Introduction
The brain, eye, and kidney are some of the organs that have glucose as the sole metabolic fuel source. Prolonged fasting or vigorous exercise depletes glycogen stores, making the body switch to de-novo glucose synthesis to maintain blood levels of this monosaccharide. Gluconeogenesis is the process that allows the body to form glucose from non-hexose precursors, particularly glycerol, lactate, pyruvate, propionate, and glucogenic amino acids.[1]
Gluconeogenesis essentially reverses glycolysis (see Image. Gluconeogenesis). Four enzymes facilitate glucose synthesis by this pathway by reversing 3 highly exergonic glycolytic steps, namely, pyruvate carboxylase, phosphoenol pyruvate carboxykinase (PEPCK), fructose-1,6-bisphosphatase, and glucose-6-phosphatase. However, these enzymes are not present in all cell types. Therefore, gluconeogenesis can only occur in specific tissues. In humans, gluconeogenesis takes place primarily in the liver and, to a lesser extent, the renal cortex.[2]
This article discusses gluconeogenesis and its clinical correlates.
Issues of Concern
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Issues of Concern
Only a moderate amount of glucose can be synthesized by gluconeogenesis, but failure of this pathway is typically fatal. Acute hypoglycemia can damage the brain and kidneys because of their dependence on glucose for fuel. Gluconeogenesis is also one of the body's main clearing mechanisms for the muscle and erythrocyte metabolite, lactic acid. Lactic acidosis arising from shock is associated with increased mortality risk.[14]
Cellular Level
Gluconeogenesis Reactions And Enzymes
The primary stimulus for gluconeogenesis is low blood glucose. Starting with pyruvate, the reactions involved in gluconeogenesis are the following:
- In the mitochondrion, pyruvate carboxylase converts pyruvate to oxaloacetate. Pyruvate carboxylase requires ATP and the coenzyme biotin for activation. This conversion is the first step that reverses the nonequilibrium reaction catalyzed by the glycolytic enzyme pyruvate kinase.
- Oxaloacetate is converted to malate before it crosses the mitochondrial membrane. Malate is converted back to oxaloacetate once in the cytosol.
- In the cytosol, PEPCK decarboxylates oxaloacetate, which then rearranges to form phosphoenol pyruvate (PEP). PEPCK requires GTP and magnesium ions for activation. This transformation is the second step that reverses the nonequilibrium reaction catalyzed by pyruvate kinase.
- Enolase hydrates PEP to form 2-phosphoglycerate.
- Phosphoglycerate mutase converts 2-phosphoglycerate to 3-phosphoglycerate.
- Phosphoglycerate kinase phosphorylates 3-phosphoglycerate to form 1,3-bisphosphoglycerate. This reaction requires ATP.
- Glyceraldehyde-3-phosphate dehydrogenase reduces 1,3-bisphosphoglycerate to glyceraldehyde 3-phosphate. Reduced nicotinamide adenine dinucleotide (NADH) is the electron donor.
- Triose phosphate isomerase isomerizes glyceraldehyde 3-phosphate to form dihydroxyacetone phosphate (DHAP).
- Aldolase combines glyceraldehyde 3-phosphate and DHAP to form fructose 1,6-bisphosphate.
- Fructose-1,6-bisphosphatase dephosphorylates fructose 1,6-bisphosphate to form fructose 6-phosphate. This step reverses the nonequilibrium reaction catalyzed by the glycolytic enzyme phosphofructokinase-1.
- Phosphohexose isomerase converts fructose 6-phosphate to glucose 6-phosphate.
- Glucose-6-phosphatase dephosphorylates glucose 6-phosphate to form glucose, which can enter the bloodstream freely. This last reaction reverses the nonequilibrium reaction catalyzed by the glycolytic enzyme hexokinase.
Substrates Of Gluconeogenesis
The major substrates of gluconeogenesis are lactate, glycerol, and glucogenic amino acids.
Lactate is a product of anaerobic glycolysis. This ATP-generating process occurs when oxygen is limited, eg, during vigorous exercise or low-perfusion states. Cells that use this pathway, such as the erythrocytes, lack mitochondria and are not equipped for oxidative phosphorylation. The liver uses lactate in the blood to produce glucose via gluconeogenesis. Glucose gets released into the bloodstream, travels back to the erythrocytes and exercising muscles, and is metabolized back into lactate. This process is called the Cori cycle.[2]
Glycerol comes from adipose tissue lipolysis. This process breaks down triglycerides to form fatty acids and glycerol molecules. In the liver, glycerol kinase phosphorylates glycerol to form glycerol phosphate. Glycerol phosphate dehydrogenase oxidizes glycerol phosphate into the glycolytic intermediate, DHAP.[3]
Glucogenic amino acids enter the gluconeogenesis pathway via the citric acid cycle (see Image. Glucogenic Amino Acids). The first step is the deamination of the glucogenic amino acids into α-ketoacids, which are substrates in the citric acid cycle. From there, these α-ketoacids are converted to oxaloacetate, the substrate for PEPCK.
Regulation Of Gluconeogenesis
Gluconeogenesis reactions are highly endergonic and, thus, easily reversible. Controls at various levels ensure that each reaction moves forward until free glucose is produced. Glucagon is the most important promoter of gluconeogenesis. Secondary ones include the catecholamines, growth hormone, cortisol, and gluconeogenic substrates.[4][5]
The pancreatic alpha cells secrete glucagon in response to falling blood glucose levels. This hormone increases the concentration of cyclic adenosine monophosphate (cAMP), which inactivates glycolytic enzymes while activating gluconeogenic ones. Catecholamines produce the same effect by enhancing intracellular cAMP concentration.[6][7]
Insulin is a potent inhibitor of gluconeogenesis.[8] Falling insulin levels during fasting activate gluconeogenesis and the processes that increase the availability of gluconeogenic substrates.[4]
Table 1. Effect of Different Hormones on Key Glycolytic and Gluconeogenic Enzymes
| Enzyme | Effect of Glucagon and Catecholamines | Effect of Insulin |
| Glycolysis | ||
|
Inhibition | Induction |
|
Inhibition | Induction |
|
Inhibition | Induction |
| Gluconeogenesis | ||
|
Induction | Inhibition |
|
Induction | Inhibition |
|
Induction | Inhibition |
|
Induction | Inhibition |
The alanine cycle, aka the Cahill cycle, augments gluconeogenic glucose production during fasting. In skeletal muscle, alanine aminotransferase (ALT) transfers an α-amino group from glutamate to pyruvate, producing alanine and α-ketoglutarate. Alanine is released from skeletal muscle and taken up by the liver for transamination back to pyruvate. Pyruvate can then be used for gluconeogenesis.[9][10]
Organ Systems Involved
During the first 18 to 24 hours of fasting, gluconeogenesis mostly occurs in the liver. Prolonged starvation forces the kidneys to assume as much as 20% of the total glucose production by this pathway. Only the liver and kidneys have the gluconeogenic enzyme glucose-6-phosphatase and, thus, have the ability to convert glucose 6-phosphate into free glucose.[1][2]
Function
Gluconeogenesis maintains blood glucose levels during starvation. Some tissues in the human body rely almost exclusively on glucose as a metabolic fuel source. The brain, for example, requires approximately 120 g of glucose per day. Ketone bodies can serve as the brain's alternative fuel source. However, the testes, renal medulla, and erythrocytes cannot survive long periods without glucose. Gluconeogenesis starts 4 to 6 hours after fasting begins, peaking after 24 when hepatic glycogen is depleted.[1][2][15]
Pathophysiology
Von Gierke disease is an autosomal recessive condition notable for deficiency of the key gluconeogenic enzyme glucose-6-phosphatase. Gluconeogenesis is impaired, resulting in fasting hypoglycemia. Glycogenolysis is also affected, as free glucose in the last step of this pathway needs the same enzyme for conversion from glucose 6-phosphate. Other metabolic abnormalities that can manifest in von Gierke disease are hyperkalemia, hyperuricemia, and lactic acidosis.[11]
Clinical Significance
Treating Hyperglycemia in Diabetes
Diabetes may result from impaired insulin production or decreased insulin sensitivity. Besides stimulating glucose uptake from the bloodstream, insulin is also a potent gluconeogenesis inhibitor. Gluconeogenesis occurs at an unusually rapid rate in insulin deficiency or insensitivity, increasing the risk of hyperglycemia in patients with diabetes mellitus.[1]
Metformin, the first-line agent for type 2 diabetes mellitus management, has been shown to suppress hepatic gluconeogenesis by various mechanisms. This drug activates adenosine monophosphate-activated protein kinase (AMPK), which, in turn, inhibits hepatic lipogenesis and increases insulin sensitivity. AMPK activation also increases cAMP breakdown, further suppressing gluconeogenesis.[1][12]
Metformin also appears to directly inhibit glycerol-3-phosphate dehydrogenase, increasing NADH levels.[1][12] High intracellular NADH favors the formation of lactate over pyruvate by the lactate dehydrogenase reaction. Gluconeogenesis using lactate as a substrate cannot proceed without this molecule's conversion back to pyruvate.
At high doses, metformin also inhibits complex I of the electron transport chain, the ATP production that drives highly endergonic processes like gluconeogenesis.[1]
Hypoglycemia as a Result of Ethanol Consumption
Ethanol clearance from the body begins with its oxidation into acetaldehyde by hepatic alcohol dehydrogenase. This enzyme uses oxidized nicotinamide adenine dinucleotide (NAD+) as an electron acceptor. Aldehyde dehydrogenase oxidizes acetaldehyde further into acetate, which is readily excreted by the body. Aldehyde dehydrogenase also requires NAD+ as a cofactor. Thus, ethanol metabolism results in NADH accumulation.[13]
As previously mentioned, high intracellular NADH increases lactic acid formation, which can inhibit gluconeogenesis. Thus, heavy ethanol consumption can lead to both lactic acidosis and hypoglycemia.[13]
Hypoglycemia in the Preterm Infant
Preterm infants are particularly at risk of developing hypoglycemia. Low-birth-weight neonates have limited glycogen and fat stores but also express gluconeogenic enzymes at suboptimal levels. Preterm infants' energy stores can diminish quickly, as they cannot mount an adequate counterregulatory response.[4]
Media
(Click Image to Enlarge)
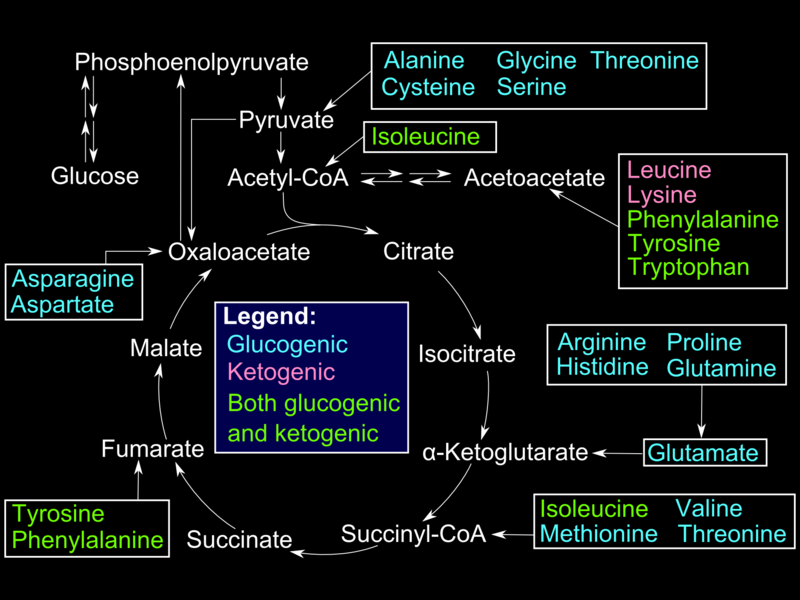
Glucogenic Amino Acids. This illustration shows how the glucogenic amino acids enter the Krebs cycle.
Image courtesy Dr Chaigasame
References
Zhang X, Yang S, Chen J, Su Z. Unraveling the Regulation of Hepatic Gluconeogenesis. Frontiers in endocrinology. 2018:9():802. doi: 10.3389/fendo.2018.00802. Epub 2019 Jan 24 [PubMed PMID: 30733709]
Chung ST, Chacko SK, Sunehag AL, Haymond MW. Measurements of Gluconeogenesis and Glycogenolysis: A Methodological Review. Diabetes. 2015 Dec:64(12):3996-4010. doi: 10.2337/db15-0640. Epub [PubMed PMID: 26604176]
da Silva IV, Rodrigues JS, Rebelo I, Miranda JPG, Soveral G. Revisiting the metabolic syndrome: the emerging role of aquaglyceroporins. Cellular and molecular life sciences : CMLS. 2018 Jun:75(11):1973-1988. doi: 10.1007/s00018-018-2781-4. Epub 2018 Feb 20 [PubMed PMID: 29464285]
Sharma A, Davis A, Shekhawat PS. Hypoglycemia in the preterm neonate: etiopathogenesis, diagnosis, management and long-term outcomes. Translational pediatrics. 2017 Oct:6(4):335-348. doi: 10.21037/tp.2017.10.06. Epub [PubMed PMID: 29184814]
Bankir L, Bouby N, Speth RC, Velho G, Crambert G. Glucagon revisited: Coordinated actions on the liver and kidney. Diabetes research and clinical practice. 2018 Dec:146():119-129. doi: 10.1016/j.diabres.2018.10.004. Epub 2018 Oct 16 [PubMed PMID: 30339786]
Droppelmann CA, Sáez DE, Asenjo JL, Yáñez AJ, García-Rocha M, Concha II, Grez M, Guinovart JJ, Slebe JC. A new level of regulation in gluconeogenesis: metabolic state modulates the intracellular localization of aldolase B and its interaction with liver fructose-1,6-bisphosphatase. The Biochemical journal. 2015 Dec 1:472(2):225-37. doi: 10.1042/BJ20150269. Epub 2015 Sep 28 [PubMed PMID: 26417114]
Borrebaek B, Bremer J, Davis EJ, Davis-Van Thienen W, Singh B. The effect of glucagon on the carbon flux from palmitate into glucose, lactate and ketone bodies, studied with isolated hepatocytes. The International journal of biochemistry. 1984:16(7):841-4 [PubMed PMID: 6468742]
Level 3 (low-level) evidenceHonma K, Kamikubo M, Mochizuki K, Goda T. Insulin-induced inhibition of gluconeogenesis genes, including glutamic pyruvic transaminase 2, is associated with reduced histone acetylation in a human liver cell line. Metabolism: clinical and experimental. 2017 Jun:71():118-124. doi: 10.1016/j.metabol.2017.03.009. Epub 2017 Mar 23 [PubMed PMID: 28521864]
Sarabhai T, Roden M. Hungry for your alanine: when liver depends on muscle proteolysis. The Journal of clinical investigation. 2019 Nov 1:129(11):4563-4566. doi: 10.1172/JCI131931. Epub [PubMed PMID: 31545302]
Hatazawa Y, Qian K, Gong DW, Kamei Y. PGC-1α regulates alanine metabolism in muscle cells. PloS one. 2018:13(1):e0190904. doi: 10.1371/journal.pone.0190904. Epub 2018 Jan 9 [PubMed PMID: 29315328]
Level 2 (mid-level) evidenceAdam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, Bali DS, El-Gharbawy A, Austin S, Pendyal S, Kishnani PS. Glycogen Storage Disease Type I. GeneReviews(®). 1993:(): [PubMed PMID: 20301489]
Hundal RS, Krssak M, Dufour S, Laurent D, Lebon V, Chandramouli V, Inzucchi SE, Schumann WC, Petersen KF, Landau BR, Shulman GI. Mechanism by which metformin reduces glucose production in type 2 diabetes. Diabetes. 2000 Dec:49(12):2063-9 [PubMed PMID: 11118008]
Tsai WW, Matsumura S, Liu W, Phillips NG, Sonntag T, Hao E, Lee S, Hai T, Montminy M. ATF3 mediates inhibitory effects of ethanol on hepatic gluconeogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2015 Mar 3:112(9):2699-704. doi: 10.1073/pnas.1424641112. Epub 2015 Feb 17 [PubMed PMID: 25730876]
Level 3 (low-level) evidence