11207355]
Kenwright S, Levi AJ. Sites of competition in the selective hepatic uptake of rifamycin-SV, flavaspidic acid, bilirubin, and bromsulphthalein. Gut. 1974 Mar:15(3):220-6 [PubMed PMID: 4842968]
Level 3 (low-level) evidenceARIAS IM,WOLFSON S,LUCEY JF,MCKAY RJ Jr, TRANSIENT FAMILIAL NEONATAL HYPERBILIRUBINEMIA. The Journal of clinical investigation. 1965 Sep [PubMed PMID: 14332157]
Grunebaum E, Amir J, Merlob P, Mimouni M, Varsano I. Breast mild jaundice: natural history, familial incidence and late neurodevelopmental outcome of the infant. European journal of pediatrics. 1991 Feb:150(4):267-70 [PubMed PMID: 2029918]
Maisels MJ, Clune S, Coleman K, Gendelman B, Kendall A, McManus S, Smyth M. The natural history of jaundice in predominantly breastfed infants. Pediatrics. 2014 Aug:134(2):e340-5. doi: 10.1542/peds.2013-4299. Epub [PubMed PMID: 25049352]
Maisels MJ, Kring E. Length of stay, jaundice, and hospital readmission. Pediatrics. 1998 Jun:101(6):995-8 [PubMed PMID: 9606225]
Level 2 (mid-level) evidenceZhang D, Chando TJ, Everett DW, Patten CJ, Dehal SS, Humphreys WG. In vitro inhibition of UDP glucuronosyltransferases by atazanavir and other HIV protease inhibitors and the relationship of this property to in vivo bilirubin glucuronidation. Drug metabolism and disposition: the biological fate of chemicals. 2005 Nov:33(11):1729-39 [PubMed PMID: 16118329]
Woodgate P, Jardine LA. Neonatal jaundice: phototherapy. BMJ clinical evidence. 2015 May 22:2015():. pii: 0319. Epub 2015 May 22 [PubMed PMID: 25998618]
Ebrahimi A, Rahim F. Crigler-Najjar Syndrome: Current Perspectives and the Application of Clinical Genetics. Endocrine, metabolic & immune disorders drug targets. 2018:18(3):201-211. doi: 10.2174/1871530318666171213153130. Epub [PubMed PMID: 29237388]
Level 3 (low-level) evidenceLampe JW, Bigler J, Horner NK, Potter JD. UDP-glucuronosyltransferase (UGT1A1*28 and UGT1A6*2) polymorphisms in Caucasians and Asians: relationships to serum bilirubin concentrations. Pharmacogenetics. 1999 Jun:9(3):341-9 [PubMed PMID: 10471066]
Level 2 (mid-level) evidenceClementi M, Di Gianantonio E, Fabris L, Forabosco P, Strazzabosco M, Tenconi R, Okolicsanyi L. Inheritance of hyperbilirubinemia: evidence for a major autosomal recessive gene. Digestive and liver disease : official journal of the Italian Society of Gastroenterology and the Italian Association for the Study of the Liver. 2007 Apr:39(4):351-5 [PubMed PMID: 17347060]
Muraca M, Fevery J. Influence of sex and sex steroids on bilirubin uridine diphosphate-glucuronosyltransferase activity of rat liver. Gastroenterology. 1984 Aug:87(2):308-13 [PubMed PMID: 6428963]
Level 3 (low-level) evidenceAuclair C, Hakim J, Boivin P, Troube H, Boucherot J. Bilirubin and paranitrophenol glucuronyl transferase activities of the liver in patients with Gilbert's syndrome An attempt at a biochemical breakdown of the Gilbert's syndrome. Enzyme. 1976:21(2):97-107 [PubMed PMID: 816648]
Bosma PJ, Chowdhury JR, Bakker C, Gantla S, de Boer A, Oostra BA, Lindhout D, Tytgat GN, Jansen PL, Oude Elferink RP. The genetic basis of the reduced expression of bilirubin UDP-glucuronosyltransferase 1 in Gilbert's syndrome. The New England journal of medicine. 1995 Nov 2:333(18):1171-5 [PubMed PMID: 7565971]
Level 2 (mid-level) evidenceErps LT, Ritter JK, Hersh JH, Blossom D, Martin NC, Owens IS. Identification of two single base substitutions in the UGT1 gene locus which abolish bilirubin uridine diphosphate glucuronosyltransferase activity in vitro. The Journal of clinical investigation. 1994 Feb:93(2):564-70 [PubMed PMID: 7906695]
Level 3 (low-level) evidenceStevenson DK, Vreman HJ, Wong RJ. Bilirubin production and the risk of bilirubin neurotoxicity. Seminars in perinatology. 2011 Jun:35(3):121-6. doi: 10.1053/j.semperi.2011.02.005. Epub [PubMed PMID: 21641484]
Sticova E, Jirsa M. New insights in bilirubin metabolism and their clinical implications. World journal of gastroenterology. 2013 Oct 14:19(38):6398-407. doi: 10.3748/wjg.v19.i38.6398. Epub [PubMed PMID: 24151358]
Level 3 (low-level) evidenceTrotman BW, Shaw L, Roy-Chowdhury J, Malet PF, Rosato EF. Effect of phenobarbital on serum and biliary parameters in a patient with Crigler-Najjar syndrome, type II and acquired cholestasis. Digestive diseases and sciences. 1983 Aug:28(8):753-62 [PubMed PMID: 6872808]
Level 3 (low-level) evidenceStrauss KA, Ahlfors CE, Soltys K, Mazareigos GV, Young M, Bowser LE, Fox MD, Squires JE, McKiernan P, Brigatti KW, Puffenberger EG, Carson VJ, Vreman HJ. Crigler-Najjar Syndrome Type 1: Pathophysiology, Natural History, and Therapeutic Frontier. Hepatology (Baltimore, Md.). 2020 Jun:71(6):1923-1939. doi: 10.1002/hep.30959. Epub 2020 Feb 5 [PubMed PMID: 31553814]
Winger J, Michelfelder A. Diagnostic approach to the patient with jaundice. Primary care. 2011 Sep:38(3):469-82; viii. doi: 10.1016/j.pop.2011.05.004. Epub [PubMed PMID: 21872092]
Hansen TWR, Maisels MJ, Ebbesen F, Vreman HJ, Stevenson DK, Wong RJ, Bhutani VK. Sixty years of phototherapy for neonatal jaundice - from serendipitous observation to standardized treatment and rescue for millions. Journal of perinatology : official journal of the California Perinatal Association. 2020 Feb:40(2):180-193. doi: 10.1038/s41372-019-0439-1. Epub 2019 Aug 16 [PubMed PMID: 31420582]
Ennever JF, Costarino AT, Polin RA, Speck WT. Rapid clearance of a structural isomer of bilirubin during phototherapy. The Journal of clinical investigation. 1987 Jun:79(6):1674-8 [PubMed PMID: 3584465]
Sherbiny HS,Youssef DM,Sherbini AS,El-Behedy R,Sherief LM, High-intensity light-emitting diode vs fluorescent tubes for intensive phototherapy in neonates. Paediatrics and international child health. 2016 May [PubMed PMID: 25844870]
Flaherman VJ, Kuzniewicz MW, Escobar GJ, Newman TB. Total serum bilirubin exceeding exchange transfusion thresholds in the setting of universal screening. The Journal of pediatrics. 2012 May:160(5):796-800.e1. doi: 10.1016/j.jpeds.2011.09.063. Epub 2011 Dec 1 [PubMed PMID: 22133423]
Level 2 (mid-level) evidenceRubaltelli FF, Da Riol R, D'Amore ES, Jori G. The bronze baby syndrome: evidence of increased tissue concentration of copper porphyrins. Acta paediatrica (Oslo, Norway : 1992). 1996 Mar:85(3):381-4 [PubMed PMID: 8696003]
Level 3 (low-level) evidenceTan KL, Jacob E. The bronze baby syndrome. Acta paediatrica Scandinavica. 1982 May:71(3):409-14 [PubMed PMID: 7136654]
Newman TB, Wu YW, Kuzniewicz MW, Grimes BA, McCulloch CE. Childhood Seizures After Phototherapy. Pediatrics. 2018 Oct:142(4):. pii: e20180648. doi: 10.1542/peds.2018-0648. Epub [PubMed PMID: 30249623]
Van Der Veere CN, Schoemaker B, Bakker C, Van Der Meer R, Jansen PL, Elferink RP. Influence of dietary calcium phosphate on the disposition of bilirubin in rats with unconjugated hyperbilirubinemia. Hepatology (Baltimore, Md.). 1996 Sep:24(3):620-6 [PubMed PMID: 8781334]
Level 3 (low-level) evidenceEvans D. Neonatal jaundice. BMJ clinical evidence. 2007 Jun 1:2007():. pii: 0319. Epub 2007 Jun 1 [PubMed PMID: 19454091]
Level 1 (high-level) evidenceSchauer R, Stangl M, Lang T, Zimmermann A, Chouker A, Gerbes AL, Schildberg FW, Rau HG. Treatment of Crigler-Najjar type 1 disease: relevance of early liver transplantation. Journal of pediatric surgery. 2003 Aug:38(8):1227-31 [PubMed PMID: 12891498]
Ambrosino G, Varotto S, Strom SC, Guariso G, Franchin E, Miotto D, Caenazzo L, Basso S, Carraro P, Valente ML, D'Amico D, Zancan L, D'Antiga L. Isolated hepatocyte transplantation for Crigler-Najjar syndrome type 1. Cell transplantation. 2005:14(2-3):151-7 [PubMed PMID: 15881424]
Level 3 (low-level) evidenceBirraux J, Menzel O, Wildhaber B, Jond C, Nguyen TH, Chardot C. A step toward liver gene therapy: efficient correction of the genetic defect of hepatocytes isolated from a patient with Crigler-Najjar syndrome type 1 with lentiviral vectors. Transplantation. 2009 Apr 15:87(7):1006-12. doi: 10.1097/TP.0b013e31819ca245. Epub [PubMed PMID: 19352119]
Level 3 (low-level) evidenceHonar N, Ghashghaei Saadi E, Saki F, Pishva N, Shakibazad N, Hosseini Teshnizi S. Effect of Ursodeoxycholic Acid on Indirect Hyperbilirubinemia in Neonates Treated With Phototherapy. Journal of pediatric gastroenterology and nutrition. 2016 Jan:62(1):97-100. doi: 10.1097/MPG.0000000000000874. Epub [PubMed PMID: 26020375]
Valaes T, Petmezaki S, Henschke C, Drummond GS, Kappas A. Control of jaundice in preterm newborns by an inhibitor of bilirubin production: studies with tin-mesoporphyrin. Pediatrics. 1994 Jan:93(1):1-11 [PubMed PMID: 8265301]
Level 1 (high-level) evidenceKappas A,Drummond GS, Control of heme metabolism with synthetic metalloporphyrins. The Journal of clinical investigation. 1986 Feb [PubMed PMID: 3511095]
Level 3 (low-level) evidenceSuresh GK, Martin CL, Soll RF. Metalloporphyrins for treatment of unconjugated hyperbilirubinemia in neonates. The Cochrane database of systematic reviews. 2003:(2):CD004207 [PubMed PMID: 12804504]
Level 1 (high-level) evidenceBhutani VK, Poland R, Meloy LD, Hegyi T, Fanaroff AA, Maisels MJ. Clinical trial of tin mesoporphyrin to prevent neonatal hyperbilirubinemia. Journal of perinatology : official journal of the California Perinatal Association. 2016 Jul:36(7):533-9. doi: 10.1038/jp.2016.22. Epub 2016 Mar 3 [PubMed PMID: 26938918]
Martinez JC, Garcia HO, Otheguy LE, Drummond GS, Kappas A. Control of severe hyperbilirubinemia in full-term newborns with the inhibitor of bilirubin production Sn-mesoporphyrin. Pediatrics. 1999 Jan:103(1):1-5 [PubMed PMID: 9917431]
Level 1 (high-level) evidenceKaplan M,Beutler E,Vreman HJ,Hammerman C,Levy-Lahad E,Renbaum P,Stevenson DK, Neonatal hyperbilirubinemia in glucose-6-phosphate dehydrogenase-deficient heterozygotes. Pediatrics. 1999 Jul; [PubMed PMID: 10390262]
Level 2 (mid-level) evidenceJohnston DE. Special considerations in interpreting liver function tests. American family physician. 1999 Apr 15:59(8):2223-30 [PubMed PMID: 10221307]
Lewis JH. Drug-induced liver disease. The Medical clinics of North America. 2000 Sep:84(5):1275-311, x [PubMed PMID: 11026929]
Singh A, Koduru B, Carlisle C, Akhter H, Liu RM, Schroder K, Brandes RP, Ojcius DM. NADPH oxidase 4 modulates hepatic responses to lipopolysaccharide mediated by Toll-like receptor-4. Scientific reports. 2017 Oct 30:7(1):14346. doi: 10.1038/s41598-017-14574-8. Epub 2017 Oct 30 [PubMed PMID: 29085012]
Kundur AR,Santhakumar AB,Bulmer AC,Singh I, Mildly elevated unconjugated bilirubin is associated with reduced platelet activation-related thrombogenesis and inflammation in Gilbert's syndrome. Platelets. 2017 Dec; [PubMed PMID: 28300459]
Lin JP, O'Donnell CJ, Schwaiger JP, Cupples LA, Lingenhel A, Hunt SC, Yang S, Kronenberg F. Association between the UGT1A1*28 allele, bilirubin levels, and coronary heart disease in the Framingham Heart Study. Circulation. 2006 Oct 3:114(14):1476-81 [PubMed PMID: 17000907]
van der Veere CN, Sinaasappel M, McDonagh AF, Rosenthal P, Labrune P, Odièvre M, Fevery J, Otte JB, McClean P, Bürk G, Masakowski V, Sperl W, Mowat AP, Vergani GM, Heller K, Wilson JP, Shepherd R, Jansen PL. Current therapy for Crigler-Najjar syndrome type 1: report of a world registry. Hepatology (Baltimore, Md.). 1996 Aug:24(2):311-5 [PubMed PMID: 8690398]
Good WV, Hou C. Visuocortical bilirubin-induced neurological dysfunction. Seminars in fetal & neonatal medicine. 2015 Feb:20(1):37-41. doi: 10.1016/j.siny.2014.12.007. Epub 2015 Jan 8 [PubMed PMID: 25577655]
Olds C,Oghalai JS, Audiologic impairment associated with bilirubin-induced neurologic damage. Seminars in fetal & neonatal medicine. 2015 Feb [PubMed PMID: 25575899]
Rose J, Vassar R. Movement disorders due to bilirubin toxicity. Seminars in fetal & neonatal medicine. 2015 Feb:20(1):20-25. doi: 10.1016/j.siny.2014.11.002. Epub 2014 Dec 16 [PubMed PMID: 25524299]
Wusthoff CJ, Loe IM. Impact of bilirubin-induced neurologic dysfunction on neurodevelopmental outcomes. Seminars in fetal & neonatal medicine. 2015 Feb:20(1):52-57. doi: 10.1016/j.siny.2014.12.003. Epub 2015 Jan 10 [PubMed PMID: 25585889]
Amin SB, Saluja S, Saili A, Orlando M, Wang H, Laroia N, Agarwal A. Chronic Auditory Toxicity in Late Preterm and Term Infants With Significant Hyperbilirubinemia. Pediatrics. 2017 Oct:140(4):. doi: 10.1542/peds.2016-4009. Epub [PubMed PMID: 28954873]
Newman TB, Liljestrand P, Jeremy RJ, Ferriero DM, Wu YW, Hudes ES, Escobar GJ, Jaundice and Infant Feeding Study Team. Outcomes among newborns with total serum bilirubin levels of 25 mg per deciliter or more. The New England journal of medicine. 2006 May 4:354(18):1889-900 [PubMed PMID: 16672700]
Level 2 (mid-level) evidence