Introduction
Gastritis is the inflammation of the gastric mucosa and is often used to describe the abnormal appearance of abnormal gastric mucosa on endoscopy or radiology. Gastritis encompasses infectious or immunological inflammation of the gastric mucosa and the host response. Histopathological evidence of inflammation in the stomach lining is essential to diagnose this condition. Gastropathy is a gastric mucosal disorder without inflammation, featuring epithelial injury and subsequent regeneration. Gastritis and gastropathy are not mutually exclusive conditions and might sometimes coexist. In clinical practice, gastritis may be accompanied by signs of mucosal injury, whereas gastropathy may present with an inflammatory reaction in the gastric mucosa.
Gastritis is classified based on the acuity of the condition (acute versus chronic), the histological features of inflammation, or the etiology. Although the categorization and classification of gastritis are not universally accepted, understanding the histological characteristics and etiological factors associated with the different types of gastritis is essential. Appropriate histological evaluation is also essential in devising management plans for this disease. The primary objective is to equip treating clinicians with the ability to improve patient outcomes through early intervention.[1]
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
Acute Gastritis
Acute gastritis is temporary stomach lining inflammation caused by stress on the gastric mucosa, manifesting as either hemorrhagic or non-hemorrhagic symptoms. This condition can develop due to various factors, including uremia, ischemia, shock, corrosive agents, medications, radiation, trauma, severe burns, sepsis, or alkaline-bile reflux. Certain infections, such as enteroviruses, can also cause a self-limited episode of gastritis. Acute gastritis may result from reduced gastric mucus secretion, mucosal barrier disruption, or decreased mucosal blood flow, depending on the underlying cause.[2]
Chronic Gastritis
Chronic gastritis is categorized into 2 forms—atrophic and non-atrophic. The primary cause of chronic gastritis is a Helicobacter pylori infection, which typically starts with a non-atrophic morphology. The non-atrophic form of chronic gastritis can progress to atrophic without treatment. The most common cause of atrophic chronic gastritis is autoimmune gastritis, though the etiology remains unclear. Autoimmune gastritis exhibits a chronic mononuclear inflammation accompanied by severe atrophic gastritis, which usually affects the corpus, along with the presence of autoantibodies against parietal cells or the intrinsic factor. However, whether autoimmune gastritis is an independent disorder or if an H pylori infection triggers the autoimmune response in susceptible individuals is unclear.[3]
Reactive Gastritis
Reactive gastritis or gastropathy has numerous causative factors with acute gastritis. Reactive gastritis may be caused by specific medications, alcohol consumption, radiation exposure, and duodenal (bile) reflux. These causative agents lead to histological mucosal lesions characterized by low-grade inflammation of the gastric mucosa. Although usually asymptomatic, they are revealed through endoscopy, often showing multiple erosions or ulcers without signs of atrophic changes. The use of immune checkpoint inhibitors to treat various malignancies has contributed to the incidence of reactive gastritis, although the condition remains considerably rare.[4]
The Sydney System of Classification for Gastritis
The Histological Division of the Sydney System was introduced in 1990 and has since become the most widely cited classification system for the morphological features of gastritis in endoscopic biopsies. This system conveys information about the type, severity, and extent of gastric pathology. The classification system conveys the topography of gastritis, which can be restricted to the antrum or corpus or involve the entire stomach (pan gastritis). If the etiology of the disease is known, this is added as a prefix to denote the topography. For instance, the label "autoimmune corpus gastritis" is used if the disease is autoimmune.
The Sydney System of Classification further delineates 5 graded morphological variables that may be added as a suffix to the core topography. These variables include the type or chronicity of inflammation, gastritis activity, intestinal metaplasia, the extent of atrophy, and the presence or absence of Heliobacter pylori. The morphological features are graded as absent, mild, moderate, or severe. The Sydney System of Classification recommends at least 2 random biopsies from both the antrum and corpus, along with an additional biopsy from the incisura angularis. Although the classification system provides a standardized and concise means of documenting the extent and severity of gastritis, the method for predicting or forecasting future morphological changes is impossible.[1][5]
Classification of Gastritis Based on Etiological Factors
An alternative approach to classifying gastritis considers the etiology and chronicity of the inflammation. This approach categorizes gastritis into 3 main subtypes—acute, chronic, and special. Infectious gastritis is most commonly attributed to the global prevalence of H pylori infection. Other types of infectious gastritis include phlegmonous gastritis (caused by pyogenic bacteria), mycobacterial gastritis (caused by Mycobacterium tuberculosis), syphilitic gastritis, viral gastritis (caused by cytomegalovirus and herpes simplex virus), parasitic gastritis (caused by Anisakis, Cryptosporidium, Ascaris lumbricoides, Giardia, Toxoplasma, and Schistosoma), and fungal gastritis (caused by Candida, Aspergillus, Mucor, Coccidioides, Histoplasma, Cryptococcus neoformans, Pneumocystis carinii, and Torulopsis glabrata).
Granulomatous gastritis is a special gastritis observed in patients with Crohn disease and sarcoidosis. Lymphocytic gastritis, collagenous gastritis, and eosinophilic gastritis are additional special subtypes of gastritis with unclear etiologies. Lymphocytic and collagenous gastritis have been associated with celiac disease, whereas eosinophilic gastritis has a strong connection to atopic conditions and food allergens.[3][4]
According to the 2015 Kyoto Consensus Conference, a classification of gastritis based on etiological factors is outlined as follows:
- Autoimmune gastritis
- Infectious gastritis
- Gastric phlegmon
- Bacterial gastritis
- H pylori-induced
- Enterococcal
- Mycobacterial
- Viral gastritis
- Cytomegaloviral
- Enteroviral
- Fungal gastritis
- Parasitic gastritis
- Gastric anisakiasis
- Cryptosporidium
- Gastric Strongyloides stercoralis
- Gastritis due to other diseases
- Crohn disease
- Sarcoidosis
- Vasculitis
- Gastritis due to external causes
- Alcoholism
- Radiation
- Chemicals
- Special gastritis
- Allergic gastritis
- Gastritis due to biliary reflux
- Lymphocytic gastritis
- Ménétrier disease
- Eosinophilic gastritis [6]
Epidemiology
Determining the incidence of acute gastritis can be challenging due to the common causes, such as enterovirus infections, which typically result in mild and self-limited episodes that go unreported. Other factors leading to acute gastritis, such as sepsis, ischemia, and caustic injury, are relatively rare compared to chronic H pylori–associated gastritis and chronic atrophic (autoimmune) gastritis. Recent data demonstrates chronic atrophic gastritis is estimated to affect approximately 25% of the global population. Furthermore, the risk of developing chronic atrophic gastritis is about 2.4 times higher in patients with H pylori.[2][7]
In Western populations, a declining incidence of infectious gastritis is thought to be caused by an increasing prevalence of autoimmune gastritis. Autoimmune gastritis is more prevalent in women and older individuals, with estimated rates ranging from 2% to 5%. However, the available data may have limited reliability.[8][9] Chronic H pylori–associated nonatrophic gastritis continues to be highly prevalent in developing countries. In Western populations, the prevalence of H pylori infection in children is approximately 10%, whereas the prevalence is 50% in developing countries.[10][11] The prevalence of H pylori infection in developing countries varies significantly based on geographical region and socioeconomic conditions. For instance, the prevalence is approximately 69% in Africa, 78% in South America, and 51% in Asia.[12]
Socioeconomic and environmental factors are crucial in the global transmission of H pylori infections. These factors encompass family hygiene practices, household overcrowding, and dietary habits. The pediatric origin of H pylori infection is currently considered the primary determinant of H pylori–associated gastritis within a community.[3] The estimated prevalence of atrophic gastritis in the United States is as high as 15%. Current estimates suggest that this prevalence is likely higher in populations with a higher baseline prevalence of H pylori infection, such as non-white racial and ethnic minorities and first-generation immigrants from countries with a high prevalence of H pylori infections.
Atrophic gastritis not associated with H pylori infection is relatively uncommon, with an estimated prevalence of 0.5% to 2%, which is likely an overestimation. The comorbidity of other autoimmune diseases increases the prevalence of this condition, with up to one-third of patients with autoimmune thyroid disease having concomitant autoimmune gastritis. In contrast to H pylori–associated atrophic gastritis, purely autoimmune gastritis does not exhibit racial or ethnic variations.[13]
Pathophysiology
H pylori–Associated Gastritis
H pylori is a flagellated gram-negative bacterium transmitted through environmental factors or the fecal-oral or oral-oral routes.[1] The pathophysiology of H pylori–induced gastritis involves a complex interaction between bacterial virulence factors and the host's immune responses. This interplay disrupts the gastric mucosal barrier and leads to chronic inflammation. H pylori possesses virulence factors that facilitate cell adhesion, cause cell damage, disrupt tight junctions, and evade the host immune response. Notably, the cytotoxin-associated gene A (CagA) is a potent inducer of inflammation and is associated with the development of gastric cancer.[14]
The survival and colonization of H pylori in the stomach depend on the urease produced by the bacterium. Urease catalyzes urea hydrolysis, releasing ammonia and forming a protective layer around the bacterium.[15] The ammonia also helps neutralize the acidic microenvironment of the stomach, allowing the bacterium to thrive in the stomach's low pH conditions.[16] Subsequently, the flagellum and other mucolytic enzymes assist the organism in penetrating the mucus layer and reaching the gastric epithelium, which attaches to the epithelial cells.[17]
The attachment of H pylori to the epithelial cells triggers an inflammatory response, which is the distinguishing characteristic of gastritis. Host macrophages and activated T-cells take up various antigenic substrates from the organism.[18] The recruited T-cells are paradoxically inhibited by the expression of B7-H1 (programmed death-1 ligand 1) on gastric epithelial cells. Studies have shown that H pylori induces B7-H1 expression on gastric epithelial cells, which is believed to contribute to the chronicity of the bacterial infection.[19] Further stimulation of inflammation occurs through an H pylori–induced increase in interleukin (IL)-8 production by the gastric epithelial cells.[20] IL-8 subsequently triggers the activation of neutrophils and the recruitment of other inflammatory cells into the mucosa. Persistent inflammation ultimately leads to the depletion of gastrin-producing (G) cells and acid-producing parietal cells in the gastric mucosa.[21] Over time, atrophy and intestinal metaplasia develop.
Autoimmune Gastritis
Autoimmune metaplastic atrophic gastritis develops due to T-cell–mediated destruction of the oxyntic mucosa and the production of autoantibodies targeting parietal cells and the intrinsic factor. A proposed mechanism for this disease in patients with H pylori-induced atrophic gastritis outlines a crossover of antigenic activity directed against the bacterium and toward host antigens in the region, including parietal cells and the intrinsic factor. In primary autoimmune atrophic gastritis, the immune response is directed against these antigens irrespective of an H pylori infection. However, the molecular factors driving the autoimmune response and initiating the pathogenesis of the condition remain unidentified. Over time, the immune-mediated destruction of the oxyntic mucosa leads to mucous cells and metaplastic glands within the gastric mucosa, including both intestinal and pseudo-pyloric types.[8]
Histopathology
Histologically, gastritis is confirmed by at least grade 2 neutrophils or mononuclear cells in at least 1 gastric biopsy site or grade 1 neutrophils or mononuclear cells in at least 2 places. The Sydney System of Classification recommends obtaining histological specimens from 5 distinct gastric locations, including the antrum (greater and lesser curvature), incisura, and corpus (greater and lesser curvature). Specimens should be individually placed into separate vials and grouped based on the location of the lesion. This approach enhances the chances of identifying H pylori and reduces the likelihood of missing the diagnosis.[22]
Normal Histology of the Gastric Mucosa
The gastric mucosa is divided into 2 main zones—the oxyntic region, comprising the fundus and corpus, and the mucous-secreting region, consisting of the antrum and incisura angularis. Within the oxyntic mucosa, specialized glands containing parietal (oxyntic) cells, chief (zymogenic) cells, and enterochromaffin cells can be observed. The parietal cells are characterized by their large size, high acidophilia, and round or pyramidal shape, whereas the chief cells are smaller and exhibit basophilia. The antral mucosa is characterized by gastric pits, or foveolae, abundant in mucous-producing cells. These foveolar cells are columnar and possess a pale eosinophilic cytoplasm. The cells located deep within the gastric pits are called mucous neck cells, and they mature as they move up the gland to replace the surface mucous cells.
Intermediate mucosa identifies the transitional boundaries between the antrum and body, body and cardia, and antrum and duodenum. The transitional zones exhibit a gradual merging of the various mucosal types. The most informative histological indicator of the transition from the body to the antrum is the absence of chief cells and a shift in the shape of the glands from simple tubular glands to branched glands. The oxyntic mucosa exhibits short gastric foveolae with long, densely packed glands. In contrast, roughly 50% of the mucosal thickness in the antral region comprises large mucus-producing glands lined by foveolar cells. In the cardia, gastric foveoli are abundant, and the presence of chief and parietal cells is infrequent. The transitional zone between the body and cardia is where the chief and parietal cells are abundant.
Histological Features of Gastritis
H pylori is a spiral-shaped bacterium observed in gastric biopsy specimens and may be detected through hematoxylin and eosin (H&E) staining (see Image. H pylori Gastritis).[4] The sensitivity and specificity of the H&E stain for detecting the bacterium are approximately 69% to 93% and 87% to 90%, respectively. The specificity of H&E stain increases from 90% to 100% when specialized staining methods, including modified Giemsa, Warthin-Starry silver, Genta, and immunohistochemical (IHC), are used. The Giemsa stain is easy and cost-effective, rendering the method preferred in many laboratories. The Genta stain effectively visualizes the inflammatory cells and H pylori by combining silver, H&E, and Alcian blue stains. The advantage of IHC stain is the ability to detect the organism in patients who have undergone partial treatment for H pylori gastritis, as the organism may assume atypical forms (such as coccoid).[23] In nearly all cases, H pylori–associated gastritis typically presents with diffuse or nodular lymphocytic inflammation.
In acute H pylori gastritis, neutrophilic infiltration of the mucous neck region and signs of mucin loss and desquamation of surface foveolar cells is common. Over time, lymphoid follicles develop, not commonly found in otherwise normal gastric mucosa.[24] Due to persistent low-grade inflammation, reactive gastritis or gastropathies are characterized by foveolar hyperplasia and vascular ectasia, edema, and muscularis mucosae hyperplasia. The early phase of autoimmune atrophic gastritis reveals nonspecific changes characterized by infiltrating lymphocytes and plasma cells into the oxyntic mucosa. Patchy destruction of oxyntic glands can be seen, and the parietal cells exhibit pseudo-hypertrophic changes.
As the disease progresses, diffuse lymphoplasmacytic infiltration of the lamina propria and significant atrophy of oxyntic glands are seen (see Image. Histopathology of Chronic Gastritis). Foveolar hyperplasia with an increase in the thickness of the foveolar component is also evident. Marked pseudopyloric metaplasia is often seen with an increasing abundance of intestinal metaplasia. These distinctive changes in the oxyntic mucosa, coupled with the absence of inflammatory and atrophic changes in the antral mucosa, are pathognomonic for autoimmune gastritis. The lack of oxyntic glands, a marked presence of foveolar hyperplasia, and the development of hyperplastic and inflammatory polyps characterize the end stage of autoimmune gastritis. Metaplastic changes, including pseudopyloric, pancreatic, and intestinal metaplasia, become evident with a significant reduction in inflammatory cells.
Pseudopyloric metaplasia, also known as spasmolytic polypeptide-expressing metaplasia, is characterized by replacing parietal and chief cells with mucus-secreting cells of the antral mucosa.[25] The precursors for these cells are the chief cells of the oxyntic glands.[26] Pseudopyloric metaplasia consists of coiled glands that are less abundant and contain considerably less mucin than normal antral glands. Another distinctive feature is the absence of endocrine cells, particularly gastrin-secreting G cells, which are typically abundant in normal antral mucosa.[27]
Intestinal metaplasia is characterized by substituting gastric mucosa, either oxyntic or antral, with intestinal epithelium. The presence of goblet cells is indicative of intestinal metaplasia.[28] Intestinal metaplasia can be complete, displaying fully formed small intestinal epithelium, or incomplete, showing features of colonic epithelium without well-defined brush border cells. Incomplete intestinal metaplasia characterized by goblet cells resembling colonic epithelium and containing sulfomucins is called type III intestinal metaplasia. This particular metaplasia has been associated with an elevated risk of gastric cancer.[29] Notably, this is not an obligatory precancerous lesion that raises questions about the usefulness of subcategorizing intestinal metaplasia in this manner.[30]
Lymphocytic gastritis is characterized by a minimum of 25 intraepithelial lymphocytes per 100 gastric epithelial cells. The predominant phenotype consists of CD3+ and CD8+ lymphocytes. Although the etiology of this condition is unclear, H pylori infection and celiac disease are associated. In eosinophilic gastritis, the diagnosis is confirmed by the presence of an average density of over 127 eosinophils per square millimeter or more than 30 eosinophils per high-power field (HPF) in at least 5 separate HPFs, with no known associated causes of eosinophilia. Ruling out H pylori infection, Crohn disease, parasitic infections, and other hematological or lymphoid disorders establish the diagnosis. Collagenous gastritis is characterized by subepithelial collagen bands, typically thicker than 10 μm, along with inflammatory mononuclear cells in the lamina propria.[4] Emphysematous gastritis is characterized by air in the gastric wall, usually caused by a bacterial infection of the gastric wall.[31] Although this condition is rare, a high mortality rate, with some reports indicating a rate of up to 60%, is expected.
History and Physical
Gastritis is difficult to diagnose based on clinical symptoms. A histologic or endoscopic examination is necessary, which may not correlate with symptom severity. Acute gastritis may manifest with a sudden onset of epigastric pain, bloating, nausea, and vomiting (dyspepsia), which typically resolves independently. Although chronic gastritis is often used synonymously with dyspeptic symptoms, the presence of symptoms poorly correlates with the actual histological or endoscopic diagnosis of gastritis. H pylori–associated chronic gastritis may remain asymptomatic and gradually progress to severe complications such as gastric cancer. However, the rate of progression is highly unpredictable.
Notably, not every patient with chronic dyspepsia is infected with H pylori. An initial or acute infection with H pylori may induce acute dyspepsia in some patients, which typically resolves independently. Although persistent colonization invariably leads to chronic gastritis, the majority of the individuals experience only transient dyspeptic symptoms. Therefore, according to the current consensus, patients are considered to have H pylori–induced dyspepsia if their symptoms improve after successful eradication therapy. However, successful eradication therapy does not immediately improve symptoms and may take up to 6 months. In contrast, functional dyspepsia is characterized by persistent dyspeptic symptoms, such as postprandial fullness, early satiation, epigastric pain, or burning sensation, without structural abnormalities that can account for these symptoms, as confirmed by upper endoscopy. As per current consensus guidelines, individuals who persistently experience chronic dyspeptic symptoms, even after successful H pylori eradication therapy, should be diagnosed with functional dyspepsia.[6]
Similar to acute or H pylori–induced gastritis, autoimmune gastritis is often asymptomatic, and patients typically seek medical attention incidentally when diagnosed with megaloblastic, pernicious, or iron-deficiency anemias. Although gastrointestinal symptoms are rare among individuals with autoimmune gastritis, most patients typically report postprandial dyspepsia when symptoms manifest. Less frequently, patients may report nonspecific epigastric pain, nausea, vomiting, functional abdominal pain syndrome, and functional bloating. Certain study results suggest that dyspeptic symptoms, such as early satiety and postprandial fullness, may correlate with autoimmune gastritis, particularly in younger patients without anemia or with a smoking history, or who are not currently smoking.[32]
Autoimmune gastritis often occurs concomitantly with other autoimmune disorders, primarily thyroid diseases. These associated autoimmune conditions may include Hashimoto thyroiditis, Addison disease, chronic spontaneous urticaria, myasthenia gravis, type 1 diabetes, vitiligo, and perioral cutaneous autoimmune disorders, especially erosive oral lichen planus.[33] The association between chronic atrophic autoimmune gastritis and autoimmune thyroid disease led to the coining of the term thyrogastric syndrome in the early 1960s to describe this correlation.
Evaluation
The diagnosis of gastritis primarily relies on histopathological examination of gastric biopsies. Although medical history and laboratory tests can provide valuable insights, endoscopy, and biopsy are the gold standards for diagnosing gastritis, as they help identify the distribution, severity, and underlying cause.[34]
Evaluation of Dyspepsia and H pylori Testing
The evaluation of gastritis should follow a stepwise process. In patients suspected of having functional dyspepsia, upper gastrointestinal endoscopy is not required to make the diagnosis. However, an evaluation for the presence of H pylori infection is indicated. According to current guidelines on the diagnosis of functional dyspepsia, patients with new-onset symptoms at an advanced age, weight loss, evidence of bleeding, dysphagia, the presence of an abdominal mass, fever, persistent vomiting, or a family history of esophageal or gastric cancer are alarm signs.
These individuals require further evaluation and testing for the presence of an H pylori infection. In most cases, this testing involves an upper gastrointestinal endoscopic evaluation and a comprehensive histological examination of biopsy samples collected during the endoscopy. However, the endoscopic evaluation is not recommended to rule out gastric malignancy in patients younger than 60, who present with dyspepsia but without a family history of gastric cancer.[35] Limited clinical utility exists in using gastrointestinal function tests to differentiate patients with organic causes of functional dyspepsia from those with true functional dyspepsia. Consequently, the American Gastroenterological Association (AGA) guidelines and the Asian consensus for functional dyspepsia do not recommend routine functional gastrointestinal testing in patients experiencing these symptoms.[36] Investigating the presence of H pylori is recommended and warrants thorough evaluation. The urea breath test and the H pylori stool antigen test are suggested for this assessment.
According to a recent Cochrane review, the authors concluded that in patients without a prior history of gastrectomy or current antibiotic or proton pump inhibitor (PPI) use, urea breath testing was found more diagnostically accurate than other noninvasive measures such as serology or stool antigen detection. As per most consensus statements, a single positive noninvasive test, such as the urea breath or stool antigen test, can initiate eradication treatment in patients with dyspepsia, even without any alarm symptoms. In addition, noninvasive testing can confirm the effectiveness of eradication treatment in patients who do not need endoscopy for other indications. Noninvasive testing to detect H pylori after eradication therapy should be performed 4 to 6 weeks after antibiotics or PPI therapy completion. In patients 60 or older with dyspepsia, endoscopy, and histological evaluation are recommended for gastritis assessment and classification.[37]
Diagnosis of Atrophic Gastritis
The diagnosis of autoimmune gastritis is based on laboratory and histological examinations, which typically involve the following criteria:
- Evidence of atrophic gastritis affecting the gastric corpus (body) and fundus
- The presence of autoantibodies against the intrinsic factor and parietal cells
- Serum pepsinogen I levels
- The ratio of pepsinogen I to pepsinogen II [38][39]
Pernicious anemia is a type of macrocytic anemia often associated with atrophic gastritis and is characterized by antibodies against parietal cells or intrinsic factors. Additional tests that may be relevant for diagnosing autoimmune gastritis include gastrin-17 levels, anti-H pylori antibodies, cytokines such as IL-8, and ghrelin, a growth hormone-releasing peptide primarily produced by the gastric fundus mucosa.[40] Recognizing pernicious anemia as a late manifestation of autoimmune gastritis is crucial.
Although anti-parietal cell antibodies represent the most sensitive serum biomarker for autoimmune atrophic gastritis, their specificity is compromised, as they may also be elevated in individuals with an H pylori infection and other autoimmune conditions. In contrast, anti-intrinsic factor antibodies have low sensitivity but high specificity. These serologic markers can often predate the clinical presentation of autoimmune gastritis; therefore, patients with a new diagnosis of pernicious anemia should undergo endoscopy with topographical biopsies to assess for the presence of atrophic gastritis. As per the AGA update on the diagnosis and management of atrophic gastritis in 2021, a histopathological assessment is necessary to confirm the diagnosis of atrophic gastritis. The presence of intestinal metaplasia on gastric histology is pathognomonic for atrophic gastritis.
From an endoscopic perspective, atrophic gastritis manifests as pale gastric mucosa, noticeable mucosal thinning indicated by enhanced vasculature visibility, and the loss of gastric folds. The use of high-definition white-light endoscopy (HD-WLE) and narrow-band imaging (NBI) demonstrates a sensitivity of 87% and specificity of 97% for detecting intestinal metaplasia without magnifying endoscopy. On HD-WLE, metaplastic areas appear slightly nodular with tubulovillous mucosal patterns. Fine, blue-white crests on the epithelial surface are diagnostic for intestinal metaplasia during endoscopy, with sensitivity and specificity approaching 90%.
Microscopic lipid accumulation within the mucosa of gastric tumors and regions with intestinal metaplasia presents as white opaque fields with a diagnostic specificity nearing 100%. Notably, the sensitivity of this endoscopic finding is relatively limited. Biopsies should be obtained from the suspected atrophic or metaplastic areas and forwarded for histopathological analysis to confirm the diagnosis and assess risk stratification. As per AGA recommendations, adhering to the updated Sydney protocol for obtaining topographical biopsies during the evaluation of atrophic gastritis is advised.
This involves taking 5 separate biopsies, each placed in appropriately labeled containers, from the following locations:
- The lesser curvature of the antrum, situated within 2 to 3 cm from the pylorus
- The greater curvature of the antrum, located within 2 to 3 cm from the pylorus
- The lesser curvature of the gastric corpus, approximately 4 cm proximal to the incisura angularis
- The greater curvature of the gastric corpus, positioned 8 cm from the cardia
- The incisura angularis
If patients with atrophic gastritis have not been previously tested for H pylori, testing is recommended upon establishing the diagnosis. In the event of a positive result, eradication treatment should be administered, followed by subsequent testing to confirm the success of eradication. Additional testing, including anti-parietal cell antibodies and anti-intrinsic factor antibodies, should be considered. In addition, further serological testing to assess the presence of anemia resulting from vitamin B-12 and iron deficiencies is advisable. For individuals with advanced atrophic gastritis, considering scheduling surveillance endoscopies every 3 years is recommended to monitor the potential development of gastric malignancies. If accessible, serological testing for pepsinogens I and II can provide valuable insights into assessing patients with atrophic gastritis. In areas with a high incidence of gastric cancer, serum pepsinogen I levels below 70 μg/L and a low pepsinogen I to pepsinogen II ratio have high sensitivity and specificity for indicating severe corpus atrophy.[13]
Treatment / Management
Approach to Treatment
As mentioned earlier, eradicating H pylori is recommended for all patients with evidence of gastritis on diagnostic testing. Furthermore, eradication therapy is the initial treatment option for patients with dyspepsia who have a documented H pylori infection. In addition, H pylori eradication therapy is indicated in patients with peptic ulcer disease, functional dyspepsia, idiopathic thrombocytopenic purpura (ITP), unexplained iron-deficiency anemia, and in cases where long-term nonsteroidal anti-inflammatory drug treatment is anticipated, particularly in patients with a history of peptic ulcer disease.[37] In patients with functional dyspepsia, eradication therapy has a limited impact on symptom relief. Nonetheless, the therapy is advantageous in mitigating the risk of peptic ulcer disease.[41] (A1)
Eradication therapy for H pylori in patients with non-atrophic chronic gastritis is highly recommended to promote healing and reduce the risk of gastric cancer. For patients with atrophic gastritis, eradication therapy targeted at the organism may result in partial regression of the gastritis and offer some potential benefits. Although the eradication therapy in patients with intestinal metaplasia does not reverse the metaplastic changes, the progression to neoplasia is slowed without reducing the overall risk of gastric cancer. Therefore, a cautious or weak recommendation is considered in this context.[42]
The management of chronic gastritis in patients who initially test negative for H pylori lacks standardized guidelines and tends to exhibit significant variability. Empirical use of proton-pump inhibitors (PPIs) has demonstrated effectiveness in alleviating symptoms for these patients.[43] According to current guidelines, empiric PPI therapy is recommended for individuals aged younger than 60 with dyspepsia if they test negative for H pylori or experience persistent symptoms despite undergoing eradication therapy. Patients who do not experience relief from these treatments may be considered for prokinetic therapy or tricyclic antidepressants. Notably, the supporting evidence is low-to-moderate quality.[35](B2)
Currently, definitive treatment does not exist for patients with atrophic gastritis. The pivotal aspect in treating patients with atrophic gastritis is the application of risk stratification systems to assess the severity of the disease and determine the risk of gastric malignancies. For this purpose, utilizing the Operative Link on Gastritis Assessment (OLGA) and Operative Link on Gastric Intestinal Metaplasia Assessment (OLGIM) grading systems is recommended. Staging gastritis using the OLGA and OLGIM systems, with a stage III or IV classification, is associated with a significantly elevated risk of gastric cancer. This approach provides an easily translated method for assessing attributable risk.[6] These systems incorporate atrophy scores obtained through histological assessment of gastric biopsies and consider atrophy topography to assign clinical stages.[44] (B2)
Histological Grading of Gastritis
In normal gastric mucosa, an acceptable range is typically 2 to 5 lymphocytes, plasma cells, or macrophages per high power field, or 2 to 3 cells located between foveolae. The degree of increase from these numbers determines the severity of gastritis, which is graded as mild (+--), moderate (++-), or marked (+++). Notably, this density measurement should be performed away from any lymphoid follicles, as they could be related to an underlying H pylori infection. Lymphocytic gastritis occurs when more than 25 lymphocytes are observed per 100 epithelial cells within the glandular epithelium. The density of neutrophils measures the activity of gastritis. The grading of gastritis activity is followed as neutrophils in the lamina propria indicate mild (+--) activity, neutrophils within the epithelium denote moderate (++-) activity, and neutrophils in the glandular lumen signify marked (+++) activity.
The discrepancy between the expected glands for the anatomical site of the gastric mucosa and what is observed represents atrophy. A reduction or complete absence of glandular units leads to collagen deposition in the lamina propria. Metaplastic changes involve the replacement of normal glandular units with metaplastic and/or dysplastic units. A score of 1 is allocated when a 1% to 30% loss of the glandular architecture or its metaplastic transformation occurs, a score of 2 is assigned for a 31% to 60% loss, and a score of 3 is designated for a loss exceeding 60%. The OLGA staging system categorizes gastritis into 5 stages, each associated with a progressively higher risk of cancer, determined by the atrophy score. In addition, an overall atrophy score based on topography is assigned, and these scores are tallied to determine the corresponding OLGA stage. Although the OLGIM staging system relies solely on intestinal metaplasia for the atrophy score, enhancing inter-observer reproducibility, a notable decrease in sensitivity for identifying high-risk patients is apparent.[4]
Patients classified as OLGA/OLGIM stage III or IV face a considerable risk of developing gastric adenocarcinoma. As a result, regular surveillance endoscopy is strongly recommended for these individuals to enhance the chances of detecting gastric cancer in the early stages, enabling surgical treatment. The AGA recommends endoscopic surveillance every 3 years in these patients. Other clinical factors that should be considered when determining the frequency of surveillance include a family history of gastric cancer, residence in regions with a high incidence of gastric cancer, a history of persistent H pylori infection, smoking history, and dietary factors.[13]
Differential Diagnosis
The differential diagnosis of gastritis encompasses the following conditions:
- Functional dyspepsia
- Peptic ulcer disease
- Gastric cancer
- Cholecystitis
- Zollinger-Ellison syndrome
- Pancreatitis
- Myocardial ischemia
- Gastric lymphoma
- Celiac disease
- Multiple endocrine neoplasias
Prognosis
The prognosis of gastritis varies based on the type, underlying cause, and patient characteristics. With appropriate management, many cases of gastritis can be effectively controlled, and the risk of complications can be minimized. Acute gastritis secondary to ischemia and emphysematous gastritis can have inferior outcomes if the underlying cause is not treated promptly and adequately. In contrast, the prognosis of atrophic gastritis, however, depends on the severity of atrophy or metaplasia. Numerous studies have reported an elevated risk of gastric malignancies in patients with atrophic gastritis.
The primary management approach for this condition is focused on staging to facilitate early detection of such complications. A study reported an annual incidence rate of 0.25% per person-year for gastric cancer and 0.68% for type I gastric carcinoids in patients with atrophic gastritis.[45] The incidence of gastric adenocarcinoma is reported to be around 14.2 cases per 1000 person-years in patients with autoimmune metaplastic atrophic gastritis compared to 0.073 cases in the general population.[46] The pattern, extent, and severity of atrophy are the most critical predictive factors for an elevated cancer risk. Conversely, the subtypes of gastrointestinal metaplasia offer limited prognostic value.
Studies from different regions worldwide present conflicting data on the effectiveness of H pylori treatment in reducing cancer risk due to significant geographic variation in malignancy risk. Nevertheless, the consensus remains that eradication treatment improves prognosis. A systematic review indicated a one-third reduction in cancer risk following successful H pylori eradication. The literature highlights a "point of no return" phenomenon, with studies showing a reduced incidence of gastric cancer in patients who were H pylori carriers without precancerous lesions, such as atrophic gastritis or gastrointestinal metaplasia.[47]
Complications
Helicobacter pylori–induced gastritis can lead to various conditions, including peptic ulcer disease, immune thrombocytopenic purpura (ITP), iron-deficiency anemia, and vitamin B12 deficiency. Recent reports have also indicated a potential association with insulin resistance, metabolic syndrome, and non-alcoholic fatty liver disease.[48] The hypothesized mechanism underlying these associations involves molecular mimicry and the chronic state of low-grade inflammation.[49] However, insufficient evidence exists to establish a causal link between gastritis and these metabolic conditions.[50] Gastric cancer represents the most severe complication of atrophic gastritis. The presence of atrophic gastritis and gastrointestinal metaplasia substantially elevates the risk of this complication compared to chronic gastritis in the absence of these lesions. The severity of atrophy also amplifies the associated risk, with a relative risk of 1.7% for gastric cancer in cases of moderate atrophic gastritis and 4.9% relative risk in cases of severe atrophic gastritis, in contrast to no elevated risk when only mild atrophic changes are present.[47]
Extranodal marginal zone B-cell lymphoma is associated with H pylori–induced gastritis, with histological evidence suggesting that lymphoma originates from B-cell clones in the site where chronic gastritis was previously sampled.[51] Recent molecular studies have shown a strong correlation between H pylori infection and gastric mucosa-associated lymphoid tissue (MALT) lymphomas. These studies demonstrate the translocation of the bacterial protein CagA into human B lymphoid cells, leading to the activation of cellular signaling pathways that inhibit apoptosis.[52] The most compelling evidence of the association between H pylori–induced gastritis and MALTomas is derived from studies documenting the complete regression of early-stage gastric B-cell lymphoma after eradication therapy.[53]
Gastric neuroendocrine tumors (NETs) represent another known complication of atrophic gastritis. The prognosis of these lesions is contingent on factors such as tumor size, mitotic activity, and the extent of invasion. Small NETs can be managed with endoscopic resection and carry a low risk of metastasis. In contrast, larger tumors exceeding 2 cm have been associated with a reported metastasis rate of nearly 20%.[13]
Both autoimmune and H pylori–induced gastritis can lead to iron-deficiency anemia.[54] Results from a retrospective analysis of premenopausal women with iron deficiency reported that 18.5% of the patients were seropositive for anti-parietal cell antibodies.[55] The association between H pylori infection and iron-deficiency anemia is particularly evident in patients with unexplained iron deficiency. Some expert guidelines advocate screening and treating H pylori in patients with recurrent or treatment-resistant iron-deficiency anemia and normal upper and lower gastrointestinal endoscopies. ITP is another recognized complication of gastritis. Consensus guidelines for managing ITP recommend screening for H pylori infection in adult patients with ITP.[50]
Deterrence and Patient Education
Education plays a crucial role in treating patients with gastritis, helping individuals take proactive measures to prevent the onset or recurrence and manage this condition.
In H pylori–induced gastritis cases, educating patients on a few critical aspects is mentioned below:
- Mode of transmission: Patients should be informed about the mode of transmission of H pylori, typically through close person-to-person contact. They should also be educated about emphasizing the importance of practicing good hygiene to reduce the risk of infection.
- Preventative measures: Patients must be educated about various preventive measures, including avoiding contaminated food and water sources, which can significantly reduce the risk of H pylori infection.
- Antibiotic treatment: Patients diagnosed with an H pylori infection should complete the entire course of antibiotics their clinician prescribes. This step is crucial to ensure the complete eradication of the bacteria and minimize the risk of recurrence.[56]
For patients with autoimmune gastritis and vitamin B12 deficiency, providing education regarding the significance of consistent vitamin B12 supplementation is important. This measure prevents anemia and other associated complications. Patients should be informed about potential complications of gastritis, such as bleeding or peptic ulcers, and the importance of seeking immediate medical care if these complications arise. Furthermore, educating patients about the potential risks associated with prolonged or excessive use of medications that can trigger or worsen gastritis is essential. As a result, patients can make informed decisions about their medication use and take steps to minimize the risk of exacerbating their gastritis.
Mass screening for H pylori, followed by eradication therapy, has garnered recent attention. National guidelines for mass screening differ from country to country, and in regions with a low prevalence of H pylori infection, most guidelines advise against mass screening.[37] In some cases, 'screen-and-treat' strategies are recommended for communities at high risk of gastric cancer and are generally considered cost-effective in these regions. A substantial population-based screen-and-treat trial conducted in a high-incidence rural area of China demonstrated good compliance and the feasibility of this approach. However, long-term follow-up data regarding the efficacy in preventing gastric cancer is still pending.
A significant concern associated with implementing these screening programs is the risk of antibiotic resistance. For instance, in a placebo-controlled trial involving healthy volunteers, the use of clarithromycin at a dosage of 500 mg twice daily for the shortest recommended duration of 7 days for H pylori eradication increases the resistance of macrolide-resistant pharyngeal Streptococcus pneumoniae. Expert guidelines strongly discourage antibiotics commonly used for treating life-threatening infections, including macrolides, penicillins, and fluoroquinolones, to eradicate H pylori in gastritis treatment. Medications such as bismuth, tetracycline, and metronidazole should be considered when strategizing public health campaigns to eradicate H pylori. Rifabutin may also be contemplated, as short-term usage is unlikely to contribute significantly to mycobacterial resistance.[50]
Enhancing Healthcare Team Outcomes
Gastritis is primarily diagnosed histologically and is prevalent globally, stemming from various causes. This condition typically presents with nonspecific symptoms, and the most severe forms are usually identified by astute clinical judgment and diligent adherence to recommended guidelines for assessing asymptomatic patients with anemia, thrombocytopenia, or vague gastrointestinal symptoms. Clinicians must familiarize themselves with the current guideline-recommended approach for evaluating patients with dyspepsia to assess iron-deficiency anemia. This approach enables the early identification of patients with gastritis, recognizing those with premalignant atrophy and the advancement to dysplasia.
Treatment approaches can significantly differ depending on the subtype and stage of gastritis, particularly when atrophic changes are evident. Close coordination and effective communication between the endoscopist and the pathologist are pivotal in arriving at an accurate diagnosis and assigning the appropriate stage to a patient's gastritis. The treating team is important in educating patients about the disease process and engaging in consistent follow-up. Also, educating patients about the pathogenesis of H pylori–associated gastritis and the risk of developing cancer is crucial. This promotes patient involvement in treatment programs, encourages adherence to antibiotic therapy, and ensures follow-up testing to confirm eradication.
The clinical pharmacist is critical in ensuring patient adherence to medication during H pylori eradication therapy and adjusting medication regimens to minimize adverse outcomes. Pharmacists specializing in infectious diseases are integral to the success of eradication therapy, as they assist clinicians in designing antibiotic regimens that align with local resistance patterns. Public health specialists also have a significant role in preventing gastritis-associated malignancies. They are responsible for developing suitable screen-and-treat programs, particularly in high-prevalence regions. A collaborative effort involving an interprofessional healthcare team of clinicians, including physicians, nurses, pharmacists, and public health experts, can substantially reduce the incidence of gastritis and enhance clinical outcomes by lowering the risk of gastric malignancies.
Media
(Click Image to Enlarge)
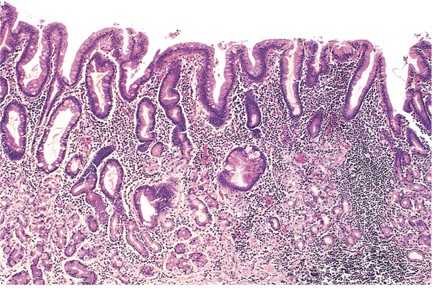
Histopathology of Chronic Gastritis. This image displays gastric mucosal atrophy with high lymphocytic infiltrate.
Contributed by A Awosika, MD, MS
(Click Image to Enlarge)
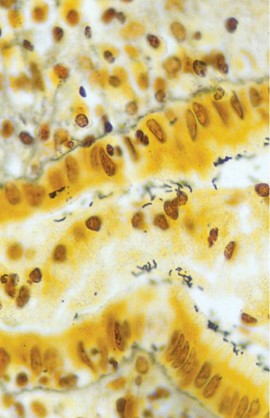
Helicobacter pylori Gastritis. The image shows the characteristic spiral shape of this gastritis-causing bacteria on hematoxylin and eosin stain.
Contributed by A Awosika, MD, MS
References
Kayaçetin S, Güreşçi S. What is gastritis? What is gastropathy? How is it classified? The Turkish journal of gastroenterology : the official journal of Turkish Society of Gastroenterology. 2014 Jun:25(3):233-47. doi: 10.5152/tjg.2014.7906. Epub [PubMed PMID: 25141310]
Chia JK, Chia AY. Acute gastritis associated with enterovirus infection. Archives of pathology & laboratory medicine. 2010 Jan:134(1):16-7 [PubMed PMID: 20073596]
Level 3 (low-level) evidenceSipponen P, Maaroos HI. Chronic gastritis. Scandinavian journal of gastroenterology. 2015 Jun:50(6):657-67. doi: 10.3109/00365521.2015.1019918. Epub 2015 Apr 22 [PubMed PMID: 25901896]
Pennelli G, Grillo F, Galuppini F, Ingravallo G, Pilozzi E, Rugge M, Fiocca R, Fassan M, Mastracci L. Gastritis: update on etiological features and histological practical approach. Pathologica. 2020 Sep:112(3):153-165. doi: 10.32074/1591-951X-163. Epub [PubMed PMID: 33179619]
Sipponen P, Price AB. The Sydney System for classification of gastritis 20 years ago. Journal of gastroenterology and hepatology. 2011 Jan:26 Suppl 1():31-4. doi: 10.1111/j.1440-1746.2010.06536.x. Epub [PubMed PMID: 21199511]
Sugano K, Tack J, Kuipers EJ, Graham DY, El-Omar EM, Miura S, Haruma K, Asaka M, Uemura N, Malfertheiner P, faculty members of Kyoto Global Consensus Conference. Kyoto global consensus report on Helicobacter pylori gastritis. Gut. 2015 Sep:64(9):1353-67. doi: 10.1136/gutjnl-2015-309252. Epub 2015 Jul 17 [PubMed PMID: 26187502]
Level 3 (low-level) evidenceYin Y, Liang H, Wei N, Zheng Z. Prevalence of chronic atrophic gastritis worldwide from 2010 to 2020: an updated systematic review and meta-analysis. Annals of palliative medicine. 2022 Dec:11(12):3697-3703. doi: 10.21037/apm-21-1464. Epub [PubMed PMID: 36635994]
Level 1 (high-level) evidenceCoati I, Fassan M, Farinati F, Graham DY, Genta RM, Rugge M. Autoimmune gastritis: Pathologist's viewpoint. World journal of gastroenterology. 2015 Nov 14:21(42):12179-89. doi: 10.3748/wjg.v21.i42.12179. Epub [PubMed PMID: 26576102]
Carmel R. Prevalence of undiagnosed pernicious anemia in the elderly. Archives of internal medicine. 1996 May 27:156(10):1097-100 [PubMed PMID: 8638997]
Mana F, Vandebosch S, Miendje Deyi V, Haentjens P, Urbain D. Prevalence of and risk factors for H. pylori infection in healthy children and young adults in Belgium anno 2010/2011. Acta gastro-enterologica Belgica. 2013 Dec:76(4):381-5 [PubMed PMID: 24592540]
Level 2 (mid-level) evidenceGoh KL, Chan WK, Shiota S, Yamaoka Y. Epidemiology of Helicobacter pylori infection and public health implications. Helicobacter. 2011 Sep:16 Suppl 1(0 1):1-9. doi: 10.1111/j.1523-5378.2011.00874.x. Epub [PubMed PMID: 21896079]
Park JS, Jun JS, Seo JH, Youn HS, Rhee KH. Changing prevalence of Helicobacter pylori infection in children and adolescents. Clinical and experimental pediatrics. 2021 Jan:64(1):21-25. doi: 10.3345/cep.2019.01543. Epub 2020 Jul 15 [PubMed PMID: 32668822]
Shah SC, Piazuelo MB, Kuipers EJ, Li D. AGA Clinical Practice Update on the Diagnosis and Management of Atrophic Gastritis: Expert Review. Gastroenterology. 2021 Oct:161(4):1325-1332.e7. doi: 10.1053/j.gastro.2021.06.078. Epub 2021 Aug 26 [PubMed PMID: 34454714]
Azuma T, Yamakawa A, Yamazaki S, Fukuta K, Ohtani M, Ito Y, Dojo M, Yamazaki Y, Kuriyama M. Correlation between variation of the 3' region of the cagA gene in Helicobacter pylori and disease outcome in Japan. The Journal of infectious diseases. 2002 Dec 1:186(11):1621-30 [PubMed PMID: 12447739]
Mobley HL. The role of Helicobacter pylori urease in the pathogenesis of gastritis and peptic ulceration. Alimentary pharmacology & therapeutics. 1996 Apr:10 Suppl 1():57-64 [PubMed PMID: 8730260]
Kolopaking MS. Urease, Gastric Bacteria and Gastritis. Acta medica Indonesiana. 2022 Jan:54(1):1-2 [PubMed PMID: 35398819]
Goodwin CS, Worsley BW. Microbiology of Helicobacter pylori. Gastroenterology clinics of North America. 1993 Mar:22(1):5-19 [PubMed PMID: 8449570]
Di Tommaso A, Xiang Z, Bugnoli M, Pileri P, Figura N, Bayeli PF, Rappuoli R, Abrignani S, De Magistris MT. Helicobacter pylori-specific CD4+ T-cell clones from peripheral blood and gastric biopsies. Infection and immunity. 1995 Mar:63(3):1102-6 [PubMed PMID: 7868233]
Das S, Suarez G, Beswick EJ, Sierra JC, Graham DY, Reyes VE. Expression of B7-H1 on gastric epithelial cells: its potential role in regulating T cells during Helicobacter pylori infection. Journal of immunology (Baltimore, Md. : 1950). 2006 Mar 1:176(5):3000-9 [PubMed PMID: 16493058]
Keates S, Hitti YS, Upton M, Kelly CP. Helicobacter pylori infection activates NF-kappa B in gastric epithelial cells. Gastroenterology. 1997 Oct:113(4):1099-109 [PubMed PMID: 9322504]
Väänänen H, Vauhkonen M, Helske T, Kääriäinen I, Rasmussen M, Tunturi-Hihnala H, Koskenpato J, Sotka M, Turunen M, Sandström R, Ristikankare M, Jussila A, Sipponen P. Non-endoscopic diagnosis of atrophic gastritis with a blood test. Correlation between gastric histology and serum levels of gastrin-17 and pepsinogen I: a multicentre study. European journal of gastroenterology & hepatology. 2003 Aug:15(8):885-91 [PubMed PMID: 12867799]
Varbanova M, Frauenschläger K, Malfertheiner P. Chronic gastritis - an update. Best practice & research. Clinical gastroenterology. 2014 Dec:28(6):1031-42. doi: 10.1016/j.bpg.2014.10.005. Epub 2014 Oct 30 [PubMed PMID: 25439069]
Lee JY, Kim N. Diagnosis of Helicobacter pylori by invasive test: histology. Annals of translational medicine. 2015 Jan:3(1):10. doi: 10.3978/j.issn.2305-5839.2014.11.03. Epub [PubMed PMID: 25705642]
Genta RM, Hamner HW, Graham DY. Gastric lymphoid follicles in Helicobacter pylori infection: frequency, distribution, and response to triple therapy. Human pathology. 1993 Jun:24(6):577-83 [PubMed PMID: 8505036]
Neumann WL, Coss E, Rugge M, Genta RM. Autoimmune atrophic gastritis--pathogenesis, pathology and management. Nature reviews. Gastroenterology & hepatology. 2013 Sep:10(9):529-41. doi: 10.1038/nrgastro.2013.101. Epub 2013 Jun 18 [PubMed PMID: 23774773]
Radyk MD, Burclaff J, Willet SG, Mills JC. Metaplastic Cells in the Stomach Arise, Independently of Stem Cells, via Dedifferentiation or Transdifferentiation of Chief Cells. Gastroenterology. 2018 Mar:154(4):839-843.e2. doi: 10.1053/j.gastro.2017.11.278. Epub 2017 Dec 14 [PubMed PMID: 29248442]
Schmidt PH, Lee JR, Joshi V, Playford RJ, Poulsom R, Wright NA, Goldenring JR. Identification of a metaplastic cell lineage associated with human gastric adenocarcinoma. Laboratory investigation; a journal of technical methods and pathology. 1999 Jun:79(6):639-46 [PubMed PMID: 10378506]
Level 3 (low-level) evidenceCorrea P, Piazuelo MB, Wilson KT. Pathology of gastric intestinal metaplasia: clinical implications. The American journal of gastroenterology. 2010 Mar:105(3):493-8. doi: 10.1038/ajg.2009.728. Epub [PubMed PMID: 20203636]
González CA, Sanz-Anquela JM, Gisbert JP, Correa P. Utility of subtyping intestinal metaplasia as marker of gastric cancer risk. A review of the evidence. International journal of cancer. 2013 Sep 1:133(5):1023-32. doi: 10.1002/ijc.28003. Epub 2013 Feb 5 [PubMed PMID: 23280711]
Level 2 (mid-level) evidenceConchillo JM, Houben G, de Bruïne A, Stockbrügger R. Is type III intestinal metaplasia an obligatory precancerous lesion in intestinal-type gastric carcinoma? European journal of cancer prevention : the official journal of the European Cancer Prevention Organisation (ECP). 2001 Aug:10(4):307-12 [PubMed PMID: 11535872]
Level 2 (mid-level) evidenceAsharaf A, Desai P, Sanati M. Emphysematous Gastritis. The Journal of the American Osteopathic Association. 2019 Dec 1:119(12):848. doi: 10.7556/jaoa.2019.140. Epub [PubMed PMID: 31790130]
Carabotti M, Lahner E, Esposito G, Sacchi MC, Severi C, Annibale B. Upper gastrointestinal symptoms in autoimmune gastritis: A cross-sectional study. Medicine. 2017 Jan:96(1):e5784. doi: 10.1097/MD.0000000000005784. Epub [PubMed PMID: 28072728]
Level 2 (mid-level) evidenceRodriguez-Castro KI, Franceschi M, Miraglia C, Russo M, Nouvenne A, Leandro G, Meschi T, De' Angelis GL, Di Mario F. Autoimmune diseases in autoimmune atrophic gastritis. Acta bio-medica : Atenei Parmensis. 2018 Dec 17:89(8-S):100-103. doi: 10.23750/abm.v89i8-S.7919. Epub 2018 Dec 17 [PubMed PMID: 30561426]
Garcés-Durán R, Llach J, Da Fieno A, Córdova H, Fernández-Esparrach G. Endoscopic diagnosis of H. pylori infection. Gastroenterologia y hepatologia. 2023 Jun-Jul:46(6):483-488. doi: 10.1016/j.gastrohep.2022.09.008. Epub 2022 Oct 3 [PubMed PMID: 36195279]
Moayyedi P, Lacy BE, Andrews CN, Enns RA, Howden CW, Vakil N. ACG and CAG Clinical Guideline: Management of Dyspepsia. The American journal of gastroenterology. 2017 Jul:112(7):988-1013. doi: 10.1038/ajg.2017.154. Epub 2017 Jun 20 [PubMed PMID: 28631728]
Miwa H, Nagahara A, Asakawa A, Arai M, Oshima T, Kasugai K, Kamada K, Suzuki H, Tanaka F, Tominaga K, Futagami S, Hojo M, Mihara H, Higuchi K, Kusano M, Arisawa T, Kato M, Joh T, Mochida S, Enomoto N, Shimosegawa T, Koike K. Evidence-based clinical practice guidelines for functional dyspepsia 2021. Journal of gastroenterology. 2022 Feb:57(2):47-61. doi: 10.1007/s00535-021-01843-7. Epub 2022 Jan 21 [PubMed PMID: 35061057]
Level 1 (high-level) evidenceFischbach W, Malfertheiner P. Helicobacter Pylori Infection. Deutsches Arzteblatt international. 2018 Jun 22:115(25):429-436. doi: 10.3238/arztebl.2018.0429. Epub [PubMed PMID: 29999489]
Venerito M, Varbanova M, Röhl FW, Reinhold D, Frauenschläger K, Jechorek D, Weigt J, Link A, Malfertheiner P. Oxyntic gastric atrophy in Helicobacter pylori gastritis is distinct from autoimmune gastritis. Journal of clinical pathology. 2016 Aug:69(8):677-85. doi: 10.1136/jclinpath-2015-203405. Epub 2016 Jan 4 [PubMed PMID: 26729016]
Alonso N, Granada ML, Soldevila B, Salinas I, Joaquin C, Reverter JL, Juncà J, Martínez Cáceres EM, Sanmartí A. Serum autoimmune gastritis markers, pepsinogen I and parietal cell antibodies, in patients with type 1 diabetes mellitus: a 5-year prospective study. Journal of endocrinological investigation. 2011 May:34(5):340-4 [PubMed PMID: 20530988]
di Mario F, Cavallaro LG. Non-invasive tests in gastric diseases. Digestive and liver disease : official journal of the Italian Society of Gastroenterology and the Italian Association for the Study of the Liver. 2008 Jul:40(7):523-30. doi: 10.1016/j.dld.2008.02.028. Epub 2008 Apr 24 [PubMed PMID: 18439884]
Du LJ, Chen BR, Kim JJ, Kim S, Shen JH, Dai N. Helicobacter pylori eradication therapy for functional dyspepsia: Systematic review and meta-analysis. World journal of gastroenterology. 2016 Mar 28:22(12):3486-95. doi: 10.3748/wjg.v22.i12.3486. Epub [PubMed PMID: 27022230]
Level 1 (high-level) evidencePimentel-Nunes P, Libânio D, Marcos-Pinto R, Areia M, Leja M, Esposito G, Garrido M, Kikuste I, Megraud F, Matysiak-Budnik T, Annibale B, Dumonceau JM, Barros R, Fléjou JF, Carneiro F, van Hooft JE, Kuipers EJ, Dinis-Ribeiro M. Management of epithelial precancerous conditions and lesions in the stomach (MAPS II): European Society of Gastrointestinal Endoscopy (ESGE), European Helicobacter and Microbiota Study Group (EHMSG), European Society of Pathology (ESP), and Sociedade Portuguesa de Endoscopia Digestiva (SPED) guideline update 2019. Endoscopy. 2019 Apr:51(4):365-388. doi: 10.1055/a-0859-1883. Epub 2019 Mar 6 [PubMed PMID: 30841008]
Gudej S, Filip R, Harasym J, Wilczak J, Dziendzikowska K, Oczkowski M, Jałosińska M, Juszczak M, Lange E, Gromadzka-Ostrowska J. Clinical Outcomes after Oat Beta-Glucans Dietary Treatment in Gastritis Patients. Nutrients. 2021 Aug 14:13(8):. doi: 10.3390/nu13082791. Epub 2021 Aug 14 [PubMed PMID: 34444949]
Level 2 (mid-level) evidenceRugge M, Meggio A, Pennelli G, Piscioli F, Giacomelli L, De Pretis G, Graham DY. Gastritis staging in clinical practice: the OLGA staging system. Gut. 2007 May:56(5):631-6 [PubMed PMID: 17142647]
Level 2 (mid-level) evidenceLahner E, Esposito G, Pilozzi E, Purchiaroni F, Corleto VD, Di Giulio E, Annibale B. Occurrence of gastric cancer and carcinoids in atrophic gastritis during prospective long-term follow up. Scandinavian journal of gastroenterology. 2015 Jul:50(7):856-65. doi: 10.3109/00365521.2015.1010570. Epub 2015 Feb 3 [PubMed PMID: 25645880]
Level 2 (mid-level) evidenceMahmud N, Stashek K, Katona BW, Tondon R, Shroff SG, Roses R, Furth EE, Metz DC. The incidence of neoplasia in patients with autoimmune metaplastic atrophic gastritis: a renewed call for surveillance. Annals of gastroenterology. 2019 Jan-Feb:32(1):67-72. doi: 10.20524/aog.2018.0325. Epub 2018 Nov 8 [PubMed PMID: 30598594]
Watari J, Chen N, Amenta PS, Fukui H, Oshima T, Tomita T, Miwa H, Lim KJ, Das KM. Helicobacter pylori associated chronic gastritis, clinical syndromes, precancerous lesions, and pathogenesis of gastric cancer development. World journal of gastroenterology. 2014 May 14:20(18):5461-73. doi: 10.3748/wjg.v20.i18.5461. Epub [PubMed PMID: 24833876]
Level 3 (low-level) evidenceWijarnpreecha K, Thongprayoon C, Panjawatanan P, Manatsathit W, Jaruvongvanich V, Ungprasert P. Helicobacter pylori and Risk of Nonalcoholic Fatty Liver Disease: A Systematic Review and Meta-analysis. Journal of clinical gastroenterology. 2018 May/Jun:52(5):386-391. doi: 10.1097/MCG.0000000000000784. Epub [PubMed PMID: 28098578]
Level 1 (high-level) evidenceTsay FW, Hsu PI. H. pylori infection and extra-gastroduodenal diseases. Journal of biomedical science. 2018 Aug 29:25(1):65. doi: 10.1186/s12929-018-0469-6. Epub 2018 Aug 29 [PubMed PMID: 30157866]
Malfertheiner P, Megraud F, O'Morain CA, Gisbert JP, Kuipers EJ, Axon AT, Bazzoli F, Gasbarrini A, Atherton J, Graham DY, Hunt R, Moayyedi P, Rokkas T, Rugge M, Selgrad M, Suerbaum S, Sugano K, El-Omar EM, European Helicobacter and Microbiota Study Group and Consensus panel. Management of Helicobacter pylori infection-the Maastricht V/Florence Consensus Report. Gut. 2017 Jan:66(1):6-30. doi: 10.1136/gutjnl-2016-312288. Epub 2016 Oct 5 [PubMed PMID: 27707777]
Level 3 (low-level) evidenceZucca E, Bertoni F, Roggero E, Bosshard G, Cazzaniga G, Pedrinis E, Biondi A, Cavalli F. Molecular analysis of the progression from Helicobacter pylori-associated chronic gastritis to mucosa-associated lymphoid-tissue lymphoma of the stomach. The New England journal of medicine. 1998 Mar 19:338(12):804-10 [PubMed PMID: 9504941]
Level 3 (low-level) evidenceLin WC, Tsai HF, Kuo SH, Wu MS, Lin CW, Hsu PI, Cheng AL, Hsu PN. Translocation of Helicobacter pylori CagA into Human B lymphocytes, the origin of mucosa-associated lymphoid tissue lymphoma. Cancer research. 2010 Jul 15:70(14):5740-8. doi: 10.1158/0008-5472.CAN-09-4690. Epub 2010 Jun 29 [PubMed PMID: 20587516]
Okame M, Takaya S, Sato H, Adachi E, Ohno N, Kikuchi T, Koga M, Oyaizu N, Ota Y, Fujii T, Iwamoto A, Koibuchi T. Complete regression of early-stage gastric diffuse large B-cell lymphoma in an HIV-1-infected patient following Helicobacter pylori eradication therapy. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America. 2014 May:58(10):1490-2. doi: 10.1093/cid/ciu111. Epub 2014 Feb 27 [PubMed PMID: 24585569]
Level 3 (low-level) evidenceHershko C, Lahad A, Kereth D. Gastropathic sideropenia. Best practice & research. Clinical haematology. 2005 Jun:18(2):363-80 [PubMed PMID: 15737896]
Kulnigg-Dabsch S, Resch M, Oberhuber G, Klinglmueller F, Gasche A, Gasche C. Iron deficiency workup reveals high incidence of autoimmune gastritis with parietal cell antibody as reliable screening test. Seminars in hematology. 2018 Oct:55(4):256-261. doi: 10.1053/j.seminhematol.2018.07.003. Epub 2018 Aug 7 [PubMed PMID: 30502855]
Bionda M, Kapoglou I, Wiest R. [H. pylori-associated gastritis: diagnostic, treatment and surveillance]. Therapeutische Umschau. Revue therapeutique. 2020:77(4):127-131. doi: 10.1024/0040-5930/a001167. Epub [PubMed PMID: 32772697]