Introduction
Dermatofibrosarcoma protuberans (DFSP) is an uncommon soft tissue sarcoma primarily found on the trunk and proximal extremities that typically appears as a slowly progressing, firm, violet-red, or blue plaque. Because DFSP is a slow-growing tumor, the diagnosis is often delayed for months to years. Most cases of DFSP are associated with a t(17;22) (q22;q13) translocation, generating the fusion protein COL1A1-PDGFB.
Extensive local infiltration, extending well beyond the visible lesion, characterizes DSFP, with distant metastasis being rare unless fibrosarcomatous transformation occurs. Surgical excision with clear margins is the preferred treatment, utilizing techniques like Mohs micrographic surgery or wide local excision.Radiation therapy can decrease local recurrence, particularly in cases where wide margins are impractical. More recently, tyrosine kinase inhibitors have shown promising response rates in patients with advanced or metastatic disease.[1] Patients require vigilant monitoring for local recurrence following curative treatment.
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
Ninety percent of patients with DFSP, including those who develop the fibrosarcomatous variant (DFSP-FS), have the t(17;22)(q22;q13) translocation.[2] An alternative translocation involving the platelet-derived growth factor subunit B (PDGFB) gene on chromosome 22 may be present in patients without the t(17;22)(q22;q13) translocation. In certain instances, the progression from DFSP to DFSP-FS is associated with genomic gain affecting the PDGFB/COL1A1 fusion gene, although this occurrence is variable.
Furthermore, researchers have noted the occurrence of microsatellite instability, characterized by alterations in the number of repeated deoxyribonucleic acid (DNA) bases within microsatellites or short, repetitive DNA sequences compared to the inherited state. Subsequently, these instances of microsatellite instability are accompanied by the acquisition of tumor protein p53 (TP53) mutations, particularly in cases displaying features of high-grade sarcoma
Epidemiology
DFSP is a rare tumor, occurring at a rate of 0.8 to 4.5 cases per million persons annually and represents 1% to 6% of all soft tissue sarcomas and 18% of cutaneous soft tissue sarcomas—affecting both genders equally. Some studies report a slight predominance in med. While most commonly diagnosed in adults between the third and fifth decades, DFSP can manifest across all age groups. Children account for 6% of all cases of DFSP. This tumor may exhibit accelerated growth during pregnancy.[3] DFSP-FS accounts for 5% to 15% of all DFSPs, and the Bednar or pigmented variant represents fewer than 5% of all DFSP cases. DFSP, including the pigmented variant, is more common in Black individuals than White.[4]
Pathophysiology
The t(17;22)(q22;q13) translocation results in the formation of supernumerary ring chromosomes from chromosome 22 and contains low-level amplified sequences from 17q22-qter and 22q10-q13.1.[4][5] Less commonly, a linear derivative of chromosome 22 may also be implicated.[6] The ring chromosomes and the translocated linear derivative of chromosome 22 harbor a fusion gene where PDGFB merges with the collagen type 1A1 (COL1A1) gene. The usually suppressed PDGFB now becomes activated by the COL1A1 promoter. This genetic rearrangement induces PDGFB upregulation, resulting in excessive platelet-derived growth factor (PDGF) production, continuous activation of the tyrosine kinase PDGFRB receptor, cellular proliferation, and tumor development.[7]
Histopathology
DFSP is poorly circumscribed and usually involves the dermis and subcutis, although rare cases can be limited to the dermis (see Image. Dermatofibrosarcoma Protruberans, Low-Power Field). Due to the possibility of distant metastasis and aggressive local invasion, DFSP is considered an intermediate tumor between a benign dermatofibroma and a frank fibrosarcoma. Transformation to a high-grade sarcoma is extremely rare. The overlying epidermis does not show any atypical histological features.[8]
The spindle-shaped tumor cells of DFSP are arranged in a storiform or woven pattern, parallel to the epidermal surface, and have little pleomorphism and scant cytoplasm (see Image. Hematoxylin and Eosin Stain Dermatofibrosarcoma Protuberans). The cells are surrounded by collagenous stroma, sometimes associated with hyaline or myxoid changes. The characteristic honeycomb appearance results from irregular tentacle-like projections infiltrating the underlying subcutaneous tissue, traversing the septa and fat, leading to fat entrapment. DFSP typically extends into subcutaneous fat but seldom involves fascia, muscle, or bone unless recurrent or long-standing. Necrosis is uncommon. While mitoses are present, no significant mitotic activity is present, and atypical mitoses are rare. A mitotic count that reaches 10 mitoses per 10 high power field (HPF) and tumor size correlates with metastatic spread. About one-fifth of all DFSPs undergo fibrosarcomatous transformation, which appears as an expansive tumor with fascicular and herringbone patterns and atypical cytological features.[9][10]
History and Physical
DFSP typically manifests as an asymptomatic, skin-colored to red-brown firm plaque, eventually progressing to multiple raised nodules with a violaceous to red-brown hue (see Image. Dermatofibrosarcoma Protuberans). The pigmented variant often presents with irregular brown pigmentation, and the atrophic variant appears as a violaceous plaque resembling morphea or scar tissue. Lesions exhibit slow growth over months to years, occasionally resembling keloids or dermatofibromas, leading to misdiagnosis, particularly in the early stages. As the tumor progresses, it may reach several centimeters in diameter, potentially accompanied by telangiectasia in the surrounding or overlying skin. The tumor is typically fixed to the dermis but can freely move in relation to underlying structures. Later in the disease, the tumor may become fixed to underlying structures. Rarely, DFSP can arise from preexisting scars or tattoos. As lesions enlarge, some may ulcerate and become painful. Nearly 50% of lesions are on the trunk, followed by 35% on the extremities, and 15% on the head and neck.[11]
Evaluation
Healthcare professionals should consider DFSP in individuals presenting with a slow-growing cutaneous nodule. Dermoscopy is the initial assessment tool, offering suggestive but not conclusive evidence. A core needle or excisional biopsy provides a definitive diagnosis. Patients should undergo a comprehensive history and physical examination, including an assessment of lymph nodes and a complete skin examination. Those with a prior treatment history may undergo a fine needle aspiration.
Typically, examining a hematoxylin and eosin-stained specimen by light microscopy can diagnose DFSP. In some cases, clinicians may find distinguishing DFSP from other neoplasms like dermatofibroma, fibrosarcoma, leiomyosarcoma, undifferentiated or unclassified soft tissue sarcoma, or atypical fibroxanthoma challenging. All cases of suspected DFSP should undergo confirmatory immunohistochemistry testing. DFSP stains positive for CD34 in 80% to 100% of cases (see Image. Cellular Dermatofibroma). Hyaluronate and vimentin are typically also positive. DFSP stains negative for factor XIIIa, smooth muscle actin, desmin, S100 proteins, and keratins.
Molecular testing using reverse transcription polymerase chain reaction (RT-PCR) or fluorescence in situ hybridization (FISH) to detect the t(17;22)(q22;q13) translocation is available. Both have a similar specificity of 100%, but FISH is more sensitive than RT-PCR for detecting the PDGFB/COL1A1 transcript at 90% versus 72%, respectively. Molecular testing is necessary when the diagnosis is unclear or to predict the likelihood of response to treatment with a tyrosine kinase inhibitor.
Because most cases of DFSP are superficial, clinicians can assess the extent of tumor and lymph node involvement by physical examination. Imaging is not a routine part of the diagnostic process. Occasionally, imaging may be helpful for large or recurrent tumors if the clinician suspects bone invasion or needs to define the extent of the disease. Magnetic resonance imaging is the imaging modality of choice.[12][13][14][15] Computed tomography (CT) is only helpful if underlying bone involvement or lung metastases are suspected. The lung is the most common site of metastasis via hematogenous spread, and regional lymph nodes are rarely involved. Due to the unlikely occurrence of lymphatic and hematogenous dissemination, an extensive staging workup is not generally necessary. Some authors suggest obtaining a radiograph or CT scan of the lungs before treatment, though the National Comprehensive Cancer Network does not make specific recommendations regarding a staging evaluation.[16][17]
Treatment / Management
The initial treatment for localized DFSP is surgical resection with negative margins.[18] The size and location of the tumor determine the best surgical approach. Regional lymph node dissection is unnecessary, given the low risk of metastasis. The risk of inadequate initial resection is high due to the delay in diagnosis and the tumor's ability to invade surrounding tissues. Nearly 50% of patients will experience local recurrence with simple excision. Recurrent tumors are more likely to invade fascia, muscle, or bone and result in distant metastases.[19][20] For these reasons, pathological negative margins are necessary.
Wide Local Excision
Wide local excision (WLE) is an option for resection. The resection margins are essential in determining the risk of local recurrence. Tentacle-like tumor projections can extend beyond 3 cm of the primary tumor. Results from one study reveal the local recurrence in patients with a resection margin less than 3 cm is 47% compared to 7% in patients with margins of 3 to 5 cm.[20][21][22] The NCCN recommends margins of 2 to 4 cm with clear pathologic margins when clinically feasible.(B2)
Mohs Micrographic Surgery
Mohs micrographic surgery (MMS) involves the progressive horizontal slicing of tissue during resection, coupled with immediate microscopic evaluation via frozen section analysis. Microscopic evaluation includes prompt immunostaining for CD34 until achieving a clear margin. Real-time microscopic examination of margins reduces the likelihood of positive margins. Some studies propose that MMS may result in lower local recurrence rates than WLE for DFSP. Notably, DFSP treated with WLE exhibits a recurrence rate of approximately 7.3%, contrasting with the 1% recurrence rate observed with MMS treatment.[23][24] However, randomized trials and comprehensive long-term data are required to confirm these findings. A significant benefit of MMS is the smaller required lateral margins, resulting in more minor wounds and less complex reconstruction. MMS is a good option for cosmetically sensitive areas, where achieving narrow margins is preferable. (B2)
Radiation Therapy
DFSP is radiosensitive tumor. Radiation therapy is rarely used alone in the treatment of DFSP.
Current NCCN guidelines recommend the use of adjuvant radiation in the following settings:
- Positive margins after surgical resection
- Negative margins, with the closest margin being less than 1 cm in patients who did not undergo MMS
- For recurrent or metastatic disease when surgical excision is not feasible [25]
The doses of radiation range from 50 to 70 Gy and typically extend 3 to 5 cm beyond the surgical margin when feasible.[26]
Molecularly Targeted Therapy
Imatinib inhibits the PDGF receptor and other receptor tyrosine kinases such as c-KIT. Imatinib has the United States Food and Drug Administration (FDA) approval for unresectable, recurrent, or metastatic DFSP.[27] Imatinib competitively inhibits adenosine triphosphate (ATP) binding to the PDGFB receptor, slowing down kinase activity, limiting tumor growth, and promoting apoptosis. Patients with the t(17;22)(q22;q13) translocation show a more significant response to imatinib, and thus, screening for this translocation should be performed before initiating therapy. Further studies are necessary to see if patients without the translocation will benefit from imatinib.(A1)
Adverse effects associated with imatinib include gastrointestinal upset, edema, fatigue, anemia, and rash. Most patients with DFSP with translocation respond favorably to imatinib therapy, with studies suggesting a response rate of approximately 65%. The duration of therapy varies. Some sources recommend 6 months of therapy, which may be extended.[28][29] Additional research is necessary to determine the potential integration of imatinib into the surgical approach for localized DFSP.
Posttreatment Surveillance
Patients should participate in regular self-examination, and the primary site should be examined every 6 months for 3 to 5 years. Lifelong examination annually follows. Radiographic surveillance, blood counts, and liver function testing are necessary only if symptoms dictate. Metastatic lesions are most likely to occur with recurrent lesions that have progressed for many years and when a fibrosarcomatous component is present. Some authors recommend limiting follow-up imaging examinations to those with recurrent DFSP or DFSP-FS.
Differential Diagnosis
The following list contains the differential diagnoses for DFSP:
- Cellular fibrous histiocytoma
- Solitary fibrous tumor
- Spindle cell lipoma
- Angiosarcoma
- Peripheral nerve sheath tumors
- Spindle cell melanoma
- Angiomyxoma
- Myxoid sarcoma
- Synovial sarcoma
- Sarcomatoid carcinoma
- Cutaneous melanoma
- Dermatofibroma
- Dermatologic metastatic carcinoma
- Epidermal inclusion cyst
- Keloid
- Morphea [17]
Prognosis
The prognosis for DFSP is generally favorable, with a 10-year survival rate of 99.1%. However, patients with metastatic disease typically survive approximately 2 years post-diagnosis. The overall risk for the development of metastatic disease is 5%, including 1% with regional lymph node metastasis and 4% with distant metastasis. While metastasis remains uncommon, local recurrence poses a more prevalent concern for morbidity. Features that increase the risk of a poor prognosis are patients older than 50, DFSP-FS, a high mitotic index, and increased cellularity.[30] A wider surgical excision improves the overall prognosis.
Complications
Complications of DFSP primarily stem from the extent of the surgical defect and issues linked to local recurrence. Common complications include wound-related problems such as infections, challenges in covering the defect necessitating skin grafting or complex plastic surgery procedures, contractures, keloid formation, and suboptimal cosmesis. Employing MMS aids in mitigating these complications. Death is an uncommon but possible complication in patients who develop metastatic disease.
Deterrence and Patient Education
DFSP is a relatively uncommon soft tissue neoplasm that is locally aggressive but rarely metastasizes. Healthcare professionals should emphasize the significance of regular skin examinations and seeking medical evaluation for any suspicious lesions, which can facilitate early detection and improve outcomes. Patients should be educated about the appearance of DFSP, emphasizing that the tumor may present as a small, asymptomatic papule or nonindurated patch that gradually enlarges over months to years. The slow progression often delays diagnosis, emphasizing the importance of promptly reporting any changes in skin lesions to healthcare professionals.
Before undergoing treatment, patients should understand that surgical excision is the preferred course of action and should be aware of potential risks, such as unsatisfactory cosmetic results. Additionally, patients should be informed about the low likelihood of metastasis but the high potential for local recurrence after resection. Clinicians should encourage patients to adhere to scheduled follow-up appointments for continuous monitoring, promoting timely intervention and optimal management of DFSP. Through comprehensive patient education and proactive deterrence strategies, clinicians can empower individuals to take an active role in their skin health and facilitate early detection and treatment of DFSP.
Enhancing Healthcare Team Outcomes
DFSP is an uncommon soft tissue sarcoma primarily found on the trunk and proximal extremities. This tumor typically presents as a slowly progressing, firm plaque with a violet-red, brown, or blue hue. Diagnosis requires a high index of suspicion as these lesions are slow-growing and may go unrecognized for long periods. Healthcare professionals caring for patients with DFSP must be proficient in recognizing the characteristics of DFSP and accurately interpreting diagnostic tests such as biopsies and imaging studies.
Characterized by extensive local infiltration, DFSP may extend well beyond visible lesions, although distant metastasis is rare unless fibrosarcomatous transformation occurs. The preferred treatment is surgical excision with clear margins, utilizing techniques like WLE and MMS. Adjunctive radiation therapy can reduce local recurrence, particularly in cases where wide margins are impractical. Emerging treatments such as tyrosine kinase inhibitors show promising response rates in patients with advanced or metastatic DFSP. Developing a comprehensive care plan tailored to each patient's unique needs is essential. This strategy should involve a combination of surgical excision, radiation therapy, or pharmacotherapy, depending on the stage, location, and extent of the DFSP lesion. Patients require vigilant monitoring for local recurrence posttreatment. Awareness of DFSP's varied presentation and treatment modalities is crucial for timely diagnosis and management.
To enhance patient-centered care, outcomes, patient safety, and team performance related to DFSP, physicians, advanced practice practitioners, nurses, pharmacists, and other healthcare professionals must collaborate effectively using a multidisciplinary approach. Effective communication is critical for coordinating care and ensuring seamless transitions between different phases of treatment. Team members should regularly communicate updates on patient status, treatment plans, and any changes in the care plan. Clear and concise communication helps prevent errors, reduces the risk of complications, and promotes patient safety. By leveraging their collective skills, implementing effective strategies, communicating openly, coordinating care seamlessly, and fostering a culture of collaboration, healthcare professionals can enhance patient-centered care, improve outcomes, ensure patient safety, and optimize team performance in the management of DFSP.
Media
(Click Image to Enlarge)
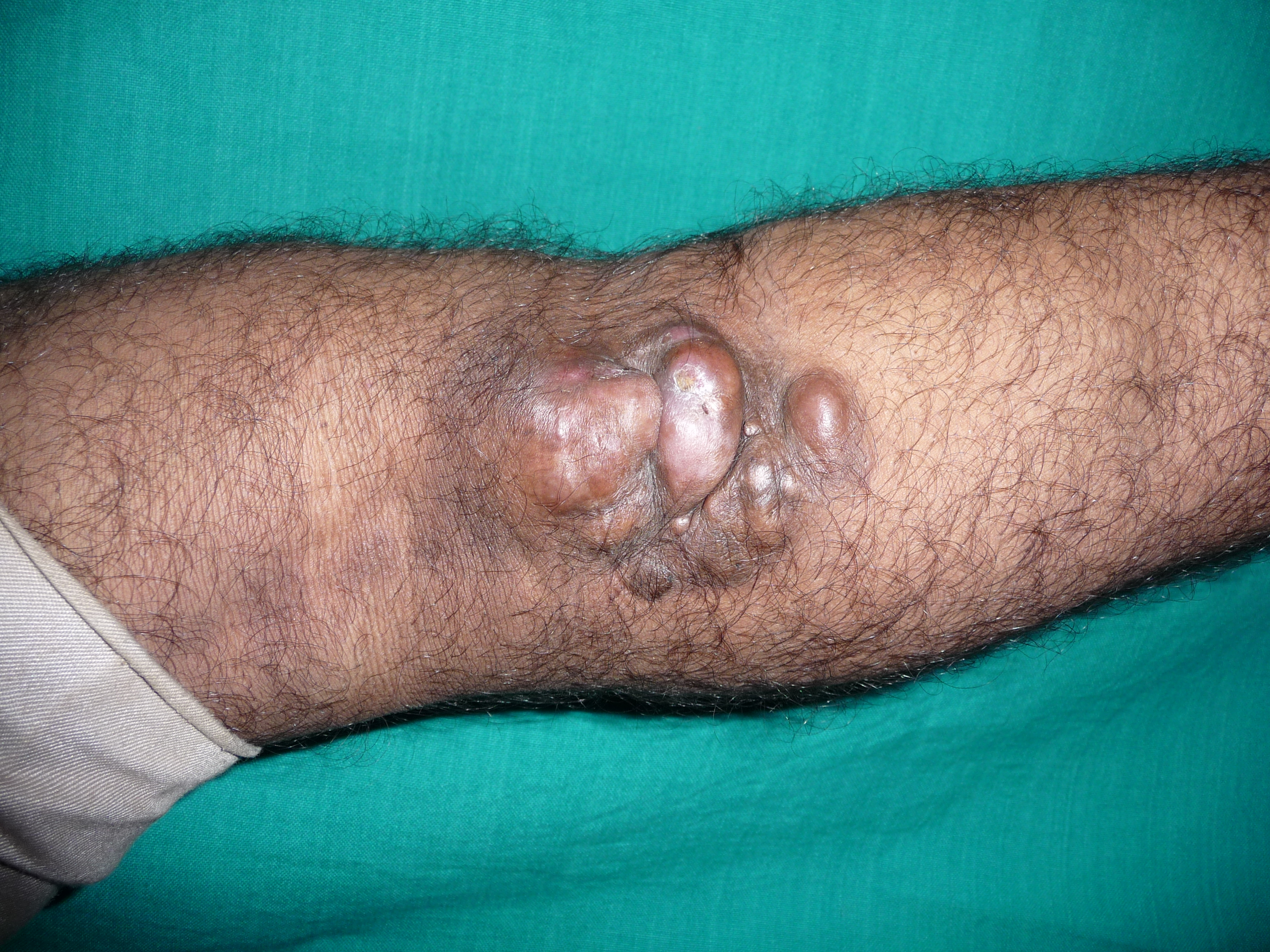
Dermatofibrosarcoma Protuberans
Contributed by S Verma, MBBS, DVD, FRCP, FAAD
(Click Image to Enlarge)
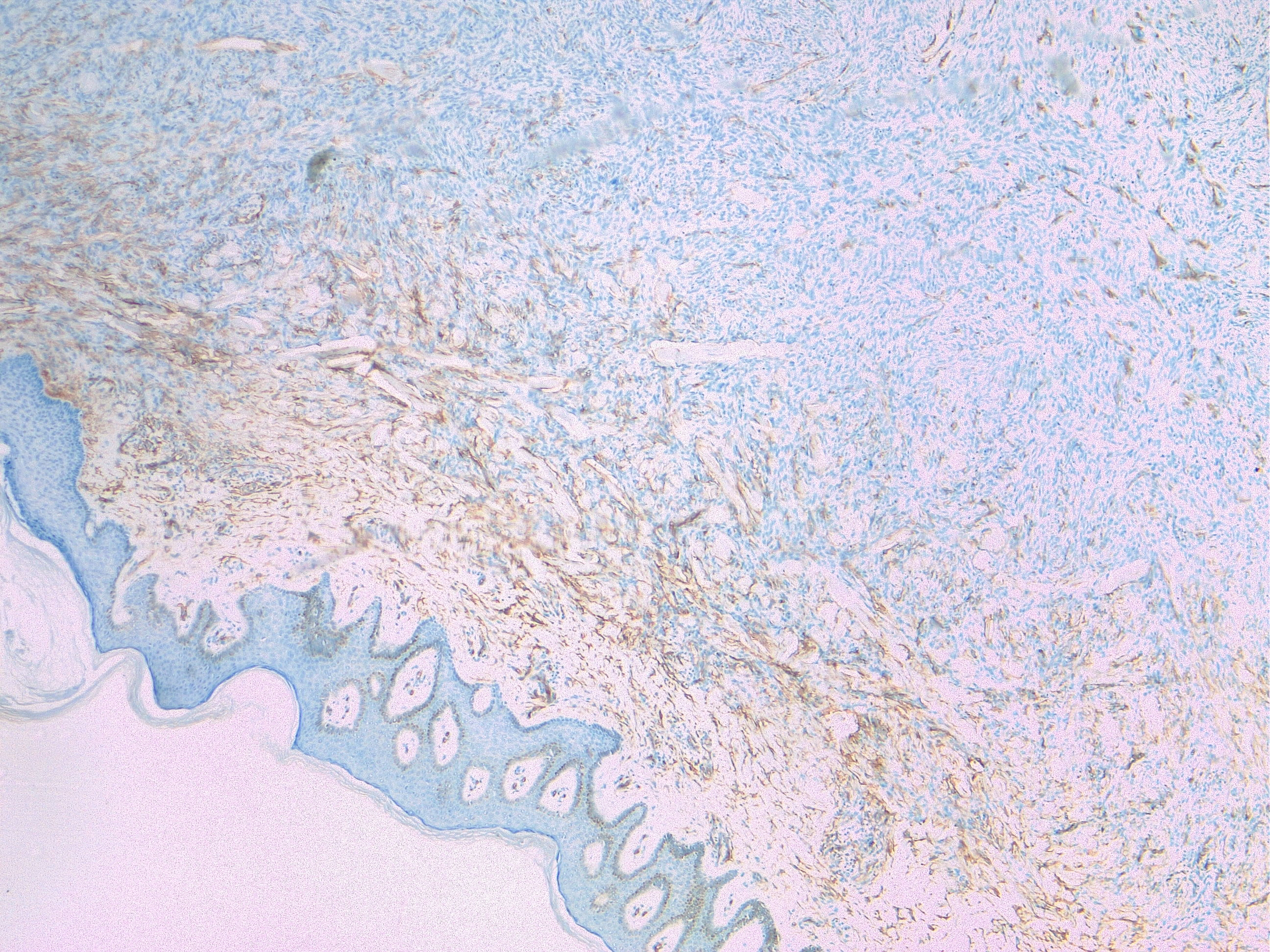
Cellular Dermatofibroma. The image depicts a dermatofibroma confirmed by the absence or limited CD34 staining in contrast to dermatofibrosarcoma protuberans that stains diffusely positive.
Contributed by F Farci, MD
(Click Image to Enlarge)
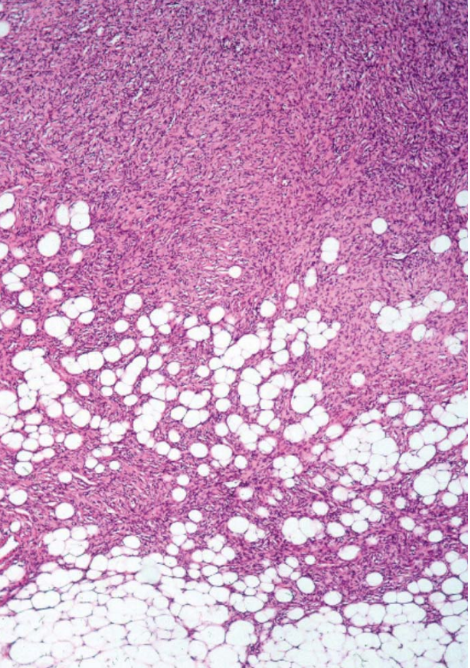
Dermatofibrosarcoma Protruberans, Low-Power Field
Contributed by D Anand, MD
(Click Image to Enlarge)
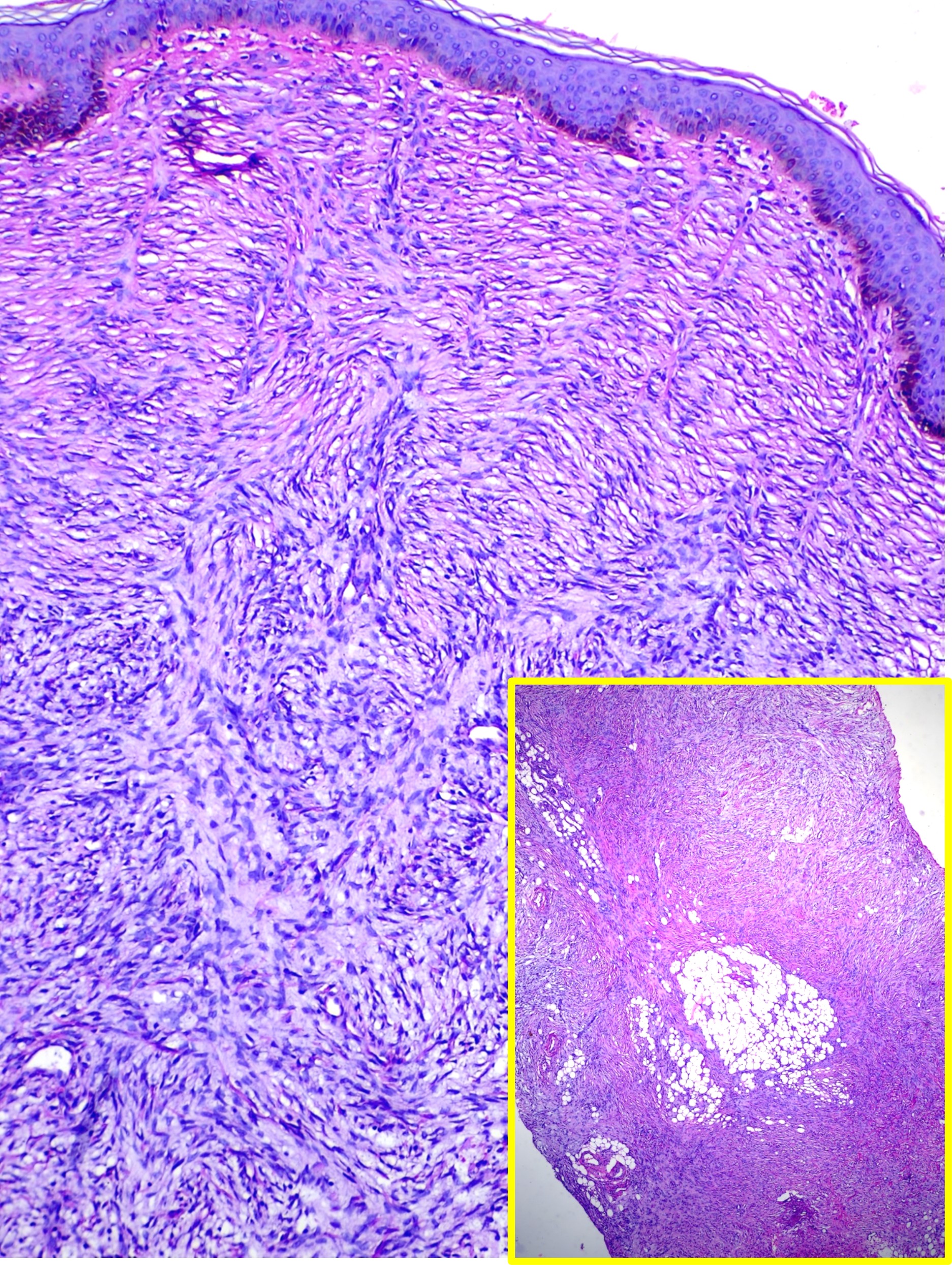
Dermatofibrosarcoma Protuberans. This image shows a spindle cell neoplasm with a storiform pattern involving the entire thickness of the dermis, findings consistent with dermatofibrosarcoma protuberans (hematoxylin and eosin stain, main high-power magnification, inset low-power magnification).
Contributed by M Abdel-Halim Ibrahim, MD
References
Rutkowski P, Van Glabbeke M, Rankin CJ, Ruka W, Rubin BP, Debiec-Rychter M, Lazar A, Gelderblom H, Sciot R, Lopez-Terrada D, Hohenberger P, van Oosterom AT, Schuetze SM, European Organisation for Research and Treatment of Cancer Soft Tissue/Bone Sarcoma Group, Southwest Oncology Group. Imatinib mesylate in advanced dermatofibrosarcoma protuberans: pooled analysis of two phase II clinical trials. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2010 Apr 1:28(10):1772-9. doi: 10.1200/JCO.2009.25.7899. Epub 2010 Mar 1 [PubMed PMID: 20194851]
Level 1 (high-level) evidenceCriscito MC, Martires KJ, Stein JA. Prognostic Factors, Treatment, and Survival in Dermatofibrosarcoma Protuberans. JAMA dermatology. 2016 Dec 1:152(12):1365-1371. doi: 10.1001/jamadermatol.2016.1886. Epub [PubMed PMID: 27262160]
Trofymenko O, Bordeaux JS, Zeitouni NC. Survival in patients with primary dermatofibrosarcoma protuberans: National Cancer Database analysis. Journal of the American Academy of Dermatology. 2018 Jun:78(6):1125-1134. doi: 10.1016/j.jaad.2017.11.030. Epub 2017 Nov 23 [PubMed PMID: 29175214]
Martin L, Piette F, Blanc P, Mortier L, Avril MF, Delaunay MM, Dréno B, Granel F, Mantoux F, Aubin F, Sassolas B, Adamski H, Dalac S, Pauwels C, Dompmartin A, Lok C, Estève E, Guillot B, French Group for Cutaneous Oncology. Clinical variants of the preprotuberant stage of dermatofibrosarcoma protuberans. The British journal of dermatology. 2005 Nov:153(5):932-6 [PubMed PMID: 16225602]
Takahira T, Oda Y, Tamiya S, Higaki K, Yamamoto H, Kobayashi C, Izumi T, Tateishi N, Iwamoto Y, Tsuneyoshi M. Detection of COL1A1-PDGFB fusion transcripts and PDGFB/PDGFRB mRNA expression in dermatofibrosarcoma protuberans. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc. 2007 Jun:20(6):668-75 [PubMed PMID: 17431412]
Simon MP, Pedeutour F, Sirvent N, Grosgeorge J, Minoletti F, Coindre JM, Terrier-Lacombe MJ, Mandahl N, Craver RD, Blin N, Sozzi G, Turc-Carel C, O'Brien KP, Kedra D, Fransson I, Guilbaud C, Dumanski JP. Deregulation of the platelet-derived growth factor B-chain gene via fusion with collagen gene COL1A1 in dermatofibrosarcoma protuberans and giant-cell fibroblastoma. Nature genetics. 1997 Jan:15(1):95-8 [PubMed PMID: 8988177]
Haycox CL, Odland PB, Olbricht SM, Piepkorn M. Immunohistochemical characterization of dermatofibrosarcoma protuberans with practical applications for diagnosis and treatment. Journal of the American Academy of Dermatology. 1997 Sep:37(3 Pt 1):438-44 [PubMed PMID: 9308560]
Allen A, Ahn C, Sangüeza OP. Dermatofibrosarcoma Protuberans. Dermatologic clinics. 2019 Oct:37(4):483-488. doi: 10.1016/j.det.2019.05.006. Epub [PubMed PMID: 31466588]
Larbcharoensub N, Kayankarnnavee J, Sanpaphant S, Kiranantawat K, Wirojtananugoon C, Sirikulchayanonta V. Clinicopathological features of dermatofibrosarcoma protuberans. Oncology letters. 2016 Jan:11(1):661-667 [PubMed PMID: 26870263]
Level 3 (low-level) evidenceThway K, Noujaim J, Jones RL, Fisher C. Dermatofibrosarcoma protuberans: pathology, genetics, and potential therapeutic strategies. Annals of diagnostic pathology. 2016 Dec:25():64-71. doi: 10.1016/j.anndiagpath.2016.09.013. Epub 2016 Sep 27 [PubMed PMID: 27806849]
Li Y, Wang C, Xiang B, Chen S, Li L, Ji Y. Clinical Features, Pathological Findings and Treatment of Recurrent Dermatofibrosarcoma Protuberans. Journal of Cancer. 2017:8(7):1319-1323. doi: 10.7150/jca.17988. Epub 2017 May 12 [PubMed PMID: 28607608]
PDQ Pediatric Treatment Editorial Board. Childhood Soft Tissue Sarcoma Treatment (PDQ®): Health Professional Version. PDQ Cancer Information Summaries. 2002:(): [PubMed PMID: 26389361]
Llombart B, Serra C, Requena C, Alsina M, Morgado-Carrasco D, Través V, Sanmartín O. Guidelines for Diagnosis and Treatment of Cutaneous Sarcomas: Dermatofibrosarcoma Protuberans. Actas dermo-sifiliograficas. 2018 Dec:109(10):868-877. doi: 10.1016/j.ad.2018.05.006. Epub 2018 Jul 4 [PubMed PMID: 30539729]
Bhatt MD, Nambudiri VE. Cutaneous Sarcomas. Hematology/oncology clinics of North America. 2019 Feb:33(1):87-101. doi: 10.1016/j.hoc.2018.08.007. Epub [PubMed PMID: 30497679]
Penel N, El Bedoui S, Robin YM, Decanter G. [Dermatofibrosarcoma: Management]. Bulletin du cancer. 2018 Nov:105(11):1094-1101. doi: 10.1016/j.bulcan.2018.08.008. Epub 2018 Oct 5 [PubMed PMID: 30297237]
Chappell AG, Doe SC, Worley B, Yoo SS, Gerami P, Alam M, Buck DW 2nd, Kim JYS, Wayne JD. Multidisciplinary surgical treatment approach for dermatofibrosarcoma protuberans: an update. Archives of dermatological research. 2021 Jul:313(5):367-372. doi: 10.1007/s00403-020-02124-8. Epub 2020 Aug 7 [PubMed PMID: 32770258]
Lyu A, Wang Q. Dermatofibrosarcoma protuberans: A clinical analysis. Oncology letters. 2018 Aug:16(2):1855-1862. doi: 10.3892/ol.2018.8802. Epub 2018 May 24 [PubMed PMID: 30008876]
Level 3 (low-level) evidenceMeguerditchian AN, Wang J, Lema B, Kraybill WG, Zeitouni NC, Kane JM 3rd. Wide excision or Mohs micrographic surgery for the treatment of primary dermatofibrosarcoma protuberans. American journal of clinical oncology. 2010 Jun:33(3):300-3. doi: 10.1097/COC.0b013e3181aaca87. Epub [PubMed PMID: 19858696]
Bowne WB, Antonescu CR, Leung DH, Katz SC, Hawkins WG, Woodruff JM, Brennan MF, Lewis JJ. Dermatofibrosarcoma protuberans: A clinicopathologic analysis of patients treated and followed at a single institution. Cancer. 2000 Jun 15:88(12):2711-20 [PubMed PMID: 10870053]
Khatri VP, Galante JM, Bold RJ, Schneider PD, Ramsamooj R, Goodnight JE Jr. Dermatofibrosarcoma protuberans: reappraisal of wide local excision and impact of inadequate initial treatment. Annals of surgical oncology. 2003 Nov:10(9):1118-22 [PubMed PMID: 14597453]
Fields RC, Hameed M, Qin LX, Moraco N, Jia X, Maki RG, Singer S, Brennan MF. Dermatofibrosarcoma protuberans (DFSP): predictors of recurrence and the use of systemic therapy. Annals of surgical oncology. 2011 Feb:18(2):328-36. doi: 10.1245/s10434-010-1316-5. Epub 2010 Sep 16 [PubMed PMID: 20844969]
Level 2 (mid-level) evidenceBehbahani R, Patenotre P, Capon N, Martinot-Duquennoy V, Kulik JF, Piette F, Pellerin P. [To a margin reduction in the dermatofibrosarcoma protuberans? Retrospective study of 34 cases]. Annales de chirurgie plastique et esthetique. 2005 Jun:50(3):179-85; discussion 186-8 [PubMed PMID: 15935539]
Level 2 (mid-level) evidenceVeronese F, Boggio P, Tiberio R, Gattoni M, Fava P, Caliendo V, Colombo E, Savoia P. Wide local excision vs. Mohs Tübingen technique in the treatment of dermatofibrosarcoma protuberans: a two-centre retrospective study and literature review. Journal of the European Academy of Dermatology and Venereology : JEADV. 2017 Dec:31(12):2069-2076. doi: 10.1111/jdv.14378. Epub 2017 Jul 3 [PubMed PMID: 28573714]
Level 2 (mid-level) evidenceLowe GC, Onajin O, Baum CL, Otley CC, Arpey CJ, Roenigk RK, Brewer JD. A Comparison of Mohs Micrographic Surgery and Wide Local Excision for Treatment of Dermatofibrosarcoma Protuberans With Long-Term Follow-up: The Mayo Clinic Experience. Dermatologic surgery : official publication for American Society for Dermatologic Surgery [et al.]. 2017 Jan:43(1):98-106. doi: 10.1097/DSS.0000000000000910. Epub [PubMed PMID: 27749444]
Bichakjian CK, Olencki T, Alam M, Andersen JS, Berg D, Bowen GM, Cheney RT, Daniels GA, Glass LF, Grekin RC, Grossman K, Ho AL, Lewis KD, Lydiatt DD, Morrison WH, Nehal KS, Nelson KC, Nghiem P, Perlis CS, Shaha AR, Thorstad WL, Tuli M, Urist MM, Wang TS, Werchniak AE, Wong SL, Zic JA, McMillian N, Hoffman K, Ho M. Dermatofibrosarcoma protuberans, version 1.2014. Journal of the National Comprehensive Cancer Network : JNCCN. 2014 Jun:12(6):863-8 [PubMed PMID: 24925197]
Dagan R, Morris CG, Zlotecki RA, Scarborough MT, Mendenhall WM. Radiotherapy in the treatment of dermatofibrosarcoma protuberans. American journal of clinical oncology. 2005 Dec:28(6):537-9 [PubMed PMID: 16317260]
Navarrete-Dechent C, Mori S, Barker CA, Dickson MA, Nehal KS. Imatinib Treatment for Locally Advanced or Metastatic Dermatofibrosarcoma Protuberans: A Systematic Review. JAMA dermatology. 2019 Mar 1:155(3):361-369. doi: 10.1001/jamadermatol.2018.4940. Epub [PubMed PMID: 30601909]
Level 1 (high-level) evidenceAmavi AK, Dossouvi T, Padaro E, Adabra K, Dosseh ED. [Management for locally advanced dermatofibrosarcoma protuberans in Togo]. Bulletin du cancer. 2018 Apr:105(4):333-334. doi: 10.1016/j.bulcan.2018.01.011. Epub 2018 Mar 2 [PubMed PMID: 29502795]
Yadav S, Verma N, Khurana N, Neogi S. Recurrent Dermatofibrosarcoma Protuberans with Pigmentation and Myoid Differentiation. Sultan Qaboos University medical journal. 2018 May:18(2):e228-e230. doi: 10.18295/squmj.2018.18.02.019. Epub 2018 Sep 9 [PubMed PMID: 30210857]
Asilian A, Honarjou N, Faghihi G, Saber M, Mozafarpoor S, Hafezi H. An experience of slow-Mohs micrographic surgery for the treatment of Dermatofibrosarcoma protuberans: A long-term cohort study. Journal of cosmetic dermatology. 2020 Oct:19(10):2701-2705. doi: 10.1111/jocd.13319. Epub 2020 Feb 10 [PubMed PMID: 32039548]