Introduction
Rapidly progressive glomerulonephritis (RPGN) is a clinical and pathological syndrome, characterized by the following:
- Rapid loss of renal function over a very short period (days to weeks)
- Nephritic urine analysis—micro- or macroscopic hematuria, dysmorphic red blood cells (RBCs), RBC casts, and proteinuria.
- Characteristic histopathological findings on renal biopsy—cellular crescent formation in the glomeruli, which is a proliferative cellular response of parietal epithelial cells within the Bowman space. Due to its crescent shape, this condition is also known as crescentic glomerulonephritis.
RPGN and crescentic glomerulonephritis are often used interchangeably, and the progression of crescent formation can be associated with irreversible renal function loss. Generally, crescentic glomerulonephritis is defined as crescents affecting over half of the glomeruli; however, even a single crescent can potentially signify an underlying pathological process. Early diagnosis and treatment are crucial to prevent further renal function loss.
RPGN is broadly classified based on histopathology and immune complex deposition.[1][2]
- Type I: Anti-glomerular basement membrane (GBM) linear antibody deposition disease.
- Type II: Granular immune complex deposition disorders.
- Type III: Pauci-immune disease (absence of deposition, generally anti-neutrophil cytoplasmic antibody (ANCA)-positive vasculitis).
- Type IV: Double antibody positive for both ANCA and anti-GBM.
Some mixed variants are also reported with features of different types.[2]
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
RPGN is broadly classified based on histopathology and immune complex deposition as follows:
Anti-Glomerular Basement Membrane Disease
Circulating immunoglobulin G (IgG) antibodies are directed against an antigen normally present in the GBM and alveolar basement membrane, specifically the non-collagenous domain of the α-3 chain of type IV collagen. This anti-GBM antibody deposition accounts for approximately 10% to 15% of diffuse crescentic glomerulonephritis cases. About 40% to 60% of these cases are associated with alveolar hemorrhage, a condition known as Goodpasture syndrome.[3] Fewer than 10% of anti-GBM disease cases present with isolated pulmonary involvement.[4]
A small percentage of patients present with mild renal dysfunction, proteinuria, and hematuria. When these patients undergo a biopsy, it confirms the presence of anti-GBM disease, with linear IgG deposition on the GBM, but without crescenting glomerulonephritis. In addition, these patients can test negative for anti-GBM antibodies.[3] Other rare cases of diffuse alveolar hemorrhage or renal insufficiency have been reported with negative anti-GBM antibodies.[4] Please see Statpearls' companion reference, "Goodpasture Syndrome," for further information.
Granular Immune Complex Glomerulonephritis
Immune complex glomerulonephritis is believed to comprise 25% to 30% of RPGN cases. Pauci-immune RPGN is more common in adults, whereas immune complex RPGN is frequently encountered in children. A pediatric population study found that the most common causes of immune complex RPGN include IgA nephropathy, lupus nephritis, cryoglobulinemia, IgA vasculitis, and postinfectious glomerulonephritis.[1] Immune complex glomerulonephritis is also more prevalent in lower-income countries due to a higher burden of infectious diseases.[5]
The general consensus favors that immune complex–mediated crescentic glomerulonephritis has a more favorable outcome compared to either anti-GBM–mediated or pauci-immune RPGN. An exception to this is lupus, typically class IV, as a cause of RPGN, which is also often associated with positive ANCA.[5] In adults, immune complex glomerulonephritis can be idiopathic or secondary to the following:
- Postinfectious glomerulonephritis, especially after a Streptococcus infection
- Collagen vascular disease
- Lupus nephritis
- IgA vasculitis, also known as Henoch-Schönlein purpura, with IgA deposits and associated systemic vasculitis
- IgA nephropathy, also known as Berger disease, without vasculitis
- Mixed cryoglobulinemia
- Membranoproliferative glomerulonephritis
- Membranous nephropathy
- Fibrillary glomerulonephritis
- Secondary glomerulonephritis associated with hepatitis B and C [6]
Pauci-Immune Glomerulonephritis or Anti-Neutrophil Cytoplasmic Antibody–Associated Vasculitis
About 40% to 50% of adult cases are considered to have pauci-immune RPGN, also called ANCA-associated vasculitis or type III crescentic glomerulonephritis, representing the most common presentation in adults with RPGN. These cases typically fall into 3 main types as follows:
- Granulomatosis with polyangiitis, formerly known as Wegener granulomatosis, with renal involvement observed in about 80% of cases.
- Microscopic polyangiitis, with renal involvement observed in about 90% of cases.
- Eosinophilic granulomatosis with polyangiitis, also referred to as Churg-Strauss syndrome, with renal involvement observed in about 45% of total cases.
Eosinophilic granulomatosis with polyangiitis is associated with ANCA positivity in about 40% to 60% of patients, typically anti-myeloperoxidase (anti-MPO). As noted above, eosinophilic granulomatosis with polyangiitis is also less likely to be associated with RPGN compared to granulomatosis with polyangiitis and microscopic polyangiitis.[7] See StatPearls' companion references, "Granulomatosis with Polyangiitis," "Microscopic Polyangiitis," and "Eosinophilic Granulomatosis With Polyangiitis (Churg-Strauss Syndrome)," for more information.
Drugs Linked to Anti-Neutrophil Cytoplasmic Antibody–Associated Crescentic Glomerulonephritis
Medications can induce an ANCA-associated RPGN. Specifically, a high MPO titer is common. Levamisole, which is often found in contaminated cocaine, can cause the elevation of both anti-MPO and anti-proteinase 3 (PR3) antibodies, along with other autoantibodies, skin lesions, and arthralgias. Treatment is the same as that of other ANCA-associated RPGN cases, but possibly with shorter induction. The most common offending medications include:
- Hydralazine
- Propylthiouracil and methimazole
- Allopurinol
- Sulfasalazine
- Minocycline
- Penicillamine
- Rifampicin
- Aminoguanidine
- Sofosbuvir
- Anti-tumor necrosis factor-alpha (TNF-α) therapy for rheumatoid arthritis and ankylosing spondylitis [8]
Double-Positive Antibody Disease
Also called dual antibody disease, this type of crescentic glomerulonephritis is associated with a positive ANCA and anti-GBM antibody. Some studies indicate that 10% to 50% of patients with anti-GBM disease have detectable ANCA, typically anti-MPO. In contrast, up to 10% of patients with ANCA-associated vasculitis also have circulating anti-GBM antibodies.[5] Generally, the positive ANCA precedes the anti-GBM antibodies; therefore, it is postulated that ANCA leads to the anti-GBM antibodies by exposing epitopes on the GBM. Renal manifestations follow an anti-GBM pattern, whereas systemic symptoms are similar to those of ANCA vasculitis.[9][10] This double-positive characteristic comes into play during treatment selection.[11][12]
Epidemiology
RPGN is a rare condition worldwide. The incidence is around 7 cases per 1 million person-years in the United States, whereas 2 cases per 1 million are reported in the United Kingdom. Reported clusters are recognized worldwide, suggesting a possible environmental influence.[13] The disease is more common among the White population and possibly in the Asian population. The prevalence is less common in the Black population. The male-to-female ratio in most studies is approximately 1:1. The disease represents a bimodal distribution related to the mean age for clinical manifestation. In most series, the mean age for the first peak is around 30, and the second peak is in the late 60s. However, cases have been reported across a wide age range, from 2 to 92, although RPGN is uncommon in the pediatric population.[14][15]
GBM disease comprises 10% to 15% of all cases of diffuse crescentic glomerulonephritis, with an annual incidence of 0.5 to 0.9 cases per million, and it is predominantly observed in White patients. The mean age of the first peak is around 30, with a slight male predominance, and the second peak in the sixth decade of life has a female predominance.[3]
Pauci-immune RPGN is the most common type of crescentic glomerulonephritis with a 40% to 50% prevalence, also primarily in White patients, with a peak age of 60 to 85, and the majority of patients are ANCA-positive. RPGN is an infrequent cause of end-stage kidney disease in various case studies.[2] PR3-ANCA positivity is predominant in the United States and Europe, whereas MPO-ANCA positivity is more common in Japan.[9]
Pathophysiology
The primary characteristics of crescentic glomerulonephritis include GBM rupture followed by extra capillary fibrin precipitation, the proliferation of parietal cells, and the formation of capsular fibrosis in a crescent shape. Glomerular damage also allows leakage of T-cells and B-cells into the Bowman space.[6] Some crescents are reversible, but once multiple layers of epithelial cells transition to a fibrotic pathology, loss of renal function becomes irreversible. Consequently, therapeutic interventions target the upstream mechanisms of fibrosis.[5]
Each disease leads to this pathway through different mechanisms. In anti-GBM disease, circulating antibodies, typically IgG (rarely IgA), are directed against an antigen in the GBM (and the alveolar basement membranes in Goodpasture disease). Specifically, these antibodies target the non-collagenous domain of the α-3 chain of type IV collagen.[16] These antibodies induce injury to the glomerular capillary wall by activating local complement and polymorphonuclear leukocytes.[17] Environmental triggers such as smoking and hydrocarbon solvents can act as precipitating factors. The HLA-DR15 allele is a risk factor for anti-GBM disease, whereas HLA-DR1 may be protective against it.[18]
The mechanism of immune complex–mediated RPGN can be either through immune complex formation in the circulation with deposition in the glomerular capillary tuft or through immune complex production in situ in the glomerular capillary wall. The causative antigen spurring the immune complex formation can be either exogenous, such as virus or bacteria, or autogenous, such as nuclear or tumor antigens. Immune complex–mediated RPGN can result from multisystem diseases, such as lupus or IgA vasculitis, or as a complication of another primary glomerulonephritis, such as membranous or complement C3 glomerulonephritis.[18]
About 40% to 50% of RPGN cases involve anti-ANCAs. These antibodies target MPO, PR3, or both. When MPO and PR3 are both involved, it often suggests drug-induced pathogenesis. ANCAs trigger neutrophils to release neutrophil extracellular traps, allowing associated elastase, MPO, cathepsins, and histones to damage the renal microvasculature. Neutrophil extracellular traps also deposit autoantigens on the vascular walls, triggering T-cell and complement activation, further damaging the glomeruli capillaries and releasing T- and B-cells into the Bowman space.[6] The activation of local and systemic complement occurs mainly through the alternative pathway. Cytokines, including TNF-α, also play a significant role in the pathogenesis. The production of anti-plasminogen and plasminogen activator autoantibodies can inhibit fibrinolysis and predispose to fibrinoid necrosis and thrombophilia.[19] HLADRB1*1501 may increase susceptibility to PR3-associated ANCA vasculitis.[20]
Ultimately, the proliferation of parietal epithelial cells, fibrin polymerization, infiltration of monocytes/macrophages and T-cells, and myofibroblast invasion from the interstitium results in glomerular crescents. The production of interleukin-1, TNF-α, macrophage chemotactic protein-1, macrophage inflammatory factor, and transforming growth factor-beta also plays a role in the inflammatory process.[5]
Histopathology
Quantitative definitions aid in the classification of ANCA-associated lesions.[5][21]
- Focal: 50% or more normal glomeruli.
- Crescentic: 50% or more glomeruli with cellular crescents.
- Mixed: less than 50% normal, less than 50% crescentic, and less than 50% globally sclerotic glomeruli.
- Sclerotic: 50% or more globally sclerotic glomeruli.
The primary lesion of crescentic glomerulonephritis results from destroying the glomerular capillary wall and accumulating parietal and visceral epithelial cells in the Bowman space, forming a crescent. This process is accompanied by an accumulation of lymphocytes, macrophages, and myofibroblasts, resulting in diffuse, proliferative, necrotizing glomerulonephritis with crescent formation.
A crescent is a hyperplastic lesion involving more than 10% of the glomerulus. If a crescent comprises more than 75% cells and fibrin, with less than 25% being the fibrous matrix, it is called cellular. If the composition comprises 25% to 75% cells/fibrin, with the remainder being the fibrous matrix, the crescent is called fibrocellular. A crescent with less than 25% cells/fibrin and more than 75% fibrous matrix is referred to as a fibrous crescent.[5]
All lesions in anti-GBM disease are generally in the same nephritic stage. Interstitial inflammation is also found mainly in the periglomerular region. In the later stages, fibrosis develops rapidly over a few days to weeks, manifesting as glomerular sclerosis and obliteration. Immunofluorescence staining in anti-GBM disease shows linear IgG deposition along the glomeruli capillary walls. Staining of the renal tubular basement membrane is also possible.[6] Although lung biopsy is not commonly performed, if conducted, similar staining of the alveolar basement membrane can also be observed.[3]
In immune complex deposition disease, immunoglobulins, such as IgG, IgM, or IgA, deposit in a granular pattern along the capillary wall. The deposits are primarily subendothelial but can also be subepithelial.[6] With this disease form, lesions can progress in different stages.
On renal biopsy, ANCA-associated vasculitic lesions can be found in various stages of crescents, such as cellular, fibrocellular, or fibrous. In pauci-immune disease, immunofluorescence typically shows scant or no IgG, IgM, or complement C3 deposits. Occasionally, fibrinoid necrosis is found along with other lesions.[22][23] Individuals with P-ANCA and primarily anti-PR3 antibodies are more likely to show renal granulomas.[24]
Please see Image. Rapidly Progressing Glomerulonephritis.
History and Physical
A rapid decline in renal function occurs over weeks to months, with early nonspecific clinical features. Occasionally, hemoptysis and shortness of breath may be the first clinical feature.[9]
- Anti-GBM disease may result in diffuse alveolar hemorrhage along with renal complications. Patients present with symptoms such as shortness of breath, cough, hemoptysis, nephritis, hematuria, and edema. Patients younger than 30 show more pulmonary involvement compared to older patients.[3]
- Extrarenal manifestations suggestive of immune complex disorders or ANCA vasculitis include the following:
- Systemic—fevers, night sweats, weight loss, and arthralgia
- Oral—mucosa ulceration
- Eyes—scleritis
- Ear, nose, and throat—hearing impairment, otitis, epistaxis, nasal drainage, sinusitis, and nasal cartilage necrosis
- Respiratory—shortness of breath, cough, hoarseness of voice, and hemoptysis
- Gastrointestinal—abdominal pain secondary to ischemia, infarction, or pancreatitis
- Central nervous system—vision changes including blurry vision or vision loss, numbness, weakness, and stroke-like symptoms
- Skin—palpable purpura, ulcers, and livedo reticularis
If not promptly addressed, rapid and progressive loss of renal function may occur, ultimately leading to renal failure. Upon the onset of renal failure, patients may experience fatigue, loss of appetite, nausea, vomiting, and oliguria.[23] The rapid drop in filtration often decreases the amount of filtered protein; therefore, signs of nephrotic syndrome are uncommon.[6]
Some studies found that in patients with double-positive antibodies (positive for anti-GBM and ANCA), the renal manifestations follow an anti-GBM pattern clinically and histologically, whereas extrarenal manifestations are similar to those of ANCA-associated vasculitis.[6][17]
Evaluation
Clinical suspicion, as outlined in the History and Physical section, is supported with laboratory testing for the major causes of RPGN:
Laboratory Testing
- Urinalysis: Dysmorphic RBCs on microscopy are characteristic of glomerular hematuria and RBC casts are highly suggestive of this diagnosis.
- The urine protein-to-creatinine ratio should be evaluated but may not be significantly elevated until later disease stages.
- Elevated serum creatinine and abnormal electrolyte levels, such as potassium, magnesium, and calcium, further support renal dysfunction.
- A complete blood count with differential may show eosinophilia in eosinophilic granulomatosis with polyangiitis.
- Serology for anti-GBM antibodies should always be performed using enzyme-linked immunosorbent assay (ELISA) or western blot. However, as noted above, approximately 10% of patients with biopsy-confirmed anti-GBM disease do not have identifiable circulating antibodies with conventional assays. Therefore, a negative anti-GBM antibody test does not rule out the disease.[17] Chemiluminescence immunoassay is also being studied as a possibly more sensitive test.[3]
- Previously, ANCA testing was performed using indirect immunofluorescence, and its qualitative assay resulted in perinuclear ANCA (P-ANCA) or cytoplasmic ANCA (C-ANCA). However, a newer method utilizes ELISA, which identifies specific antigens and provides a quantitative titer for anti-PR-3 and anti-MPO. C-ANCA (a cytoplasmic antineutrophilic nuclear antibody with extranuclear cytoplasmic staining) is 90% PR-3 reactive and 10% MPO reactive; P-ANCA (a perinuclear antineutrophilic nuclear antibody with staining around the nucleus) is 90% MPO reactive and 10% PR-3 reactive.[25]
- Serologies for postinfectious causes include antistreptolysin titer for streptococcal infection, HIV, and hepatitis B and C.
- Complement C3 and C4 levels may be low in some forms of granular immune complex disorders causing RPGN, such as lupus, cryoglobulinemia, and primary membranoproliferative glomerulonephritis.
- Serologies for lupus include antinuclear antibodies, anti-double–stranded DNA, and anti-Smith.
- Rheumatoid factor and cryoglobulin levels should be checked in suspected cases of cryoglobulinemia.
- Two new antibodies have been identified and are associated with ANCA disease.
- Lysosome-associated membrane protein-2 (LAMP-2) antibodies: These antibodies are positive in more than 90% of ANCA-positive patients and also more than 90% of ANCA-negative pauci-immune crescentic glomerulonephritis patients. LAMP-2 antibodies activate neutrophils and also directly injure endothelium.[26][27][28]
- Anti-plasminogen antibodies: These antibodies range between 22% and 43% for PR3-positive ANCA-associated vasculitis and 6% to 27% for MPO-positive ANCA-associated vasculitis.
- Both correlate with venous thromboembolic events.[29]
- Other tests: Lactate dehydrogenase, reticulocyte count, and anticardiolipin antibodies often rule out other disease processes. A blood smear should also be performed to rule out other causes mimicking RPGN, such as thrombotic thrombocytopenic purpura or antiphospholipid syndrome.[9]
Imaging and Invasive Diagnostic Testing
- Chest x-ray and computed tomography scan can assess for signs of diffuse alveolar hemorrhage or cavitary lesions of vasculitis.
- Bronchoscopy can be performed if diffuse alveolar hemorrhage is suspected.
- Otolaryngologic evaluation and skin biopsy can be performed for vasculitis.
- Histological diagnosis through renal biopsy is the mainstay for a definitive diagnosis.
Treatment / Management
Untreated RPGN progresses to renal function loss over weeks to months. Initiating treatment as soon as possible is crucial to preserve renal function. Immunosuppression therapy is similar for all 3 disease etiologies; therefore, general therapeutic categories are addressed first, followed by disease-specific therapeutics.
Glucocorticoids are the longest-used and most-studied drugs. Glucocorticoid receptor inhibition helps reduce cellular crescent formation and proliferation and the migration of parietal epithelial cells into the Bowman space.[5] Significant evidence shows that initial treatment with corticosteroids along with immunosuppression results in greater effectiveness and lower rates of relapse in patients with ANCA-associated vasculitis, anti-GBM disease, and lupus-related RPGN; for these conditions, it is considered the standard of care. In cases where immunosuppression is contraindicated, plasmapheresis can be considered as an alternative. The coadministration of intravenous pulse corticosteroids and cyclophosphamide has been the most commonly used regimen to help reduce autoantibody production.[3][9](A1)
Other medications used include azathioprine, methotrexate, cyclosporine, and mycophenolate mofetil. Due to the rarity of the disease, randomized controlled trials are not common, and many protocols depend on clinician and institution experience.
Although immunosuppression along with corticosteroids is the preferred treatment, it may be contraindicated in situations of active infection, leukopenia, or liver dysfunction. Dose adjustments should also be made based on age and renal function. Cyclophosphamide can be given orally at a dose of 25 to 100 mg daily or as monthly pulses at a dose of 250 to 750 mg/m2 intravenously.
One such protocol is outlined in the table below.[9](A1)
Pulsed Cyclophosphamide Reductions for Renal Function and Age
|
Age (years) |
Creatinine, 1.7–3.4 mg/dL |
Creatinine, 3.4–5.7 mg/dL |
| <60 | 15 mg/kg/pulse | 12.5 mg/kg/pulse |
| 60-70 | 12.5 mg/kg/pulse | 10 mg/kg/pulse |
| >70 | 10 mg/kg/pulse | 7.5 mg/kg/pulse |
When serology or kidney biopsy results are not immediately available, empirical treatment is suggested before a definitive diagnosis is made. This empirical treatment includes a pulse intravenous dose of methylprednisolone, 500 mg or 1 g, for a minimum of 3 doses. Plasmapheresis may be considered empirically if the patient has hemoptysis, suggesting a severe form of Goodpasture disease. Subsequently, more specific treatment can be considered when more information is available.
Anti-Glomerular Basement Membrane Disease
Plasmapheresis, in combination with immunosuppressive agents, is the preferred treatment for anti-GBM disease to remove the autoantibodies. Initiating treatment as early as possible is crucial for preventing progressive renal failure and pulmonary damage.
Recommendations from the 2012 guidelines for Kidney Disease Improving Global Outcomes are initiating immunosuppression with cyclophosphamide plus corticosteroids and plasma exchange in all patients with Goodpasture syndrome, except in cases of patients that are dialysis-dependent at presentation and have 100% crescents on a biopsy without pulmonary hemorrhage. Rituximab can be substituted for cyclophosphamide in cases of adverse effects of cyclophosphamide or concerns for fertility in younger patients.[30]
The dose of plasmapheresis is 4 L of an exchange over 2 to 4 weeks. Typically, 5% albumin is the replacement fluid, but fresh frozen plasma (0.3-2 L) should be administered in cases of invasive procedures or when pulmonary hemorrhage is also noted. Plasmapheresis typically continues until antibody levels are suppressed or for 14 days. Treatment may be considered for an extended period if active pulmonary disease is apparent or the antibody level does not decline as expected. Plasmapheresis is always followed by immunosuppressive therapy—typically glucocorticoids and cyclophosphamide. Recent studies indicate the potential benefits of immunoadsorption agents as part of plasmapheresis in treating anti-GBM disease.[3][31]
Many protocols recommend initiating treatment with 1 gram of methylprednisolone given daily for 3 days, followed by oral glucocorticoid treatment with prednisolone at a dosage of 1 mg/kg/d (up to a maximum of 60 mg) given orally. The dosage should be reduced weekly to 20 mg by 6 weeks, then gradually tapered until entirely stopped by 6 months.[3] The dosage of cyclophosphamide is 1 to 2 mg/kg/d orally but not more than 100 mg/d for patients older than 60. Rituximab or mycophenolate mofetil is recommended for patients experiencing adverse effects from cyclophosphamide, such as gross hematuria. Of note, rituximab is removed by plasmapheresis, especially if plasmapheresis is performed within a 3-day window after rituximab infusion, so this must be taken into account when dosing both modalities.[32][33]
Recently, the anti-lymphocytic agent alemtuzumab has been studied to halt the progression of anti-GBM disease. Another promising medication is imlifidase, an IgG protease approved for severe anti-GBM disease. Studies show a reduction in antibody levels, but limited data are available at this time.[3]
The optimal duration of treatment is unclear. Anti-GBM antibody levels should be monitored for 1 to 2 weeks after initiating the treatment until 2 consecutive levels a week apart are negative. Following this remission phase, maintenance treatment with an agent associated with fewer adverse effects, such as low-dose prednisone or azathioprine, should be considered.
Immune Complex Crescentic Glomerulonephritis
Treatment depends on the underlying cause of the associated condition and should be treated accordingly. For example, IgA glomerulonephritis, lupus nephritis, and cryoglobulinemia each mandate their own systemic treatment. Immunosuppressive treatment is generally recommended for all causes except infectious agents, which are the most common etiology. Poststreptococcal glomerulonephritis typically recovers spontaneously, but glucocorticoids are occasionally used in cases of severely crescentic RPGN. See StatPearls' companion reference, "Post-streptococcal Glomerulonephritis," for more information.
Anti-Neutrophil Cytoplasmic Antibody–Positive Pauci-Immune Crescentic Glomerulonephritis
Treatment involves the following:
- An intravenous dose of methylprednisolone followed by oral prednisone
- Intravenous or oral cyclophosphamide or rituximab
- Plasmapheresis
- The duration of the therapy is 3 to 4 months
- Maintenance therapy is mandatory
The recommended initial treatment involves glucocorticoids along with either cyclophosphamide or rituximab with or without plasmapheresis. Plasmapheresis is indicated if renal function rapidly deteriorates or severe renal involvement is observed during presentation. Serum creatinine levels greater than 4 mg/mL, the requirement for dialysis, the presence of pulmonary hemorrhage, or coexisting anti-GBM antibodies are also indications of plasmapheresis. Once the treatment is completed, ANCA levels should be monitored at least every 1 to 3 months to assess for any signs of relapse. However, ANCA levels are not the sole guides to treatment and should be interpreted in a clinical context. Both ANCA subtypes are treated similarly, and treatment is determined by disease severity rather than antibody type.[9][34][35][36](A1)
B-cell modulators, such as rituximab, may effectively inhibit ANCA-producing plasma cells and decrease antigen presentation.[5] The RAVE study, a randomized control trial, demonstrated that rituximab is as effective as cyclophosphamide for disease remission and may be more effective compared to cyclophosphamide in preventing relapse.[37] Many clinicians prefer rituximab due to its ease of administration and fewer adverse effects.(A1)
Drugs Associated With Anti-Neutrophil Cytoplasmic Antibody Vasculitis
After discontinuing the medication or drug, the RPGN typically resolves. However, if renal function does not improve, treatment may be necessary for pauci-immune glomerulonephritis, as described above.
Double-Positive Antibody Crescentic Glomerulonephritis
Treatment for double-positive antibody crescentic glomerulonephritis follows the same approach as for pauci-immune glomerulonephritis, but plasmapheresis should be included. As mentioned earlier, renal manifestations follow an anti-GBM pattern, whereas systemic symptoms are similar to those of ANCA vasculitis.[9] Prolonged immunosuppression and long-term monitoring are crucial, as relapses can occur.[11][17][11](A1)
Differential Diagnosis
When diagnosing RPGN, other causes of reversible acute kidney injury, proteinuria, and hematuria should be ruled out, including the following:
- Prerenal acute kidney injury
- Acute kidney injury due to acute tubular necrosis
- Obstructive uropathy
- Nephrotic syndrome caused by focal segmental glomerulonephritis, minimal change disease, or membranous nephropathies
- Hematuria due to a urologic etiology
- Antiphospholipid antibody syndrome
- Thrombotic microangiopathy
Pertinent Studies and Ongoing Trials
Finerenone is a first-in-class selective nonsteroidal mineralocorticoid receptor antagonist approved by the Food and Drug Administration for treating patients with type 2 diabetes and chronic kidney disease. This medication has been shown to reduce cardiovascular mortality and proteinuria. Although preclinical animal studies suggest its effectiveness in slowing the progression of RPGN, further clinical studies are required to confirm these findings.[38][39]
Other novel therapies being studied for possible use include eculizumab and stem cell therapy.[31]
Prognosis
Early treatment initiation of RPGN is pivotal to halting rapidly progressive, irreversible damage. Age and gender do not significantly affect the overall prognosis; however, children typically do better after treatment compared to older adults.[40] The extent of proteinuria has not been shown to affect short-term prognosis. However, persistent proteinuria, despite treatment, indicates poor long-term outcomes. Renal function at the time of presentation reflects the severity of the disease, and higher serum creatinine at presentation, anuria, or dialysis requirement is associated with a poor outcome after the treatment and progression to renal failure.[40]
The extent of crescentic involvement in microscopic findings is indicative of the prognosis. Typically, a disease pattern of focal lesions with more than 50% normal glomeruli has a more favorable prognosis—over 90% of these cases show renal recovery at a 5-year follow-up after treatment. When more than 50% of glomeruli are globally sclerosed, the renal recovery is less than 25% at a 5-year follow-up period.[40]
Pretreatment antibody levels also affect the prognosis. The higher the anti-GBM antibody levels are at the time of diagnosis, the worse the renal outcome. ANCA levels that do not decrease with treatment or increase post-treatment suggest a more severe disease form.[9][40]
Relapse in anti-GBM disease is rare, possibly due to suppressor T-cell activity, with a study reporting a relapse rate of less than 3%.[17]
Without treatment, pauci-immune RPGN, the most common RPGN in Western countries, has a mortality rate of 80%. However, aggressive immunosuppression increases survival rates to 75% at 5 years. About 25% of patients with pauci-immune RPGN eventually progress to end-stage kidney disease, and worse outcomes are found in older patients, those who require dialysis, and those with pulmonary hemorrhage. About 40% of patients have a disease relapse; therefore, close monitoring is crucial.[24]
Mortality in patients with ANCA-associated disease is typically related to pulmonary involvement, especially in younger individuals. With immunosuppressive treatment, infection is the most common cause of mortality.
Crescentic glomerulonephritis is a known complication of IgA nephropathy. There is significant controversy surrounding the inclusion or exclusion of crescents in the Oxford-MEST clinical criteria; however, it is widely accepted that if crescents are present in more than 25% of glomeruli, the prognosis tends to be worse. Patients in this category are less likely to return to baseline renal function compared to those with fewer than 25% of the glomeruli showing crescents. Some patients diagnosed with IgA nephropathy are also ANCA-positive or have other signs of autoimmunity.[5]
Complications
Complications can be categorized into 2 forms—those arising directly from the disease and those resulting from the treatment.
Disease-Related Complications
- Pulmonary hemorrhage is often observed in cases of anti-GBM disease.
- In lupus-related RPGN, various immune complex deposits causing the renal RPGN pattern can also present with extrarenal manifestations, such as serositis, cerebritis, and skin lesions. See StatPearls' companion reference, "Lupus Nephritis," for more information.
Treatment-Related Complications
The primary complications associated with immunosuppressive therapy are various opportunistic infections, which can sometimes be life-threatening. Cyclophosphamide has specific other complications, including cystitis and hematuria. Older patients are particularly prone to infections and complications arising from cyclophosphamide use.[9] Plasmapheresis is associated with removing clotting factors, increasing the patient's susceptibility to bleeding.
Deterrence and Patient Education
Timely diagnosis and treatment significantly improve outcomes of RPGN. This high-risk population should be educated about the presenting symptoms of RPGN and instructed to seek medical help in case of decreased urine output, blood in the urine, shortness of breath, hemoptysis, and other systemic involvement. All patients should be educated about the importance of therapy compliance and treatment complications and should be instructed to seek medical help before stopping any medications. All patients should be educated about the significant adverse effects of immunosuppressive drugs, including various infections, teratogenic effects, and the importance of antibiotic prophylaxis and appropriate vaccinations to decrease opportunistic infections.
Pearls and Other Issues
The following points elucidate key aspects of RPGN, encompassing its terminology, pathophysiology, classification, treatment modalities, and prognostic implications.
- The terms RPGN and crescentic glomerulonephritis are used interchangeably.
- The underlying pathology involves GBM rupture followed by extra capillary fibrin precipitation, the proliferation of parietal cells, and the formation of capsular fibrosis in a crescent shape.
- The 4 main types include anti-glomerular membrane disease, immune complex–mediated RPGN, ANCA-associated vasculitis, and double antibody-positive RPGN.
- Treatment approaches vary by indication, but plasmapheresis is typically performed for anti-GBM disease and occasionally for ANCA-associated vasculitis.
- Rituximab may be more effective compared to cyclophosphamide in treating ANCA-associated vasculitis.
- About 40% of patients with ANCA-associated RPGN have a disease relapse; therefore, close post-treatment monitoring is crucial.
- RPGN associated with drugs and infection is often linked to a better outcome compared to autoimmune disease–associated RPGN.
- Early treatment is crucial. Once significant layers of crescents and fibrosis form, treatment is no longer effective.
Enhancing Healthcare Team Outcomes
Delivering patient-centered care for individuals with RPGN requires a collaborative effort among healthcare professionals, including clinicians, advanced practice practitioners, pharmacists, and other healthcare providers. First and foremost, healthcare providers must possess the necessary clinical skills and expertise when diagnosing, evaluating, and treating this condition. Proficiency in interpreting laboratory findings, recognizing potential complications, and understanding the need for urgent intervention are paramount. Moreover, a strategic approach involving evidence-based guidelines and individualized care plans tailored to each patient's unique circumstances is vital.
Primary care clinicians should refer patients to nephrologists promptly. Dialysis and critical care facilities are necessary for treating RPGN patients. A comprehensive interprofessional team approach is crucial for managing extrarenal manifestations and may also involve hematology for plasmapheresis and interventional radiology for catheter placement. Pharmacists and clinicians play pivotal roles in patient care and education. After discharge, patients may receive immunosuppressive therapy and require close follow-up to monitor recovery and for opportunistic infections. Lifelong dialysis is necessary for patients with poor outcomes. Timely referrals for kidney transplants are indicated in cases of end-stage kidney disease.
Ethical considerations come into play when determining treatment options and respecting patient autonomy in decision-making. Responsibilities within the interprofessional team should be clearly defined, with each member contributing their specialized knowledge and skills to optimize patient care. Effective interprofessional communication fosters a collaborative environment where information is shared, questions are encouraged, and concerns are addressed promptly.
Lastly, care coordination is pivotal in ensuring seamless and efficient patient care. Clinicians, advanced practitioners, pharmacists, and other healthcare providers must work together to streamline the patient's journey, from diagnosis through treatment and follow-up. This coordination minimizes errors, reduces delays, and enhances patient safety, ultimately leading to improved outcomes and patient-centered care that prioritizes the well-being and satisfaction of those affected by RPGN.
Media
(Click Image to Enlarge)
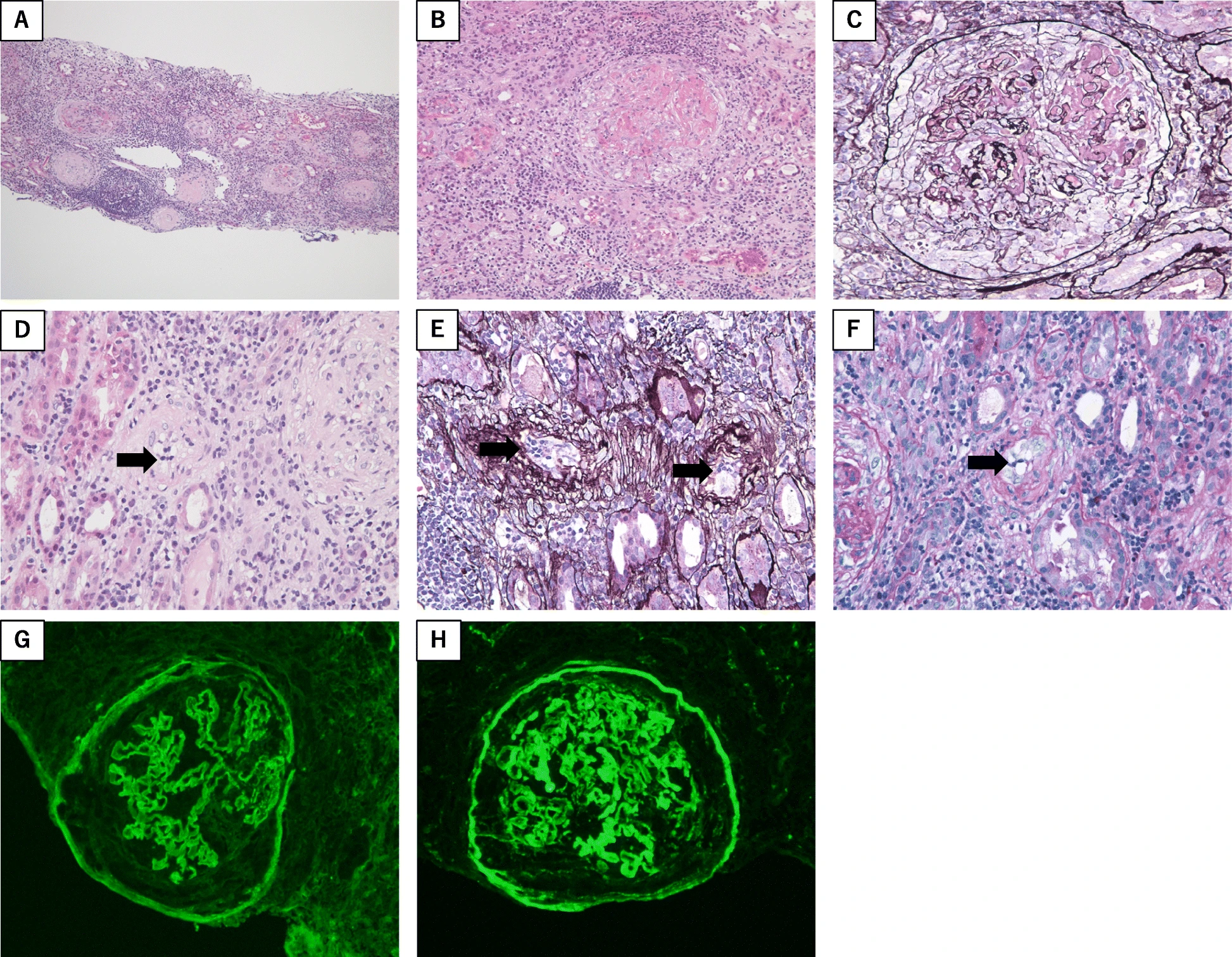
Rapidly Progressive Glomerulonephritis. Renal biopsy findings of this case.
Light microscopy showed (A) global sclerosis, tubulitis, interstitial inflammation (B and C) cellular crescents with segmental fibrinoid necrosis (D, E, and F) endarteritis of arterioles showing the accumulation of mononuclear cells in the arteriole walls (indicated by arrows)
Immunofluorescence microscopy showed linear deposits of (G) IgG and (H) C3c along the glomerular basement membrane
Staining with hematoxylin and eosin stain (A) low-power field; (B and D) high-power field
Periodic acid methenamine silver (C and E) high-power field
Periodic acid-Schiff stain (E and F) high power field
Kato M, Wakiya R, Kameda T, et al. The development of rapidly progressive glomerulonephritis associated with both antineutrophil cytoplasmic antibody-associated vasculitis and anti-glomerular basement membrane nephritis in the course of nontuberculous mycobacterium infection: a case report. BMC Rheumatol. 2020;4(1):68.
CC BY 4.0
References
Mohamed ON, Ibrahim SA, Saleh RK, Issa AS, Setouhi A, Rabou AAA, Mohamed MR, Kamel SF. Clinicopathological characteristics and predictors of outcome of rapidly progressive glomerulonephritis: a retrospective study. BMC nephrology. 2024 Mar 18:25(1):103. doi: 10.1186/s12882-024-03532-y. Epub 2024 Mar 18 [PubMed PMID: 38500101]
Level 2 (mid-level) evidenceMoroni G, Ponticelli C. Rapidly progressive crescentic glomerulonephritis: Early treatment is a must. Autoimmunity reviews. 2014 Jul:13(7):723-9. doi: 10.1016/j.autrev.2014.02.007. Epub 2014 Mar 19 [PubMed PMID: 24657897]
Reggiani F, L'Imperio V, Calatroni M, Pagni F, Sinico RA. Goodpasture syndrome and anti-glomerular basement membrane disease. Clinical and experimental rheumatology. 2023 Apr:41(4):964-974. doi: 10.55563/clinexprheumatol/tep3k5. Epub 2023 Mar 30 [PubMed PMID: 36995324]
Vries TB, Boerma S, Doornebal J, Dikkeschei B, Stegeman C, Veneman TF. Goodpasture's Syndrome with Negative Anti-glomerular Basement Membrane Antibodies. European journal of case reports in internal medicine. 2017:4(8):000687. doi: 10.12890/2017_000687. Epub 2017 Jul 13 [PubMed PMID: 30755961]
Anguiano L, Kain R, Anders HJ. The glomerular crescent: triggers, evolution, resolution, and implications for therapy. Current opinion in nephrology and hypertension. 2020 May:29(3):302-309. doi: 10.1097/MNH.0000000000000596. Epub [PubMed PMID: 32132388]
Level 3 (low-level) evidenceGluhovschi C, Gadalean F, Velciov S, Nistor M, Petrica L. Three Diseases Mediated by Different Immunopathologic Mechanisms-ANCA-Associated Vasculitis, Anti-Glomerular Basement Membrane Disease, and Immune Complex-Mediated Glomerulonephritis-A Common Clinical and Histopathologic Picture: Rapidly Progressive Crescentic Glomerulonephritis. Biomedicines. 2023 Nov 6:11(11):. doi: 10.3390/biomedicines11112978. Epub 2023 Nov 6 [PubMed PMID: 38001978]
Grayson PC, Ponte C, Suppiah R, Robson JC, Craven A, Judge A, Khalid S, Hutchings A, Luqmani RA, Watts RA, Merkel PA, DCVAS Study Group. 2022 American College of Rheumatology/European Alliance of Associations for Rheumatology Classification Criteria for Eosinophilic Granulomatosis with Polyangiitis. Annals of the rheumatic diseases. 2022 Mar:81(3):309-314. doi: 10.1136/annrheumdis-2021-221794. Epub 2022 Feb 2 [PubMed PMID: 35110334]
Hogan JJ, Markowitz GS, Radhakrishnan J. Drug-induced glomerular disease: immune-mediated injury. Clinical journal of the American Society of Nephrology : CJASN. 2015 Jul 7:10(7):1300-10. doi: 10.2215/CJN.01910215. Epub 2015 Jun 19 [PubMed PMID: 26092827]
Arimura Y, Muso E, Fujimoto S, Hasegawa M, Kaname S, Usui J, Ihara T, Kobayashi M, Itabashi M, Kitagawa K, Hirahashi J, Kimura K, Matsuo S. Evidence-based clinical practice guidelines for rapidly progressive glomerulonephritis 2014. Clinical and experimental nephrology. 2016 Jun:20(3):322-41. doi: 10.1007/s10157-015-1218-8. Epub [PubMed PMID: 27099135]
Level 1 (high-level) evidencePacheco M, Silva JE, Silva C, Soares N, Almeida J. Double-Positive Anti-GBM and ANCA-MPO Vasculitis Presenting With Crescentic Glomerulonephritis. Cureus. 2021 May 2:13(5):e14806. doi: 10.7759/cureus.14806. Epub 2021 May 2 [PubMed PMID: 34094762]
Walsh M, Merkel PA, Peh CA, Szpirt W, Guillevin L, Pusey CD, De Zoysa J, Ives N, Clark WF, Quillen K, Winters JL, Wheatley K, Jayne D, PEXIVAS Investigators. Plasma exchange and glucocorticoid dosing in the treatment of anti-neutrophil cytoplasm antibody associated vasculitis (PEXIVAS): protocol for a randomized controlled trial. Trials. 2013 Mar 14:14():73. doi: 10.1186/1745-6215-14-73. Epub 2013 Mar 14 [PubMed PMID: 23497590]
Level 1 (high-level) evidenceClerte M, Philip R, Levi C, Cornec-Le Gall E, Audard V, Huart A, Puéchal X, Touzot M, Rabot N, Thervet É, Aouba A, Karras A. Renal and overall outcomes of double-positive (ANCA and anti-GBM antibodies) patients compared to ANCA-associated vasculitis patients with severe renal involvement: A multicenter retrospective study with systematic renal pathology analysis. Scandinavian journal of rheumatology. 2022 May:51(3):205-213. doi: 10.1080/03009742.2021.1920120. Epub 2021 Jun 25 [PubMed PMID: 34169779]
Level 1 (high-level) evidenceBerti A, Cornec-Le Gall E, Cornec D, Casal Moura M, Matteson EL, Crowson CS, Ravindran A, Sethi S, Fervenza FC, Specks U. Incidence, prevalence, mortality and chronic renal damage of anti-neutrophil cytoplasmic antibody-associated glomerulonephritis in a 20-year population-based cohort. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 2019 Sep 1:34(9):1508-1517. doi: 10.1093/ndt/gfy250. Epub [PubMed PMID: 30102330]
Moutzouris DA, Herlitz L, Appel GB, Markowitz GS, Freudenthal B, Radhakrishnan J, D'Agati VD. Renal biopsy in the very elderly. Clinical journal of the American Society of Nephrology : CJASN. 2009 Jun:4(6):1073-82. doi: 10.2215/CJN.00990209. Epub 2009 May 14 [PubMed PMID: 19443626]
Level 2 (mid-level) evidenceLingaraj U, Mallappa SS, Neminah RE, Mohan SM, Venkatesh L, Gurusiddaiah SC, Rachaiah NM. A "Mini-Epidemic" of anti-glomerular basement membrane disease: Clinical and epidemiological study. Saudi journal of kidney diseases and transplantation : an official publication of the Saudi Center for Organ Transplantation, Saudi Arabia. 2017 Sep-Oct:28(5):1057-1063. doi: 10.4103/1319-2442.215128. Epub [PubMed PMID: 28937063]
Cui Z, Zhao MH, Jia XY, Wang M, Hu SY, Wang SX, Yu F, Brown KL, Hudson BG, Pedchenko V. Antibodies to α5 chain of collagen IV are pathogenic in Goodpasture's disease. Journal of autoimmunity. 2016 Jun:70():1-11. doi: 10.1016/j.jaut.2016.04.001. Epub 2016 Apr 23 [PubMed PMID: 27117167]
McAdoo SP, Pusey CD. Anti-Glomerular Basement Membrane Disease. Clinical journal of the American Society of Nephrology : CJASN. 2017 Jul 7:12(7):1162-1172. doi: 10.2215/CJN.01380217. Epub 2017 May 17 [PubMed PMID: 28515156]
Ooi JD, Petersen J, Tan YH, Huynh M, Willett ZJ, Ramarathinam SH, Eggenhuizen PJ, Loh KL, Watson KA, Gan PY, Alikhan MA, Dudek NL, Handel A, Hudson BG, Fugger L, Power DA, Holt SG, Coates PT, Gregersen JW, Purcell AW, Holdsworth SR, La Gruta NL, Reid HH, Rossjohn J, Kitching AR. Dominant protection from HLA-linked autoimmunity by antigen-specific regulatory T cells. Nature. 2017 May 11:545(7653):243-247. doi: 10.1038/nature22329. Epub 2017 May 3 [PubMed PMID: 28467828]
Jennette JC, Falk RJ, Hu P, Xiao H. Pathogenesis of antineutrophil cytoplasmic autoantibody-associated small-vessel vasculitis. Annual review of pathology. 2013 Jan 24:8():139-60. doi: 10.1146/annurev-pathol-011811-132453. Epub [PubMed PMID: 23347350]
Level 3 (low-level) evidenceCao Y, Schmitz JL, Yang J, Hogan SL, Bunch D, Hu Y, Jennette CE, Berg EA, Arnett FC Jr, Jennette JC, Falk RJ, Preston GA. DRB1*15 allele is a risk factor for PR3-ANCA disease in African Americans. Journal of the American Society of Nephrology : JASN. 2011 Jun:22(6):1161-7. doi: 10.1681/ASN.2010101058. Epub 2011 May 26 [PubMed PMID: 21617122]
Level 2 (mid-level) evidenceBerden AE, Ferrario F, Hagen EC, Jayne DR, Jennette JC, Joh K, Neumann I, Noël LH, Pusey CD, Waldherr R, Bruijn JA, Bajema IM. Histopathologic classification of ANCA-associated glomerulonephritis. Journal of the American Society of Nephrology : JASN. 2010 Oct:21(10):1628-36. doi: 10.1681/ASN.2010050477. Epub 2010 Jul 8 [PubMed PMID: 20616173]
Sethi S, Fervenza FC. Standardized classification and reporting of glomerulonephritis. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 2019 Feb 1:34(2):193-199. doi: 10.1093/ndt/gfy220. Epub [PubMed PMID: 30124958]
Alchi B, Griffiths M, Sivalingam M, Jayne D, Farrington K. Predictors of renal and patient outcomes in anti-GBM disease: clinicopathologic analysis of a two-centre cohort. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 2015 May:30(5):814-21. doi: 10.1093/ndt/gfu399. Epub 2015 Jan 20 [PubMed PMID: 25609740]
Level 2 (mid-level) evidenceSyed R, Rehman A, Valecha G, El-Sayegh S. Pauci-Immune Crescentic Glomerulonephritis: An ANCA-Associated Vasculitis. BioMed research international. 2015:2015():402826. doi: 10.1155/2015/402826. Epub 2015 Nov 25 [PubMed PMID: 26688808]
Radice A, Sinico RA. Antineutrophil cytoplasmic antibodies (ANCA). Autoimmunity. 2005 Feb:38(1):93-103 [PubMed PMID: 15804710]
Kain R, Matsui K, Exner M, Binder S, Schaffner G, Sommer EM, Kerjaschki D. A novel class of autoantigens of anti-neutrophil cytoplasmic antibodies in necrotizing and crescentic glomerulonephritis: the lysosomal membrane glycoprotein h-lamp-2 in neutrophil granulocytes and a related membrane protein in glomerular endothelial cells. The Journal of experimental medicine. 1995 Feb 1:181(2):585-97 [PubMed PMID: 7836914]
Kain R, Exner M, Brandes R, Ziebermayr R, Cunningham D, Alderson CA, Davidovits A, Raab I, Jahn R, Ashour O, Spitzauer S, Sunder-Plassmann G, Fukuda M, Klemm P, Rees AJ, Kerjaschki D. Molecular mimicry in pauci-immune focal necrotizing glomerulonephritis. Nature medicine. 2008 Oct:14(10):1088-96. doi: 10.1038/nm.1874. Epub 2008 Oct 5 [PubMed PMID: 18836458]
Level 3 (low-level) evidenceKain R, Tadema H, McKinney EF, Benharkou A, Brandes R, Peschel A, Hubert V, Feenstra T, Sengölge G, Stegeman C, Heeringa P, Lyons PA, Smith KG, Kallenberg C, Rees AJ. High prevalence of autoantibodies to hLAMP-2 in anti-neutrophil cytoplasmic antibody-associated vasculitis. Journal of the American Society of Nephrology : JASN. 2012 Mar:23(3):556-66. doi: 10.1681/ASN.2011090920. Epub 2012 Feb 9 [PubMed PMID: 22323643]
Level 2 (mid-level) evidenceGöçeroğlu A, Grenmyr E, Berden AE, Hagen EC, Bunch D, Sommarin Y, Bruijn JA, Bajema IM, Wieslander J. Anti-plasminogen antibodies in ANCA-associated vasculitis: An optimized anti-plasminogen assay. PloS one. 2018:13(11):e0207064. doi: 10.1371/journal.pone.0207064. Epub 2018 Nov 12 [PubMed PMID: 30419041]
Heitz M, Carron PL, Clavarino G, Jouve T, Pinel N, Guebre-Egziabher F, Rostaing L. Use of rituximab as an induction therapy in anti-glomerular basement-membrane disease. BMC nephrology. 2018 Sep 20:19(1):241. doi: 10.1186/s12882-018-1038-7. Epub 2018 Sep 20 [PubMed PMID: 30236081]
Greenhall GH, Salama AD. What is new in the management of rapidly progressive glomerulonephritis? Clinical kidney journal. 2015 Apr:8(2):143-50. doi: 10.1093/ckj/sfv008. Epub 2015 Feb 19 [PubMed PMID: 25815169]
Apaydin S, The treatment of ANCA-associated rapidly-progressive glomerulonephritis and Goodpasture syndrome with therapeutic apheresis. Transfusion and apheresis science : official journal of the World Apheresis Association : official journal of the European Society for Haemapheresis. 2018 Feb [PubMed PMID: 29503131]
Puisset F, White-Koning M, Kamar N, Huart A, Haberer F, Blasco H, Le Guellec C, Lafont T, Grand A, Rostaing L, Chatelut E, Pourrat J. Population pharmacokinetics of rituximab with or without plasmapheresis in kidney patients with antibody-mediated disease. British journal of clinical pharmacology. 2013 Nov:76(5):734-40. doi: 10.1111/bcp.12098. Epub [PubMed PMID: 23432476]
McGregor JG, Hogan SL, Hu Y, Jennette CE, Falk RJ, Nachman PH. Glucocorticoids and relapse and infection rates in anti-neutrophil cytoplasmic antibody disease. Clinical journal of the American Society of Nephrology : CJASN. 2012 Feb:7(2):240-7. doi: 10.2215/CJN.05610611. Epub 2011 Dec 1 [PubMed PMID: 22134625]
Faurschou M, Westman K, Rasmussen N, de Groot K, Flossmann O, Höglund P, Jayne DR, European Vasculitis Study Group. Brief Report: long-term outcome of a randomized clinical trial comparing methotrexate to cyclophosphamide for remission induction in early systemic antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis and rheumatism. 2012 Oct:64(10):3472-7. doi: 10.1002/art.34547. Epub [PubMed PMID: 22614882]
Level 1 (high-level) evidenceGeetha D, Hruskova Z, Segelmark M, Hogan J, Morgan MD, Cavero T, Eriksson P, Seo P, Manno RL, Dale J, Harper L, Tesar V, Jayne DR. Rituximab for treatment of severe renal disease in ANCA associated vasculitis. Journal of nephrology. 2016 Apr:29(2):195-201. doi: 10.1007/s40620-015-0208-y. Epub 2015 May 19 [PubMed PMID: 25986390]
Stone JH, Merkel PA, Spiera R, Seo P, Langford CA, Hoffman GS, Kallenberg CG, St Clair EW, Turkiewicz A, Tchao NK, Webber L, Ding L, Sejismundo LP, Mieras K, Weitzenkamp D, Ikle D, Seyfert-Margolis V, Mueller M, Brunetta P, Allen NB, Fervenza FC, Geetha D, Keogh KA, Kissin EY, Monach PA, Peikert T, Stegeman C, Ytterberg SR, Specks U, RAVE-ITN Research Group. Rituximab versus cyclophosphamide for ANCA-associated vasculitis. The New England journal of medicine. 2010 Jul 15:363(3):221-32. doi: 10.1056/NEJMoa0909905. Epub [PubMed PMID: 20647199]
Level 1 (high-level) evidenceBärfacker L, Kuhl A, Hillisch A, Grosser R, Figueroa-Pérez S, Heckroth H, Nitsche A, Ergüden JK, Gielen-Haertwig H, Schlemmer KH, Mittendorf J, Paulsen H, Platzek J, Kolkhof P. Discovery of BAY 94-8862: a nonsteroidal antagonist of the mineralocorticoid receptor for the treatment of cardiorenal diseases. ChemMedChem. 2012 Aug:7(8):1385-403. doi: 10.1002/cmdc.201200081. Epub 2012 Jul 12 [PubMed PMID: 22791416]
Ma FY, Han Y, Nikolic-Paterson DJ, Kolkhof P, Tesch GH. Suppression of Rapidly Progressive Mouse Glomerulonephritis with the Non-Steroidal Mineralocorticoid Receptor Antagonist BR-4628. PloS one. 2015:10(12):e0145666. doi: 10.1371/journal.pone.0145666. Epub 2015 Dec 23 [PubMed PMID: 26700873]
Salmela A, Törnroth T, Poussa T, Ekstrand A. Prognostic Factors for Survival and Relapse in ANCA-Associated Vasculitis with Renal Involvement: A Clinical Long-Term Follow-Up Study. International journal of nephrology. 2018:2018():6369814. doi: 10.1155/2018/6369814. Epub 2018 Oct 16 [PubMed PMID: 30410799]