Introduction
The ear is an incredible organ of hearing and equilibrium divided into three anatomic parts: the external, middle, and internal ear. The external ear, or outer ear, consists of the auricle or pinna, and the tubular external auditory canal ending at the tympanic cavity. The external ear resonates and amplifies sound, and it directs sound towards the tympanic membrane. The middle ear's tympanic membrane converts energy from sound waves into mechanical energy as vibrations. The middle ear is essentially an air-filled cavity that houses three auditory ossicles: the malleus, incus, and stapes. The ossicles transmit sound vibrations from the tympanic membrane to the internal or inner ear. The internal ear, or labyrinth of the ear, houses the organs of hearing and balance. The internal ear is composed of the vestibule, semicircular canals, and cochlea. The vestibule functions to sense linear acceleration, while the semicircular canals sense rotational movements. The cochlea's organ of Corti functions to transduce auditory signals into neuronal impulses that reach the brain via the vestibulocochlear nerve. The delicate structures of the internal, middle, and external ear must function in concert to transmit sound and sense movement.
The development of the ear requires contributions from all three germ layers and involves a sophisticated process with intricate embryologic patterning. Each anatomic division of the ear has a distinct origin and unique developmental processes resulting in their typical form.[1] While the development of the ear continues post-birth, a fetus can functionally hear by about 26 weeks of development. Notably, several anatomic variants and congenital conditions can arise from deviations in the typical developmental processes.[2]
Development
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Development
Internal Ear
The internal ear is derived from ectoderm, and it is the first of the three anatomic parts of the ear to form. Development begins as a pair of short-lived thickenings of the surface ectoderm, the otic placode or otic disc, appear dorsolateral to the hindbrain around the fourth week of development—the otic placode forms due to the induction of surface ectoderm by the nearby notochord and paraxial mesoderm. The otic placode is one of the first sensory placodes involved in the formation of special sensory organs to develop.
The otic placode invaginates into the mesenchyme adjacent to the rhombencephalon to form an otic pit. The sides of the otic pit fold together and fuse to form a hollow piriform structure lined with columnar epithelium, called the otic vesicle. Rapidly, the otic vesicle moves deep to the surface ectoderm and is instead enveloped in mesenchyme to form the otic capsule. The statoacoustic, or vestibulocochlear, ganglion arises as neurons delaminate during the formation of the otic vesicle and, later, the ganglion splits into cochlear and vestibular portions.
The otic vesicle forms two visible regions: a ventral saccular portion and a dorsal utricular portion. The ventral saccular portion gives rise to internal ear structures involved in hearing, including the cochlear ducts and saccules. The dorsal utricular portion gives rise to the vestibular system, including the utricle, semicircular canals, and endolymphatic tube.[3][4] Ultimately, the otic vesicle will differentiate to form all of the components of the membranous labyrinth and the internal ear structures associated with hearing and balance.
The otic vesicle elongates within the first four weeks to form a tube-like structure called the endolymphatic appendage. Soon after, a groove-like indentation forms and demarcates a tubular diverticulum on the medial side of the endolymphatic appendage. This diverticulum differentiates into the endolymphatic duct and sac and continues to grow until around the age of four.[5]
Internal Ear: Ventral Saccular Component
The ventral saccular component of the otic vesicle forms a tubular cochlear duct, the primordial cochlea, within the mesenchyme by the sixth week. The cochlear duct grows and spirals two and a half times to produce the membranous cochlea. Rapidly, the saccule connects to the utricle via a duct called the ductus reuniens.
Mesenchyme surrounding the otic vesicle is induced to form a cartilaginous otic capsule, which will ossify to produce the internal ear's bony labyrinth later in development. The cartilaginous otic capsule then forms vacuoles that coalesce into the fluid-filled perilymphatic space of the cochlea. The fluid, or perilymph, resides within the perilymphatic space and surrounds the membranous labyrinth. The perilymphatic space then separates into two divisions: the scala vestibule and the scala tympani. Two membranes separate the cochlear duct from the perilymphatic divisions. The basilar membrane demarcates the cochlear duct from the scala tympani, while the vestibular membrane separates the cochlear duct from the scala vestibule. Cells in the lateral aspect of the cochlear duct differentiate to form the organ of Corti, or spiral organ or spiral organ of Corti, within the scala media of the cochlear duct. The cochlear duct also develops an attachment to the surrounding cartilage via connective tissue, the spiral ligament.
The organ of Corti is formed when ridges of epithelial cells from the cochlear duct produce rows of mechanosensory hair cells that are covered by the tectorial membrane. The spiral ganglion forms when ganglion cells derived from the vestibulocochlear nerve (CN VIII) migrate along the spirals of the membranous cochlea. Nervous processes then extend from the spiral ganglion to hair cells of the organ of Corti.
The cartilaginous otic capsule surrounding the membranous labyrinth ossifies by about 23 weeks to form the true bony labyrinth.[6] Around this time, the internal ear has reached its adult size and form.
Internal Ear: Dorsal Utricular Component
The dorsal utricular portion of the otic vesicle forms the utricle and semicircular canals, the organs of balance. During the sixth week, disc-like epithelial outpouchings extend dorsolaterally from the dorsal utricular portion of the primordial membranous labyrinth. The central portions of these discs approach each other, and their epithelium joins to form fusion plates which ultimately regress via programmed cell death. The peripheral unfused portions of the discs that fail to regress form incipient semicircular ducts that attach to the utricle. Later, the semicircular ducts are incorporated within the anterior, posterior, and lateral semicircular canals.
At one end of each semicircular duct, a dilatation of the duct develops and is called an ampulla. The ampullae contain sensory hair cells that form crests with specialized receptor areas, the cristae ampullares. Similar specialized areas form in the walls of the saccule and utricle. These regions sense changes in angular acceleration and serve as the sensory organ of rotation. Sensory cells of the cristae ampullares generate impulses that reach the brain via vestibular fibers of the vestibulocochlear nerve.[7]
Middle Ear
The middle ear is composed of the tympanic cavity and the Eustachian, also known as the auditory or pharyngotympanic, tube. Structures of the middle ear are derived from the tubotympanic sulcus, or tubotympanic recess, an endodermal extension from the first pharyngeal pouch. Around the 5th week of development, the tubotympanic sulcus extends laterally to approach the floor of the first pharyngeal groove but remains separated by mesenchyme. During development, the endoderm of the tubotympanic sulcus and the ectoderm of the first pharyngeal groove further approach each other, but they continue to maintain a layer of mesoderm between them. The end result is a trilaminar tympanic membrane made up of tissues derived from all three germ layers: ectoderm, mesoderm, and endoderm.
The tympanic cavity develops as an expansion of the distal portion of the tubotympanic sulcus. Anatomically, the tympanic cavity divides into upper (attic) and lower (atrium) chambers and gradually surrounds the ossicles, their attachments, and the chorda tympani. The Eustachian tube is formed from the proximal portion of the tubotympanic sulcus. The Eustachian tube is more horizontal, short, and narrow at birth than in later adulthood, which is a major reason infants have recurrent ear infections. Despite an endodermal origin, both the tympanic cavity and the Eustachian tube are ultimately lined by epithelium-derived from endoderm and neural crest cells. The Eustachian tube demonstrates the most growth during weeks 16 to 28 of the fetal period.[8]
The middle ear ossicles initially form around six weeks of development. They first appear in a cartilaginous form that arises from neural crest-derived mesenchymal cells within the first and second pharyngeal arches that condense at the dorsal end of the tubotympanic sulcus. The malleus and incus develop from Meckel's cartilage of the first pharyngeal arch. The stapes have a complex origin, partly arising from both neural crest cells and Reichert's cartilage of the second pharyngeal arch. As the tympanic cavity develops, the ossicular cartilages go through endochondral ossification that continues throughout the entire fetal period. Late in the fetal period, the mesenchyme that fills the tympanic cavity and surrounds the ossicles is resorbed to produce an air-filled tympanic cavity with ossicles suspended inside. Eventually, the tympanic cavity expands and forms the mastoid antrum.
The tensor tympani muscle arises from the mesoderm of the first pharyngeal arch and is innervated by the mandibular branch of the trigeminal nerve. The stapedius muscle originates from the mesoderm of the second pharyngeal arch and is innervated by the facial nerve.[9][10] The middle ear continues to develop post-birth and through puberty.
External Ear
The external ear first develops in the lower cervical region, but it gradually moves posterolaterally during development to reach its typical location.[11] The external ear's auricle develops from the mesenchymal proliferation of the first and second pharyngeal arches at the end of the fourth week of development. Six prominences, or auricular hillocks, form around the external auditory meatus and eventually fuse to form the auricle. Three auricular hillocks, hillocks 1 to 3, arise from the first pharyngeal arch to form the tragus, helix, and cymba concha; and three auricular hillocks, hillocks 4 to 6, arise from the second pharyngeal arch to form the concha, antihelix, and antitragus.
The external auditory meatus arises from the dorsal portion of the first pharyngeal groove. The meatus first develops as an invagination of ectoderm between the first and second pharyngeal arches that extends toward the developing middle ear structures. Around the fifth week, the ectodermal diverticulum extends toward the pharynx and houses proliferating ectodermal cells that produce an epithelial plug, the meatal plug, that will fills its entire lumen. At approximately ten weeks of development, the end of the meatal plug expands circumferentially to create a disc-like structure. Eventually, the disc-like meatal plug contacts the primordial malleus, divides, and leaves behind a thin ectodermal layer forming an incipient tympanic membrane. A continuation of the thin skin of the pinna lines the entire external auditory meatus and the outer surface of the tympanic membrane. By 18 weeks, the external auditory meatus is completely patent and expands to produce its typical morphology.
Cellular
The auricle and external auditory canal are both lined with keratinized squamous epithelium. The external auditory canal is formed partly of cartilage and partly of bone. The internal bony segment has tiny hairs and cerumen-producing apocrine glands along its lining.
The tympanic membrane separates the external ear from the tympanic cavity and has a trilaminar structure with contributions from all three germ layers. The outer layer of the tympanic membrane is composed of keratinized stratified squamous epithelium and is continuous with the surrounding external skin. The epithelium of the outer layer originates from the ectoderm of the first pharyngeal groove. The middle layer of the tympanic membrane is a thin fibrous connective tissue layer derived from mesoderm and composed of collagen and elastic fibers called the lamina propria. The inner mucosal layer of the tympanic membrane is derived from the endoderm of the first pharyngeal pouch. The mucous membrane is composed of a non-keratinized squamous epithelium that is continuous with the lining of the tympanic cavity.
The utricle and saccule are otolith organs located in the vestibule that detect movement in different planes. The utricle and saccule consist of sensory areas called maculae composed of supporting cells and hair cells covered in a gelatinous acellular matrix called the otolithic membrane. The otolithic membrane is embedded with calcium carbonate crystals called otoliths. The crista ampullaris of the semicircular ducts have a sensory epithelium similar to that of the macula, also consisting of hair cells and supporting cells. The hair cells of the cristae project into a gelatinous material called the cupula, which does not contain otoliths, and serves to detect rotational acceleration.
The organ of Corti is located on the basilar membrane and consists of a variety of supporting cells and two groups of hair cells: inner hair cells and outer hair cells. The inner hair cells account for approximately 95% of the sensory input into the auditory system and arrange in one line along the entire basilar membrane. The outer hair cells account for about 5% of sensory input and serve primarily as acoustical pre-amplifiers. The outer hair cells receive efferent input and contract when stimulated, resulting in amplified sound waves. The supporting cells include Hensen cells, Corti pillars, Deiters cells, and Claudius cells. The supporting cells play essential roles in the function and maintenance of the internal ear and primarily serve structural and homeostatic functions.[12]
Molecular Level
Proper formation and axial positioning of the components of the ear occur through complex reciprocal interactions between the epithelium and mesenchyme of the pharyngeal arches and hindbrain. These complex interactions involve a wide variety of essential genes, morphogens, and transcription factors that ultimately determine the fate of cells in the internal ear. Members of the Wnt, Sonic Hedgehog (SHH), and fibroblast-growth-factor (FGF) families, combined with retinoic acid signals, regulate transcription factor genes within the primordial internal ear to regionalize neurogenic activity and establish the axial identity of the ear.
Otic placode induction is dependent on Wnts and FGFs provided by the hindbrain and surrounding head mesenchyme. After induction, the otic placode continues to be influenced by signaling information from surrounding tissues that determine its positional identity along the dorsal-ventral, anterior-posterior, and medial-lateral axes. The anterior-posterior axis is the first axis to be specified. It requires retinoic acid, a key morphogen, to confer proper anterior and posterior identities of the internal ear. Somites express high levels of Raldh2, a retinoic acid synthesizing enzyme that serves as the primary source of retinoic acid for patterning the internal ear. Retinoic acid signaling results in proper anterior-posterior patterning of the internal ear and establishes the neural-sensory-competent domain (NSD) in the anterior otic cup.
The neural-sensory-competent domain gives rise to neurons of the cochleovestibular ganglion, as well as prosensory cells of the internal ear that differentiate into supporting cells or sensory hair cells. Neurogenin1 is a proneural gene that encodes a basic helix-loop-helix region (bHCH) transcription factor and is one of the earliest molecular markers determining the neurogenic fate of cells in the internal ear. The anterior portion of the NSD contains Ngn1-positive cells that ultimately leave the otic epithelium and coalesce to become neurons of the cochleovestibular ganglion. The remaining sensory epithelium of the NSD develops into supporting cells, sensory hair cells, and some nonsensory cells.[13][14][15]
Proper patterning of the internal ear dorsal-ventral axis involves the secretion of Wnts transcription factors from the dorsal hindbrain and the release of Sonic Hedgehog from the notochord and ventral floor plate. The patterning of the medial-lateral axis of the internal ear has not been well studied. It is thought to involve hindbrain signaling mediated by the transcription factor Gbx2 from the otic epithelium.
Sonic Hedgehog is not only imperative in determining the dorsal-ventral axis of the internal ear, but it is also responsible for regulating and determining auditory cell fates within the internal ear. Sonic Hedgehog is released from the notochord and ventral hindbrain and allows for proper cochlear duct and semicircular canal development. The mesenchyme encasing the developing internal ear is also essential for shaping the semicircular canals and cochlear duct into their final form through both structural and molecular means.
Although the mechanisms and molecules involved in the process of semicircular canal formation are largely unexplored, studies have implicated a variety of mesenchymal genes in canal formation, such as Prx and Pou3f4. Proper extension and outgrowth of the cochlear duct are dependent on Sonic Hedgehog secretion from the notochord and the release of transcription factors called Tbx1 and Pou3f4 from the otic mesenchyme. Studies have shown that an absence of Pou3f4 or Tbx1 in the otic mesenchyme results in abnormal shortening or coiling of the cochlear duct.[16][17][18]
Clinical Significance
Congenital anomalies involving the ear may be of significant physical and psychosocial concern to patients and the parents of afflicted children, given that these conditions may affect physical appearance, hearing, and balance.[19] In addition, the financial cost of such conditions can be significant given the potential for long-term special education, healthcare, and accessibility needs. Around 15 to 20% of neonates are estimated to be born with congenital abnormalities of the ear, and around 30% of these will resolve without intervention by six weeks of age.
While a wide variety of congenital anomalies of the ear exist, those that impact hearing are particularly concerning.[2] Neonatal hearing loss may be complete or partial, and approximately 1 in 1,000 neonates is estimated to have "significant" congenital hearing loss. Developmental anomalies of the ear that result in conductive hearing loss tend to involve the external and/or middle ear, while those that result in sensorineural hearing loss often involve the inner ear. Additional congenital causes of sensorineural hearing loss impact anatomic structures outside of the ear, including the vestibulocochlear nerve and auditory regions of the brain. The various developmental anomalies of the ear may result from genetic and/or environmental factors, the latter often caused by viral infections, neonatal exposures, or noise.
Internal Ear
Neonatal hearing loss is sometimes due to developmental anomalies of the neurosensory components of the internal ear. The most common cause, Enlarged Vestibular Aqueduct Syndrome (EVA), is an autosomal recessive condition in which there is a bilateral enlargement of the endolymphatic duct and vestibular aqueduct.[20]
Maternal infection with rubella is another source of neonatal hearing loss that may hinder the development of the organ of Corti in the fourth week of development, resulting in its malformation. Similarly, maternal infection with cytomegalovirus is another potential cause of congenital sensorineural hearing loss. Other relatively common congenital anomalies of the internal ear include Mondini dysplasia and autosomal dominant nonsyndromic hearing loss.
Middle Ear
Congenital anomalies of the middle ear are relatively rare and include congenital fixation of one or more of the ossicles, a rare primary bone dysplasia called familial expansile osteolysis, and a cyst-like abnormal accumulation of skin cells called cholesteatoma. Developmental malformations of the middle ear structures responsible for sound conversion and transmission contribute to neonatal hearing loss.
External ear
Numerous congenital anomalies of the external ear have been recorded in the literature.[21][22] Congenital anomalies of the external ear can potentially impact physical appearance or hearing. Given the role of the pharyngeal arches in the development of the external ear, anomalies of the external ear are associated with other pharyngeal arch anomalies and a variety of chromosomal disorders.
Preauricular tags, or simply ear tags, are common and usually benign findings in neonates that involve cutaneous, fatty, or cartilaginous growths. Occasionally, preauricular tags may be associated with other pharyngeal arch anomalies or genetic syndromes. Developmentally, accessory auricular hillocks sometimes produce auricular appendages, preauricular tags, or an accessory auricle.
Microtia is a developmental anomaly of the external ear involving an under-development of the typical mesenchymal proliferations that form the external ear. This condition presents at birth as an unusually small and sometimes misshapen external ear and is highly variable in its degree of severity. Microtia is associated with conductive hearing loss due to the possibility of middle and external ear malformations and the potential for complete agenesis of the external auditory canal.[19] Bilateral microtia is a classic indicator of Treacher-Collins Syndrome (TCS) and is present in approximately 85% of patients with TCS.[23]
Cryptotia is a malformation of the cartilage of the external ear that involves part of the external ear, usually the superior portion, being buried under the adjacent skin.[21]
Another external ear congenital anomaly, unilateral or bilateral atresia of the external acoustic meatus, occurs in individuals who retain the meatal plug due to a failure of canalization. In most cases, the external acoustic meatus is only superficially obstructed by fibrous or bony tissue. Given its relationship to the first pharyngeal groove, atresia of the external acoustic meatus has been associated with various malformations. Finally, the complete absence of the external auditory meatus is a rare congenital anomaly of the external ear; this condition occurs due to a failure in the mesenchymal proliferation arising from the first pharyngeal groove.
Media
(Click Image to Enlarge)
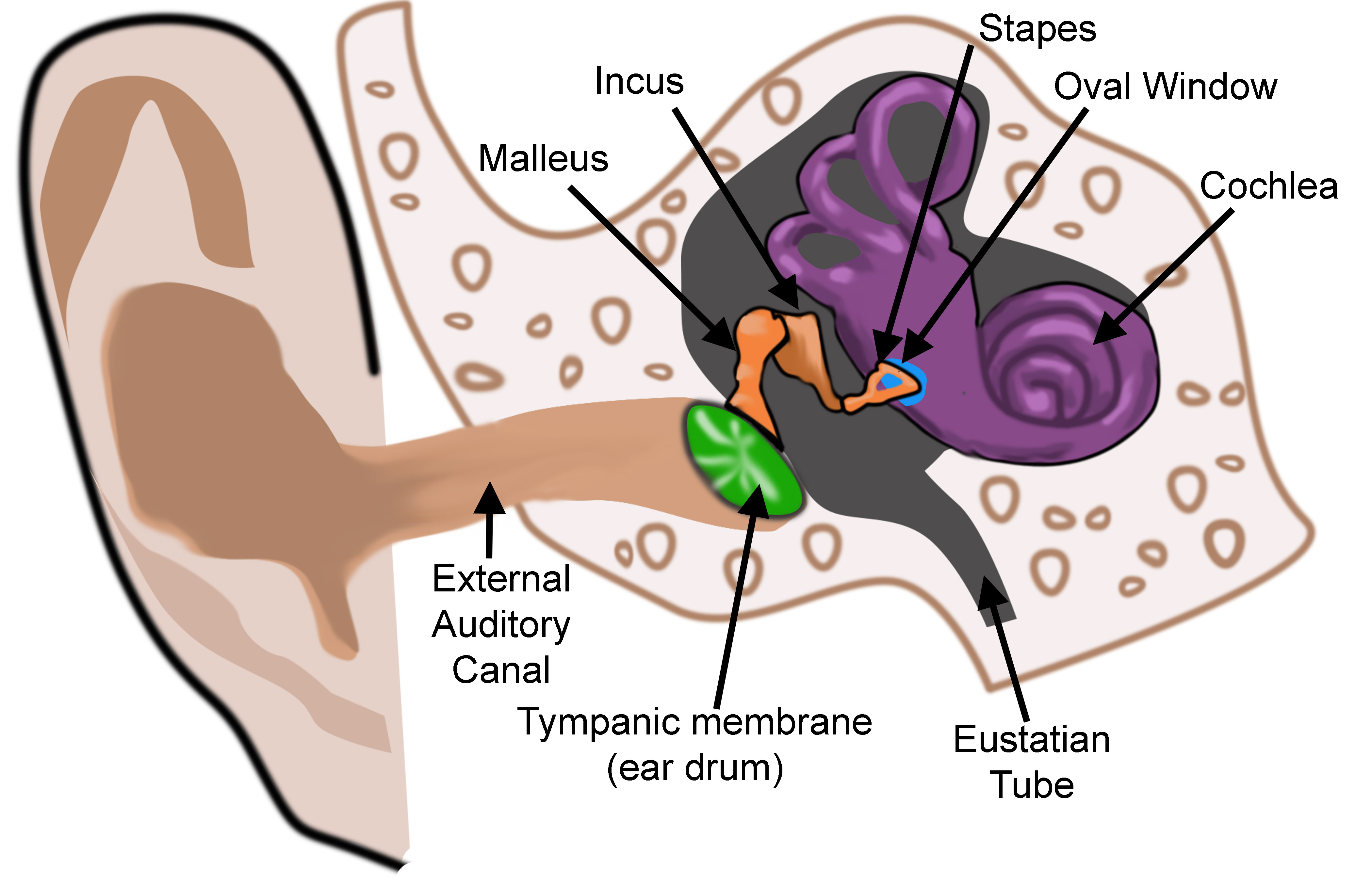
Anatomy of the outer, middle, and inner ear. Image created for publication by Diana Peterson.
References
Cheatham MA. Cochlear function reflected in mammalian hair cell responses. Progress in brain research. 1993:97():13-9 [PubMed PMID: 8234739]
Level 3 (low-level) evidenceCurtin HD. Congenital malformations of the ear. Otolaryngologic clinics of North America. 1988 May:21(2):317-36 [PubMed PMID: 3282212]
Giraldez F, Fritzsch B. The molecular biology of ear development - "Twenty years are nothing". The International journal of developmental biology. 2007:51(6-7):429-38 [PubMed PMID: 17891706]
Level 3 (low-level) evidenceSolomon KS,Kwak SJ,Fritz A, Genetic interactions underlying otic placode induction and formation. Developmental dynamics : an official publication of the American Association of Anatomists. 2004 Jul; [PubMed PMID: 15188428]
Level 3 (low-level) evidenceChristophorou NA, Mende M, Lleras-Forero L, Grocott T, Streit A. Pax2 coordinates epithelial morphogenesis and cell fate in the inner ear. Developmental biology. 2010 Sep 15:345(2):180-90. doi: 10.1016/j.ydbio.2010.07.007. Epub 2010 Jul 17 [PubMed PMID: 20643116]
Level 3 (low-level) evidenceWu DK, Kelley MW. Molecular mechanisms of inner ear development. Cold Spring Harbor perspectives in biology. 2012 Aug 1:4(8):a008409. doi: 10.1101/cshperspect.a008409. Epub 2012 Aug 1 [PubMed PMID: 22855724]
Level 3 (low-level) evidenceChang W, Brigande JV, Fekete DM, Wu DK. The development of semicircular canals in the inner ear: role of FGFs in sensory cristae. Development (Cambridge, England). 2004 Sep:131(17):4201-11 [PubMed PMID: 15280215]
Level 3 (low-level) evidenceAnthwal N, Thompson H. The development of the mammalian outer and middle ear. Journal of anatomy. 2016 Feb:228(2):217-32. doi: 10.1111/joa.12344. Epub 2015 Jul 30 [PubMed PMID: 26227955]
Hall JW 3rd. Development of the ear and hearing. Journal of perinatology : official journal of the California Perinatal Association. 2000 Dec:20(8 Pt 2):S12-20 [PubMed PMID: 11190691]
Level 3 (low-level) evidenceMaier W, Ruf I. Evolution of the mammalian middle ear: a historical review. Journal of anatomy. 2016 Feb:228(2):270-83. doi: 10.1111/joa.12379. Epub 2015 Sep 23 [PubMed PMID: 26397963]
Fuchs JC, Tucker AS. Development and Integration of the Ear. Current topics in developmental biology. 2015:115():213-32. doi: 10.1016/bs.ctdb.2015.07.007. Epub 2015 Oct 1 [PubMed PMID: 26589927]
Tos M, Anatomy and histology of the middle ear. Clinical reviews in allergy. 1984 Nov; [PubMed PMID: 6388791]
Bok J, Zenczak C, Hwang CH, Wu DK. Auditory ganglion source of Sonic hedgehog regulates timing of cell cycle exit and differentiation of mammalian cochlear hair cells. Proceedings of the National Academy of Sciences of the United States of America. 2013 Aug 20:110(34):13869-74. doi: 10.1073/pnas.1222341110. Epub 2013 Aug 5 [PubMed PMID: 23918393]
Level 3 (low-level) evidenceBarrionuevo F, Naumann A, Bagheri-Fam S, Speth V, Taketo MM, Scherer G, Neubüser A. Sox9 is required for invagination of the otic placode in mice. Developmental biology. 2008 May 1:317(1):213-24. doi: 10.1016/j.ydbio.2008.02.011. Epub 2008 Feb 21 [PubMed PMID: 18377888]
Level 3 (low-level) evidenceUrness LD, Paxton CN, Wang X, Schoenwolf GC, Mansour SL. FGF signaling regulates otic placode induction and refinement by controlling both ectodermal target genes and hindbrain Wnt8a. Developmental biology. 2010 Apr 15:340(2):595-604. doi: 10.1016/j.ydbio.2010.02.016. Epub 2010 Feb 18 [PubMed PMID: 20171206]
Level 3 (low-level) evidenceHans S, Christison J, Liu D, Westerfield M. Fgf-dependent otic induction requires competence provided by Foxi1 and Dlx3b. BMC developmental biology. 2007 Jan 19:7():5 [PubMed PMID: 17239227]
Level 3 (low-level) evidenceOhyama T, Mohamed OA, Taketo MM, Dufort D, Groves AK. Wnt signals mediate a fate decision between otic placode and epidermis. Development (Cambridge, England). 2006 Mar:133(5):865-75 [PubMed PMID: 16452098]
Level 3 (low-level) evidenceRiccomagno MM, Martinu L, Mulheisen M, Wu DK, Epstein DJ. Specification of the mammalian cochlea is dependent on Sonic hedgehog. Genes & development. 2002 Sep 15:16(18):2365-78 [PubMed PMID: 12231626]
Level 3 (low-level) evidenceBhatti SL, Daly LT, Mejia M, Perlyn C. Ear Abnormalities. Pediatrics in review. 2021 Apr:42(4):180-188. doi: 10.1542/pir.2019-0167. Epub [PubMed PMID: 33795464]
Ruthberg JS, Kocharyan A, Farrokhian N, Stahl MC, Hicks K, Scarborough J, Murray GS, Wu S, Manzoor N, Otteson T. Hearing loss patterns in enlarged vestibular aqueduct syndrome: Do fluctuations have clinical significance? International journal of pediatric otorhinolaryngology. 2022 May:156():111072. doi: 10.1016/j.ijporl.2022.111072. Epub 2022 Feb 22 [PubMed PMID: 35276529]
Joukhadar N, McKee D, Caouette-Laberge L, Bezuhly M. Management of Congenital Auricular Anomalies. Plastic and reconstructive surgery. 2020 Aug:146(2):205e-216e. doi: 10.1097/PRS.0000000000006997. Epub [PubMed PMID: 32740598]
Bartel-Friedrich S. Congenital Auricular Malformations: Description of Anomalies and Syndromes. Facial plastic surgery : FPS. 2015 Dec:31(6):567-80. doi: 10.1055/s-0035-1568139. Epub 2015 Dec 14 [PubMed PMID: 26667631]
Chang CC, Steinbacher DM. Treacher collins syndrome. Seminars in plastic surgery. 2012 May:26(2):83-90. doi: 10.1055/s-0032-1320066. Epub [PubMed PMID: 23633935]