Introduction
Epidermolytic hyperkeratosis is a rare autosomal dominant pathology of cornification caused by mutations in keratins 1 and 10. It was originally termed bullous congenital ichthyosiform erythroderma owing to the hallmark features of erythroderma, blistering and skin denudation present at birth and subsequent development of marked hyperkeratosis. This presentation occurs with or without palmoplantar keratoderma. Epidermolytic hyperkeratosis is easily distinguishable from other forms of congenital ichthyoses via its highly characteristic histologic findings. In more recent literature, epidermolytic hyperkeratosis has the designation as a pathologic term, and the disease entity has the name of epidermolytic ichthyosis.[1][2][3]
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
Epidermolytic hyperkeratosis is primarily the result of point or missense mutations in the genes encoding keratin 1 (KRT1) and keratin 10 (KRT10).[1][4][2][5][6] The majority of cases transmit through an autosomal dominant pattern of inheritance.[1][2][7] A severe form of epidermolytic hyperkeratosis exists in three consanguineous pedigrees due to autosomal recessive loss-of-function mutations in keratin 10.[2][8][9] Sporadic mutations occur in up to 50% of cases.[1][2][7] Mutations in keratin 1 have been associated with epidermolytic hyperkeratosis with severe palmoplantar keratoderma, while mutations in keratin 10 cause a phenotype lacking palmoplantar keratoderma.[7][10]
The offspring of parents with epidermal nevi may have generalized epidermolytic hyperkeratosis due to genetic mosaicism. The mosaic form of epidermolytic hyperkeratosis, variably labeled ichthyosis hystrix or linear epidermolytic hyperkeratosis, is caused by postzygotic mutations in keratin 1 and keratin 10 during embryogenesis. If these mutations involve gonadal cells, they can be transmitted to offspring and cause phenotypical epidermolytic hyperkeratosis.[7][11] Currently, epidermolytic hyperkeratosis is the only known human keratin disease to exhibit genetic mosaicism.[7]
Epidemiology
Epidermolytic hyperkeratosis has been reported to affect 1 in 200000 to 300000 infants.[1][12] The disorder is predominantly autosomal dominant, and therefore, there is no gender predilection.[1]
Pathophysiology
Epidermolytic hyperkeratosis is caused by mutations that disrupt the keratin network of the epidermis. Keratin proteins are the most important proteins necessary for the structural development of the epidermis.[1][13] The epidermis protects the skin from environmental conditions by forming a massive cytoskeletal structure of keratin filaments. Genes encoding keratin 5 and keratin 14 get expressed in the basal layer of the epidermis. As the epidermal cells continue to terminally differentiate, the suprabasal cells are made up of keratin 1 and keratin 10 filaments, which are thicker than the tonofilament bundles of basal cells. Thicker tonofilament bundles allow for enhanced survival of keratins.[1][14][15]
Mutations in epidermolytic hyperkeratosis occur in the highly conserved a-helical rod domains of keratin 1 and keratin 10. Mutations in helix boundary sequence motifs affect helix initiation and termination motifs and lead to significantly disrupted filament assembly, tonofilament aggregation and generally, more severe phenotypes. The compromised epidermis is in turn prone to cytolysis and blistering, leading to disruption in the skin barrier function, causing increased transepidermal water loss and bacterial colonization.[7] Hyperkeratosis results from hyperproliferation in the basal cells and decreased desquamation.[1]
Chipev et al. discovered via electron microscopy a leucine to a proline point mutation in the H1 sub-domain of the intermediate keratin 1 filament that significantly disrupted the structure and organization of keratin filaments.[4]. Snyder et al. identified an arginine to histidine mutation in the amino end of the a-helical rod domain of KRT10 and tyrosine to cysteine mutation in the carboxy-domain of KRT1.[16] Severe forms of epidermolytic hyperkeratosis have associations with deletion of exon 6 of KRT1[6] and missense mutations (c.475T>C;p.Ser159Pro and c.562A>C;p.N188H) in KRT10 and KRT1, respectively.[1][17]
Histopathology
Characteristic histopathologic findings distinguish epidermolytic hyperkeratosis from other congenital ichthyoses. Prominent features on histology include dense orthohyperkeratosis, hypergranulosis, coarse keratohyaline granules and cytolysis of the suprabasal and granular layers. Keratinocytes demonstrate marked intracellular vacuolar degeneration and dense clumps of keratin intermediate filaments. Varying degrees of dyskeratosis and a mild perivascular lymphohistiocytic infiltrate also occur. Electron microscopy shows clumped keratin intermediate filaments at the suprabasal layer, and immunohistochemistry shows defects in keratins 1 or 10.[12] Patients with the mosaic form of epidermolytic hyperkeratosis exhibit focal areas of involvement with skip areas of normal epidermis.[1][2]
History and Physical
Epidermolytic hyperkeratosis appears at birth with generalized erythroderma. Skin fragility causes blisters, peeling/exfoliation, erosions, and widespread areas of denuded skin, even with minor trauma. After several months, the erythema and blistering decrease and marked hyperkeratosis develop. Occasionally, skin fragility persists, and patients periodically shed large plates of the superficial epidermis. Hyperkeratosis characteristically resembles “corrugated cardboard” when overlying flexural areas or "cobblestoning" when overlying the extensor surfaces of joints. Occasionally, severe scalp and neck involvement occur, leading to encasement of hair shafts and alopecia. Bacterial colonization of the macerated scales causes a distinctive foul odor resembling rotten eggs. Commonly associated symptoms include xerosis, pruritus, painful fissuring, anhidrosis, and decreased range of motion of joints.[1][12]
Researchers have identified several different phenotypes of epidermolytic hyperkeratosis. In 1994, DiGiovanna and Bale described two main clinical categories: with palmoplantar keratoderma and without palmoplantar keratoderma.[18] Each clinical category has three subtypes with varying degrees of erythroderma, blistering, scaling (non-palmoplantar type), and truncal involvement (palmoplantar type).[12] Digital contractures from palmoplantar involvement can lead to functional impairment. The mosaic form of epidermolytic hyperkeratosis features unilateral or bilateral streaks of hyperkeratosis in a Blaschkoid distribution.[7][11] Those with more extensive involvement and lesions with protruding, porcupine-like spines have been termed “ichthyosis hystrix.”[7]
Evaluation
The diagnostic basis is on clinical, histopathologic, and laboratory findings. Genetic mutation analysis for keratin defects with multigene-panel screening is the current gold standard and is usable for prenatal screening.[1][12] Prenatal diagnosis is obtainable via chorionic villus sampling, amniocentesis, and fetoscopy with a fetal skin biopsy.[1][12] However, these studies have limitations of low sensitivity or genetic heterogeneity. Optical and spectroscopic techniques such as the Raman spectroscopy and optical coherence tomography have been used in in-vivo identification of nonmelanoma skin cancers and may be a potential diagnostic technique for epidermolytic hyperkeratosis.[1]
Treatment / Management
Treatment is predominantly symptomatic and depends on the patient’s age and presentation. Infants should be monitored in the intensive care setting to manage dehydration, electrolyte imbalance, and cutaneous superinfection. Sepsis treatment should be with broad-spectrum intravenous antibiotics. Topical emollients and protective padding should be used for skin protection and to heal denuded areas.[12](B3)
The goal of therapy in children and adults is a reduction in hyperkeratosis. Topical emollients and keratolytic agents containing glycerin, lactic acid, urea, and a-hydroxy acids have demonstrated improvement in hyperkeratosis but are often not well tolerated due to burning and stinging. Clinicians should avoid widespread use of higher concentration salicylic acid topical preparations because of the risk of systemic salicylism.[1][12] Topical retinoids, N-acetylcysteine, liarozole, and calcipotriol have been shown to affect corneocyte function and decrease epidermal hyperproliferation. However, these agents may also cause skin irritation.[1] In severe cases of epidermolytic hyperkeratosis, oral retinoids may dramatically improve hyperkeratosis and frequency of superinfection but can paradoxically increase skin fragility and exacerbate blistering. Thus, it is recommended to use low initial doses and then gradually increase with careful monitoring.[19] Of note, patients with keratin 10 mutations respond better to topical or systemic retinoids compared to patients with keratin 1 mutations.[1][12] Antibacterial soaps, chlorhexidine, and dilute sodium hypochlorite baths can decrease bacterial colonization. Topical or systemic antibiotics are needed when bacterial skin infection occurs.[1][12](B3)
Differential Diagnosis
The differential diagnosis of epidermolytic hyperkeratosis includes other causes of erythroderma, bullae/blisters, or exfoliation in childhood.
Other congenital ichthyoses:
- Superficial epidermolytic ichthyosis
- Lamellar ichthyosis
- Congenital ichthyosiform erythroderma
Vesiculobullous and erosive disorders in childhood:
- Epidermolysis bullosa
- Staphylococcal scalded skin syndrome
- Bullous impetigo
- Herpes simplex
- Congenital erosive and vesicular dermatosis
- Autoimmune blistering diseases
Genodermatoses:
- Sjogren-Larsson syndrome
- Neutral lipid storage disease
- Trichothiodystrophy
- Netherton syndrome
- Steroid sulfatase deficiency
- Peeling skin syndromes
- Conradi-Hunermann-Happle syndrome
- CHILD syndrome
- KID syndrome
Prognosis
The severity of epidermolytic hyperkeratosis is variable. Neonates with this disorder are at increased risk of sepsis, dehydration and ultimately, death. Due to the rarity of this disorder, statistics on mortality rate are lacking. For those that survive the neonatal period, episodes of blistering and skin infections occur intermittently throughout life. As a result, patients often have significant disfigurement and pungent body odors, causing severe psychological and social distress.[1][12]
Complications
Neonates born with epidermolytic hyperkeratosis are at higher risk of dehydration, electrolyte imbalances, and sepsis due to increased transepidermal water loss and ineffective skin barrier function. Without proper treatment, these complications are fatal. Also, severe palmoplantar involvement can lead to digital contractures, which can negatively affect joint mobility, gait, and posture.[12]
Deterrence and Patient Education
Patients with epidermolytic hyperkeratosis have increased skin fragility throughout their life, and therefore, it is essential to educate patients on ways to minimize mechanical trauma. This is possible by wearing loose-fitting, comfortable clothes, and well-fitting shoes.
Enhancing Healthcare Team Outcomes
Epidermolytic hyperkeratosis usually presents soon after birth. It has no cure, and the disorder is progressive. Treatment is primarily symptomatic and depends on the patient’s age and presentation. Physician management involves the family doctor, pediatrician, and dermatologist, as well as the NP and PA. Infants should be monitored in the intensive care setting to manage dehydration, electrolyte imbalance, and cutaneous superinfection. Sepsis should receive treatment with broad-spectrum intravenous antibiotics, which should have input from the pharmacist for coverage, dosing, and monitoring for drug interactions. The nurse should educate the caregiver or parent on the use of topical emollients and protective padding for skin protection and heal denuded areas, which can also have pharmacy input. Because the skin is fragile, the nurse practitioner should also educate patients on ways to minimize mechanical trauma, which is achievable by wearing loose-fitting, comfortable clothes, and well-fitting shoes.
Because of the considerable psychosocial morbidity caused by long-term sequelae of epidermolytic hyperkeratosis, it is necessary to coordinate care with an interprofessional team including intensivists, infectious disease specialists, mental health nurse, and psychiatrists. Delays in diagnosis and management can produce negative outcomes and possible death. All these members of the interprofessional healthcare team need to collaborate and communicate to bring about positive patient outcomes.[1] [Level 5]
Media
(Click Image to Enlarge)
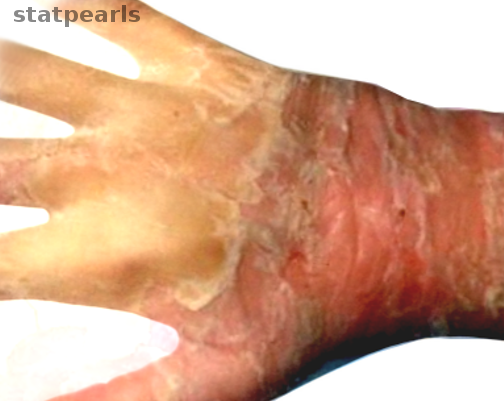
Skin Fragility in Epidermolytic Hyperkeratosis. This image shows marked exfoliation in a patient with epidermolytic hyperkeratosis.
Image courtesy S Bhimji MD
(Click Image to Enlarge)
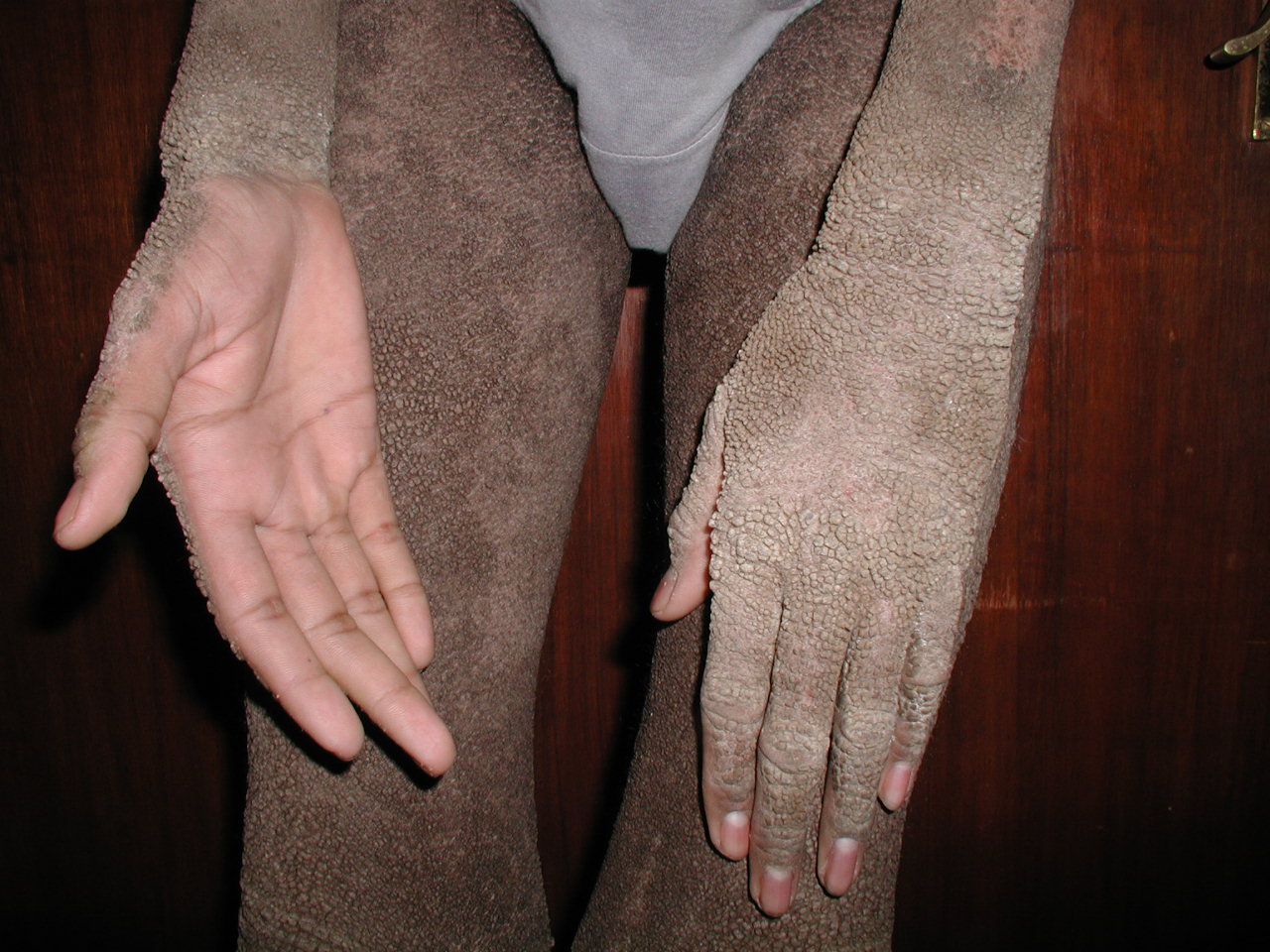
Hyperkeratosis in Epidermolytic Hyperkeratosis. Severe hyperkeratosis in a young patient with epidermolytic hyperkeratosis.
Contributed by Dr. Shyam Verma, MBBS, DVD, FRCP, FAAD, Vadodara, India
References
Peter Rout D, Nair A, Gupta A, Kumar P. Epidermolytic hyperkeratosis: clinical update. Clinical, cosmetic and investigational dermatology. 2019:12():333-344. doi: 10.2147/CCID.S166849. Epub 2019 May 8 [PubMed PMID: 31190940]
Covaciu C, Castori M, De Luca N, Ghirri P, Nannipieri A, Ragone G, Zambruno G, Castiglia D. Lethal autosomal recessive epidermolytic ichthyosis due to a novel donor splice-site mutation in KRT10. The British journal of dermatology. 2010 Jun:162(6):1384-7. doi: 10.1111/j.1365-2133.2010.09665.x. Epub 2010 Mar 10 [PubMed PMID: 20302579]
Level 3 (low-level) evidenceOji V, Tadini G, Akiyama M, Blanchet Bardon C, Bodemer C, Bourrat E, Coudiere P, DiGiovanna JJ, Elias P, Fischer J, Fleckman P, Gina M, Harper J, Hashimoto T, Hausser I, Hennies HC, Hohl D, Hovnanian A, Ishida-Yamamoto A, Jacyk WK, Leachman S, Leigh I, Mazereeuw-Hautier J, Milstone L, Morice-Picard F, Paller AS, Richard G, Schmuth M, Shimizu H, Sprecher E, Van Steensel M, Taïeb A, Toro JR, Vabres P, Vahlquist A, Williams M, Traupe H. Revised nomenclature and classification of inherited ichthyoses: results of the First Ichthyosis Consensus Conference in Sorèze 2009. Journal of the American Academy of Dermatology. 2010 Oct:63(4):607-41. doi: 10.1016/j.jaad.2009.11.020. Epub [PubMed PMID: 20643494]
Level 3 (low-level) evidenceChipev CC, Korge BP, Markova N, Bale SJ, DiGiovanna JJ, Compton JG, Steinert PM. A leucine----proline mutation in the H1 subdomain of keratin 1 causes epidermolytic hyperkeratosis. Cell. 1992 Sep 4:70(5):821-8 [PubMed PMID: 1381288]
Ishida-Yamamoto A, McGrath JA, Judge MR, Leigh IM, Lane EB, Eady RA. Selective involvement of keratins K1 and K10 in the cytoskeletal abnormality of epidermolytic hyperkeratosis (bullous congenital ichthyosiform erythroderma). The Journal of investigative dermatology. 1992 Jul:99(1):19-26 [PubMed PMID: 1376754]
Virtanen M, Smith SK, Gedde-Dahl T Jr, Vahlquist A, Bowden PE. Splice site and deletion mutations in keratin (KRT1 and KRT10) genes: unusual phenotypic alterations in Scandinavian patients with epidermolytic hyperkeratosis. The Journal of investigative dermatology. 2003 Nov:121(5):1013-20 [PubMed PMID: 14708600]
Irvine AD, McLean WH. Human keratin diseases: the increasing spectrum of disease and subtlety of the phenotype-genotype correlation. The British journal of dermatology. 1999 May:140(5):815-28 [PubMed PMID: 10354017]
Level 3 (low-level) evidenceMüller FB, Huber M, Kinaciyan T, Hausser I, Schaffrath C, Krieg T, Hohl D, Korge BP, Arin MJ. A human keratin 10 knockout causes recessive epidermolytic hyperkeratosis. Human molecular genetics. 2006 Apr 1:15(7):1133-41 [PubMed PMID: 16505000]
Level 3 (low-level) evidenceTsubota A, Akiyama M, Kanitakis J, Sakai K, Nomura T, Claudy A, Shimizu H. Mild recessive bullous congenital ichthyosiform erythroderma due to a previously unidentified homozygous keratin 10 nonsense mutation. The Journal of investigative dermatology. 2008 Jul:128(7):1648-52. doi: 10.1038/sj.jid.5701257. Epub 2008 Jan 24 [PubMed PMID: 18219278]
DiGiovanna JJ, Bale SJ. Clinical heterogeneity in epidermolytic hyperkeratosis. Archives of dermatology. 1994 Aug:130(8):1026-35 [PubMed PMID: 8053700]
Level 2 (mid-level) evidencePaller AS, Syder AJ, Chan YM, Yu QC, Hutton E, Tadini G, Fuchs E. Genetic and clinical mosaicism in a type of epidermal nevus. The New England journal of medicine. 1994 Nov 24:331(21):1408-15 [PubMed PMID: 7526210]
Kwak J, Maverakis E. Epidermolytic hyperkeratosis. Dermatology online journal. 2006 Sep 8:12(5):6 [PubMed PMID: 16962021]
Level 3 (low-level) evidenceFuchs E, Coulombe P, Cheng J, Chan YM, Hutton E, Syder A, Degenstein L, Yu QC, Letai A, Vassar R. Genetic bases of epidermolysis bullosa simplex and epidermolytic hyperkeratosis. The Journal of investigative dermatology. 1994 Nov:103(5 Suppl):25S-30S [PubMed PMID: 7525738]
Nelson WG, Sun TT. The 50- and 58-kdalton keratin classes as molecular markers for stratified squamous epithelia: cell culture studies. The Journal of cell biology. 1983 Jul:97(1):244-51 [PubMed PMID: 6190820]
Roop DR, Huitfeldt H, Kilkenny A, Yuspa SH. Regulated expression of differentiation-associated keratins in cultured epidermal cells detected by monospecific antibodies to unique peptides of mouse epidermal keratins. Differentiation; research in biological diversity. 1987:35(2):143-50 [PubMed PMID: 2450799]
Level 3 (low-level) evidenceSyder AJ, Yu QC, Paller AS, Giudice G, Pearson R, Fuchs E. Genetic mutations in the K1 and K10 genes of patients with epidermolytic hyperkeratosis. Correlation between location and disease severity. The Journal of clinical investigation. 1994 Apr:93(4):1533-42 [PubMed PMID: 7512983]
Chen PJ, Li CX, Wen J, Peng YS, Zeng K, Zhang SQ, Tian X, Zhang XB. S159P mutation of keratin 10 gene causes severe form of epidermolytic hyperkeratosis. Journal of the European Academy of Dermatology and Venereology : JEADV. 2016 Oct:30(10):e102-e104. doi: 10.1111/jdv.13345. Epub 2015 Sep 15 [PubMed PMID: 26373619]
DiGiovanna JJ, Bale SJ. Epidermolytic hyperkeratosis: applied molecular genetics. The Journal of investigative dermatology. 1994 Mar:102(3):390-4 [PubMed PMID: 7509838]
Lacour M, Mehta-Nikhar B, Atherton DJ, Harper JI. An appraisal of acitretin therapy in children with inherited disorders of keratinization. The British journal of dermatology. 1996 Jun:134(6):1023-9 [PubMed PMID: 8763418]