Introduction
Cystic fibrosis is a chronic genetic disorder that has affected children and adults since ancient times, primarily affecting the respiratory, digestive, and reproductive systems. Important chronic manifestations of cystic fibrosis include chronic lung infections, pancreatic insufficiency, sinusitis, and infertility. Cystic fibrosis is often associated with shortened lifespans, and the most common cause of death is end-stage lung disease.[1][2][3][4]
Historically misunderstood, cystic fibrosis gained recognition in folklore through the "salty skin" symptom, which symbolized an incurable condition. In medieval Europe, these children were believed to be cursed by witches and doomed to die. The curse that became folklore pronounced, "Woe to the child who tastes salty from a kiss on the brow, for he is cursed and soon will die." Salty skin was a sign of an impending illness without cause or cure. Until relatively modern times, cystic fibrosis was poorly understood.
Modern understanding began in 1949 when scientists identified the autosomal recessive inheritance pattern of cystic fibrosis and linked the disease to a defect in the CFTR protein, a chloride channel crucial for regulating salt and water balance in the body. Quinton postulated that sweat ducts in these patients were impermeable to chloride. Further studies led to the hypothesis that the faulty chloride channel must be situated in the apical membranes of the lung surface or glandular epithelium to explain the respiratory and systemic organ failure associated with cystic fibrosis.[5]
Symptoms may appear in infancy, childhood, or adulthood, with newborn screening and genetic testing aiding early diagnosis. The disease is caused by over 2,000 possible mutations in the CFTR gene, each disrupting chloride ion transport, leading to thick mucus accumulation, organ dysfunction, and electrolyte imbalances.
Advancements in treatment include CFTR modulators like ivacaftor and lumacaftor, which address underlying protein defects, and supportive therapies targeting nutrition, airway clearance, and infection control. Despite progress, cystic fibrosis remains life-limiting, with lung transplantation serving as an option for end-stage disease. Interprofessional care focusing on improving lung function, managing complications, and optimizing quality of life is essential for those with cystic fibrosis.
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
Cystic fibrosis is caused by a genetic mutation in a gene on chromosome 7 that codes for the cystic fibrosis transmembrane conductance regulator (CFTR) protein. This protein functions as a transmembrane cAMP-activated chloride channel.[6] In clinical disease, both copies of the gene are mutated. More than 2000 different mutations in the CFTR gene have been identified that can cause cystic fibrosis. These mutations are divided into the following 5 classes:
Epidemiology
Researchers now know that cystic fibrosis is an autosomal recessive disorder of exocrine gland function most commonly affecting persons of Northern European descent at a rate of 1 in 3500 and Black patients at a rate of 1 in 15,000.[8] However, for unknown reasons, the prevalence is much lower in Asia (1:30,000).[9] The most common mutation is [delta]F508 in exon 11, found in 70% of White patients in the United States with cystic fibrosis and two-thirds of all cases worldwide. This mutation is a class II mutation of abnormal folding of CFTR, leading to premature destruction within the Golgi apparatus. The [delta]F508 mutation commonly results in exocrine pancreatic insufficiency and a higher likelihood of meconium ileus.[10]
Pathophysiology
CFTR disease-causing mutations have been categorized into the following dysfunctional variants, classes I through V:[11]
- Class I dysfunction results from nonsense, frameshift, or splice-site mutations that terminate the mRNA sequence prematurely. Translational failure results in the complete absence of CFTR expression. Class I mutations account for approximately 2% to 5% of cystic fibrosis cases.
- Class II dysfunction results in abnormal or dysfunctional posttranslational processing of CFTR that prohibits normal intracellular protein transit. CFTR cannot be moved to the correct cellular location.
- Class III dysfunction is characterized by diminished protein activity in response to intracellular signaling. The result is a fully-formed but nonfunctional protein channel in the cellular membrane.
- Class IV dysfunction results in a protein that correctly localizes to the cell surface. However, the rate of chloride ion flow and the duration of channel activation after stimulation is decreased from normal.
- Class V dysfunction is the net decreased concentration of CFTR channels in the cellular membrane due to rapid degradation by cellular processes. Class V dysfunction includes mutations that alter the stability of mRNA and those mutations that alter the stability of the mature CFTR protein.[11]
Systemic Pathophysiologic Manifestations of Cystic Fibrosis
All mutations result in decreased chloride secretion and, consequently, increased sodium resorption into the cellular space. The increased sodium reabsorption leads to increased water resorption and manifests as thicker mucus secretions on epithelial linings and more viscous secretions from exocrine tissues. Thickened mucus secretions in nearly every affected organ system result in mucous plugging with obstruction pathologies.[12] The most commonly affected organs include the sinuses, lungs, pancreas, biliary and hepatic systems, intestines, and sweat glands.
Sinus disease
Sinus disease occurs when secretion viscosity increases, obstructing the sinus ostia. Additional processes often coexist. These coexisting processes include ciliary dysfunction, increased inflammatory mediators, and increased pathogenic bacterial colonization, including Pseudomonas aeruginosa. The result of this syndrome is impaired sinus secretion clearance. Subsequently, chronic sinusitis occurs, and secondary structural damage may occur.
Pulmonary disease
Lung disease occurs as a spectrum of obstruction from thickened mucous secretion. Notably, the lungs of a cystic fibrosis patient are normal in utero, at birth, and after birth. Disease results as a cascade effect following infection and the subsequent inflammatory process. Mucus plugging in the bronchioles results in a clinical picture of obstructive lung disease. As a result of obstruction, an environment optimal for bacterial growth is created within the airways. Bronchiectasis and further thick purulent sputum production occur.
Part of the inflammatory reaction includes the production of the neutrophil interleukin 8 from epithelial cells, which functions as a secretagogue, increasing mucous secretion, thereby creating a positive feedback loop of mucous secretion with the persistence of inflammation, infection, and structural damage. The result of this cascade is obstruction of the airways, which results in lung ventilation failure. Poorly managed pulmonary manifestations are the primary cause of death in patients with cystic fibrosis.[13]
Pancreatic and hepatic disease
Pancreatic manifestations of cystic fibrosis are primarily due to pancreatic ductular obstruction by thickened secretions.[14] Coinciding with the passage of gastric contents into the proximal duodenum, the pancreatic exocrine glands are triggered to excrete pancreatic enzymes into the luminal space of the small intestines. However, increased viscosity of excretions and obstruction of the pancreatic ductules inhibits this process. The net pH of the secretions lessens due to decreased sodium bicarbonate composition, leading to a reduced neutralization of the acidic chyme. The lower pH chyme effectively degrades what pancreatic enzymes reach the intestinal lumen. As a result, intestinal chyme is not enzymatically processed, resulting in pathognomonic greasy stools, colicky abdominal pain, and malabsorption of nutrients from ingested foods. In particular, fat-soluble vitamins A, D, E, and K are notably deficient.
Furthermore, autodigestion of the pancreas may occur as these enzymes target the pancreatic tissues, resulting in pancreatitis. In severe, chronic cases, this can lead to endocrine pancreatic failure when the islets of Langerhans begin to be digested by trapped pancreatic enzymes. The lasting impacts of this spectrum of disease mimic type 1 diabetes.
Increased secretion viscosity does not spare the biliary and hepatic systems. The biliary ductules may be plugged with secretions. Obstructive cirrhosis and posthepatic hyperbilirubinemia can occur. Secondary esophageal varices, splenomegaly, and hypersplenism may occur due to increased hepatic portal vein pressures. Gallbladder disease is more likely to occur as a spectrum of this manifestation of cystic fibrosis; up to 15% of patients with cystic fibrosis have gallstones.
Intestinal disease
Intestinal involvement is typically seen in children with meconium ileus at birth and intestinal obstruction later in life. The cause of meconium ileus is multifactorial but likely secondary to increased fluid absorption due to the faulty CFTR channel. Dehydration of the intestinal contents leads to constipation and an abnormal change in luminal contents secondary to pancreatic insufficiency. Chronic mechanical obstruction leads to inflammation and eventual scarring and stricture formation. Intestinal strictures and scarring may lead to further intestinal obstruction by fecal impaction or intussusception later in life.
Sweat gland manifestations
Sweat glands offer an interesting contrast to all other tissues containing CFTR channels in that the flow of chloride is reversed. Typically, sweat glands move chloride from the extracellular space into the intracellular space. Thus, sodium and water are reabsorbed from the sweat gland tissues into the body. However, failure of the chloride channel to reabsorb chloride leads to a loss of sodium onto the skin surface and a subsequent fluid loss, causing the pathognomonic salty skin of cystic fibrosis. In prolonged or warm environments or more severe cases, sodium and fluid loss may lead to hyponatremic dehydration.
In addition to acting as a chloride transport protein, other interactions of CFTR have been postulated. In the apical plasma membrane, CFTR is part of a multiprotein assembly where 3 of its amino acids, threonine, arginine, and leucine, act to root the protein to a region known as PDZ-type receptors. These PDZ regions have also been observed to occur in multiple intracellular signaling proteins associated with the plasma membrane. This also roots CFTR closely to other transporters, ion channels, receptors, kinases, phosphatases, signaling molecules, and cytoskeletal elements. These interactions between CFTR and its binding proteins are critically involved in regulating CFTR-mediated transepithelial ion transport in vitro and in vivo. These close associations seem to allow CFTR to play an essential role in epithelial cells beyond its function as an ion channel.
While not yet fully understood, modulation of CFTR protein in animal studies proved that inflammatory responses, maturational processing, nonchloride ion transport, and intracellular signaling are related to its function. These other interacting proteins are potential modifiers of the cystic fibrosis phenotype and may help explain the substantial differences in clinical severity between similar genotypic patients with cystic fibrosis.[15][16][17]
History and Physical
Newborns with cystic fibrosis may be identified through screening without clinical symptoms.[18] A subgroup of newborns, however, can present with gastrointestinal symptoms such as meconium ileus or prolonged neonatal jaundice, difficulty to thrive,[19] or an early lung infection.[20] Infants and children with cystic fibrosis may present with failure to thrive and poor weight gain, anemia, undescended testicles in boys, recurrent sinopulmonary infections, and a distal intestinal obstructive syndrome with or without pancreatic insufficiency. The median age of diagnosis is 6 to 8 months, although individuals may not exhibit clinical signs and symptoms until later.
Adults with cystic fibrosis often present with exacerbations of 1 or more symptoms in multiple organ systems [21], including:
- Lung manifestations: chronic bronchitis, abnormal pulmonary function tests, bronchiectasis, atypical asthma, allergic bronchopulmonary aspergillosis, and colonization with P aeruginosa [22]
- Sinus manifestations: chronic rhinosinusitis, chronic postnasal drip, nasal polyposis, and panopacification of the paranasal sinuses [23]
- Pancreatic manifestations: pancreatic insufficiency, recurrent pancreatitis, and early-onset diabetes with a mean age of 20 years [24]
- Hepatobiliary manifestations: focal biliary cirrhosis, cholelithiasis, periportal fibrosis, liver cirrhosis, portal hypertension, and variceal bleeding [25]
- Musculoskeletal manifestations: kyphoscoliosis, osteopenia or osteoporosis, and arthropathy [26]
- Hematologic manifestations: iron-deficiency anemia or anemia of chronic disease leading to splenomegaly [27]
- Nephrogenic manifestations: nephrolithiasis, nephrocalcinosis, hyperoxaluria, and hypocitraturia
- Dermatologic manifestations: “salty sweat,” digital clubbing, cyanosis, and additional dermatologic conditions due to malabsorption, including palm wrinkling following a brief immersion in the water test,[28] acrodermatitis enteropathica due to zinc deficiency, and scaly dermatitis associated with fatty acid deficiency [29]
- Infertility issues: absent vas deferens in males and thickened cervical mucus in females [30]
- Electrolyte abnormalities: low serum sodium, low chloride, and potassium levels with possible metabolic alkalosis [31]
- Cardiovascular disease: myocardial infarction, left-sided heart failure, and atrial fibrillation [32][21][32]
Evaluation
In the United States, newborns are screened for cystic fibrosis as part of a standard newborn screening panel. Additionally, some cases of cystic fibrosis are discovered during prenatal ultrasonography, which may demonstrate meconium peritonitis, bowel dilation, or absent gallbladder. Such findings often lead to prenatal cystic fibrosis carrier screening.[33][34][35][36] Criteria of at least 1 clinical symptom and 1 laboratory finding must be met to diagnose cystic fibrosis.
Clinical Symptoms of Cystic Fibrosis Criteria
The following symptoms and positive screenings are clinical criteria for cystic fibrosis:
- Sibling with cystic fibrosis
- Positive newborn screening
- Clinical symptoms consistent with cystic fibrosis in 1 or more organ systems:
- Chronic sinopulmonary disease
- Gastrointestinal or nutritional abnormalities
- Salt loss syndromes
- Obstructive azoospermia
Laboratory Criteria for CFTR Dysfunction
The following laboratory findings meet the criteria for cystic fibrosis diagnosis:
- Elevated sweat chloride greater than 60 mEq/L
- Two disease-causing CFTR mutations
- Abnormal nasal potential difference
The diagnostic approach for cystic fibrosis begins with a sweat chloride test. Furthermore, if a sweat chloride test is normal but the patient remains symptomatic, a repeat sweat chloride test is indicated. If the initial sweat test is abnormal, DNA testing is indicated. If ≤1 CFTR mutations are found, expanded DNA analysis is indicated. However, the finding of 2 cystic fibrosis-related mutations confirms the diagnosis.
The test for immunoreactive trypsinogen (IRT), a pancreatic enzyme, increases sensitivity and specificity in screening newborns with meconium ileus for cystic fibrosis. IRT monitoring can correlate with the severity of cystic fibrosis; when it drops below detectable levels, pancreatic enzyme replacement initiation may be indicated.
Additional diagnostics may be indicated depending on the presenting symptoms. A chest radiograph may help identify hyperinflation, bronchiectasis, abscesses, or atelectasis (see Image. Periapical Chest Radiograph, Cystic Fibrosis). Sinus radiography may demonstrate panopacification of the paranasal sinuses. Abdominal radiology may be helpful in neonates who present with meconium ileus. Bronchoalveolar lavage typically reveals many neutrophils, and microbiology is commonly positive for Haemophilus influenza, Staphylococcus aureus, P aeruginosa, Burkholderia cepacia, Escherichia coli, or Klebsiella pneumoniae.
Pulmonary function testing is a significant tool for evaluating and monitoring disease state and progression in cystic fibrosis. Spirometry is the most commonly used pulmonary function test and measures the volume of air exhaled during a forceful and complete exhalation after a maximal inhalation. The total exhaled volume, known as the forced vital capacity (FVC), the volume exhaled in the first second, known as the forced expiratory volume in 1 second (FEV1), and their ratio (FEV1/FVC) are the most critical variables reported. These values allow for interpretation of the status of the lung ventilation function. These values are compared to an expected normal for age, height, and gender to generate an expected normal value. The measured value is then calculated as a percent of normal, where normal equals 100%. A normal or high FEV1 and a low FVC may indicate restrictive lung disease. A low FEV1 with a high FVC indicates obstructive lung disease with airway trapping. Cystic fibrosis can be expected to show air-trapping patterns with low FEV1 values proportional to the severity of the disease.[37]
Acute Exacerbation of Cystic Fibrosis
A pulmonary exacerbation is the worsening of lung function due to an infection. Often, this is characterized by shortness of breath, fatigue, productive cough, and fever. Pulmonary function testing measures will usually worsen from baseline during an acute cystic fibrosis exacerbation.[38] Pulmonary disease is the most common cause of mortality in cystic fibrosis. As such, having a low threshold for diagnosis and intervention in pulmonary illness exacerbations is vital. Any acute illness should prompt admission to a hospital facility familiar with cystic fibrosis management.
Treatment / Management
Cystic fibrosis is a systemic illness that has broad implications for both quality and quantity of life when poorly controlled. Therefore, management should focus on optimizing function to avoid acute illness events. This approach should target maintaining lung function by aggressively controlling respiratory infection and clearing airways of mucus, optimizing nutritional status with pancreatic enzyme supplements and multivitamins, and treating any other health complications that may arise. This is best achieved through a team approach of specialists experienced in managing cystic fibrosis.[39][40][41][42](A1)
Pulmonary Management
Pulmonary illnesses responsible for acute cystic fibrosis exacerbation should be managed with 2 primary goals: treating the infection and improving oxygenation. P aeruginosa is the typical infectious etiologic agent, and antibiotic therapy should have spectrum coverage against this pathogen. However, a sputum culture should be obtained, and a sensitivity profile should be obtained for the pathogens present.
A recent Cochrane database study found that inhaled antibiotics alone or with oral antibiotics were better than no treatment for P aeruginosa infections. At the same time, new evidence does not support the superiority of intravenous antibiotics in eradicating pseudomonal infections.[43] A major clinical investigation revealed that combining inhaled antibiotics with oral ciprofloxacin is equally effective as the intravenous administration of ceftazidime plus tobramycin. However, oral antibiotics are more cost-effective.[44](A1)
Inhaled bronchodilators, including albuterol and ipratropium bromide, should support ventilation and oxygenation. Furthermore, agents, such as inhaled dornase alfa or inhaled hypertonic saline (7%), are prescribed to promote airway secretion clearance in conjunction with chest physiotherapy.[45] However, the efficacy of hypertonic saline administration during hospitalization for acute cystic fibrosis exacerbation lacks substantial evidence-based support. A randomized controlled study of 132 adult cystic fibrosis patients randomized to 7% saline or control evaluated the impact of hypertonic saline during respiratory exacerbations and found no significant benefit in reducing the length of stay.[46] (A1)
The efficacy of inhaled corticosteroids for cystic fibrosis is unclear. A Cochrane review deemed existing evidence insufficient and low in quality, not confirming benefits for cystic fibrosis patients. Long-term effects on lung inflammation and survival improvement remain unproven. Thus, prescribing inhaled corticosteroids for cystic fibrosis should follow asthma treatment guidelines reserved for patients with recurrent wheezing unresponsive to bronchodilators alone. The withdrawal or step-down of inhaled steroids is safe and usually based on symptom control.[47](A1)
Optimizing breathing with nasal cannula oxygen is advisable when suitable. However, long-term oxygen supplementation for advanced cystic fibrosis-related lung disease lacks substantial evidence. Benefits include enhanced exercise endurance, sleep quality, and regular school or work attendance.[48] Nighttime oxygen levels increased during REM and non-REM sleep with low-flow oxygen. Participants using nocturnal oxygen spent less time in REM sleep and fell asleep more quickly.(A1)
Noninvasive ventilation (NIV), including bilevel positive airway pressure (BPAP) ventilation, may be necessary to overcome air trapping. NIV offers multiple advantages in treating respiratory disorders associated with cystic fibrosis. Beyond enhancing airway clearance for patients struggling with sputum expectoration, NIV can also improve nighttime gas exchange more effectively than oxygen therapy alone when combined with it. Nevertheless, there is a lack of evidence supporting the long-term efficacy of NIV in cystic fibrosis patients.[49] Invasive mechanical ventilation is an option, and although it has been associated with increased mortality, recent studies showed that it can serve as a bridge for transplant with decreased mortality over time.[50](A1)
Supportive Therapy
Chronic, supportive therapy for patients with cystic fibrosis includes regular physical exercise. Studies suggest that exercise programs for cystic fibrosis patients can boost their exercise capacity compared to no exercise regimen. However, these interventions show no measurable effects on pulmonary function or overall life quality.[51](A1)
Although supportive therapy includes supplementation with pancreatic enzymes, fat-soluble vitamins, mucolytics, bronchodilators, antibiotics, and anti-inflammatory agents, no substantial evidence exists to recommend it. Likewise, insufficient evidence has been found to recommend respiratory muscle training for cystic fibrosis.[52](A1)
CFTR Modulator Therapies
A new class of medications known as CFTR modulator therapies is designed to correct the dysfunction by improving the production, intracellular processing, or function of the CFTR protein caused by the mutated gene. Each medication targets a specific dysfunction caused by a specific gene mutation.
Ivacaftor treats class III dysfunctions, where the primary aberration is a G551D mutation (c.1652G>A).[53] This CFTR activator acts by binding the defective CFTR protein at the cell surface and opening the chloride channel, thus restoring the proper function of the protein. Ivacaftor was the first medication to directly impact the protein channel rather than treat the effects of cystic fibrosis. Dosing for patients older than 6 years is 150 mg by mouth every 12 hours.[54] Younger patients should receive weight-based dosing; those weighing less than 14 kg receive 50 mg orally every 12 hours, and those weighing more than 14 kg should receive 75 mg orally every 12 hours.
Lumacaftor is a chaperone molecule designed to move the defective CFTR from the intracellular organelles for processing onto the cell surface. As such, lumacaftor is effective in [delta]F508 homozygous mutation genotypes.[55] This medicine has no clinical benefit when administered in isolation. However, when combined as lumacaftor/ivacaftor, the 92-week PROGRESS study showed improvement in pulmonary function testing by approximately 30% to 40%.[55] This medicine combination was subsequently approved for use in patients with cystic fibrosis older than 6. Dosing is 2 tablets containing lumacaftor 200 mg/ivacaftor 125 mg taken orally every 12 hours.
Another medication combination currently in clinical trials is tezacaftor/ivacaftor. Tezacaftor, as a medicine, is very similar to lumacaftor in that it is a CFTR chaperone that improves intracellular processing and trafficking.[56] Likewise, its clinical efficacy is only seen when combined with ivacaftor. This medicine demonstrates a good safety profile, and preliminary EVOLVE and EXPAND trials indicate fewer side effect profiles than lumacaftor/ivacaftor.[56]
Lung Transplant
Despite significant advancements in medical therapies for cystic fibrosis, the disease process continues to advance, and the lungs will ultimately fail prematurely from the disease burden without surgical intervention. Lung transplant is the treatment of choice for end-stage lung disease.[57] The timing of the transplant is multifactorial.(B3)
The International Society of Heart and Lung Transplantation published a list of conditions[58] to be used when considering transplant referral and takes into consideration the 5-year predicted survival of <50%, the FEV1 that has fallen to 30% of predicted values, rapidly falling FEV1 despite optimal therapy, a 6-minute walk distance of less than 400 meters, the development of pulmonary hypertension in the absence of a hypoxemic exacerbation, clinical decline characterized by increasing frequency of exacerbations including acute respiratory failure requiring noninvasive ventilation, a pattern of poor clinical recovery from successive exacerbations, worsening nutritional status despite supplementation, pneumothorax, or life-threatening hemoptysis despite bronchial artery embolization.(B3)
Virtually all lung transplants for cystic fibrosis will need the replacement of both lungs because a native, diseased lung acts as a source of infected secretions that would threaten the transplanted lung and possibly induce respiratory failure. Notably, transplantation is not a cure for cystic fibrosis, but it confers a prolongation of life and offers significant symptomatic relief.
Diet and Exercise
Individuals with cystic fibrosis are encouraged to consume a high-fat diet with supplemental fat-soluble vitamins to compensate for malabsorption. Additionally, patients living with cystic fibrosis are encouraged to consume a high-calorie diet to maintain a healthy weight and combat chronic inflammation and frequent infections that are commonly encountered. According to the Cystic Fibrosis Foundation, women should consume 2500 to 3000 calories daily, while men should consume 3000 to 3700 calories daily.[59](A1)
Those living in hot climates or participating in activities that cause sweating are encouraged to consume additional sodium. Oral feedings are preferred; if the intake does not meet metabolic demand as determined by continued decreases in body mass index, enteral (tube) feedings should be considered. These are typically in the form of gastric or jejunal tube feedings. Multiple control studies of enteral nutrition in patients with cystic fibrosis have shown benefits in improved or neutral lung function following exacerbations of illness that directly correlate with body mass index. With that noted, however, no randomized studies of enteral nutrition have been performed in patients with cystic fibrosis to date. Parenteral nutrition may be considered only when oral or enteral nutrition is not meeting metabolic needs. Parenteral nutrition has been linked to increased risk for sepsis events and should be used sparingly. Regular exercise is encouraged in patients with cystic fibrosis to maintain and support lung function.
Differential Diagnosis
Differential diagnoses of cystic fibrosis include the following:
- Asthma
- Bronchiolitis
- Bronchiectasis
- Celiac disease
- Nutritional considerations in failure to thrive
- Pediatric aspergillosis
- Primary ciliary dyskinesia
- Sinusitis
Prognosis
Patients with cystic fibrosis are estimated to live until about the fourth decade of life before requiring lung transplantation. Lung transplantation confers a median survival of 8.5 years.
Deterrence and Patient Education
Cystic fibrosis is a genetic disorder that cannot currently be prevented, but early detection through newborn screening and genetic counseling can significantly improve outcomes. Educating patients and families about cystic fibrosis's inheritance patterns, including its autosomal recessive nature, helps identify carriers and inform reproductive choices. Prenatal and carrier screening can guide at-risk families in understanding the likelihood of cystic fibrosis in their offspring, allowing for early intervention or alternative family planning options.
Ongoing patient education is crucial to managing cystic fibrosis effectively. Patients and caregivers should be informed about the importance of routine airway clearance, proper use of medications, and maintaining a high-calorie diet supplemented with pancreatic enzymes and fat-soluble vitamins. They should also recognize early signs of pulmonary exacerbations, eg, increased cough, fatigue, or shortness of breath, to ensure timely medical attention. Promoting adherence to multidisciplinary care, including regular follow-ups with specialized cystic fibrosis teams, is essential for managing complications and optimizing quality of life. Additionally, fostering an understanding of advances in cystic fibrosis therapies (eg, CFTR modulators) empowers patients to make informed decisions about their care.
Enhancing Healthcare Team Outcomes
Managing cystic fibrosis effectively requires an interprofessional approach that leverages the skills and expertise of various healthcare professionals to optimize patient-centered care, improve outcomes, ensure patient safety, and enhance team performance. Physicians, particularly pulmonologists, infectious disease specialists, and gastroenterologists, play a critical role in diagnosing and managing cystic fibrosis's systemic manifestations. They coordinate the overall treatment strategy, focusing on maintaining lung function, preventing respiratory infections, and addressing complications such as pancreatic insufficiency. Advanced practitioners and nurses monitor patients closely, track disease progression, and facilitate communication between specialists to ensure a cohesive treatment plan. Nurses also educate patients and caregivers, empowering them to adhere to therapies and recognize early signs of exacerbations.
Pharmacists are integral to medication management, ensuring the safety and efficacy of treatments by checking for drug-drug interactions and educating patients about adherence and potential side effects. Since lung infections are the leading cause of mortality in cystic fibrosis, the team must work collaboratively to prevent and treat infections aggressively, often with tailored antibiotic regimens. For some patients, lung transplantation may be necessary to extend survival, particularly in children with advanced disease. Care coordination and interprofessional communication are essential in these cases to optimize outcomes. Regular team meetings, shared decision-making, and the use of electronic health records ensure seamless collaboration. This team-based strategy not only improves the quality and quantity of life for patients but also provides comprehensive support to manage cystic fibrosis's complex and systemic challenges.
Media
(Click Image to Enlarge)
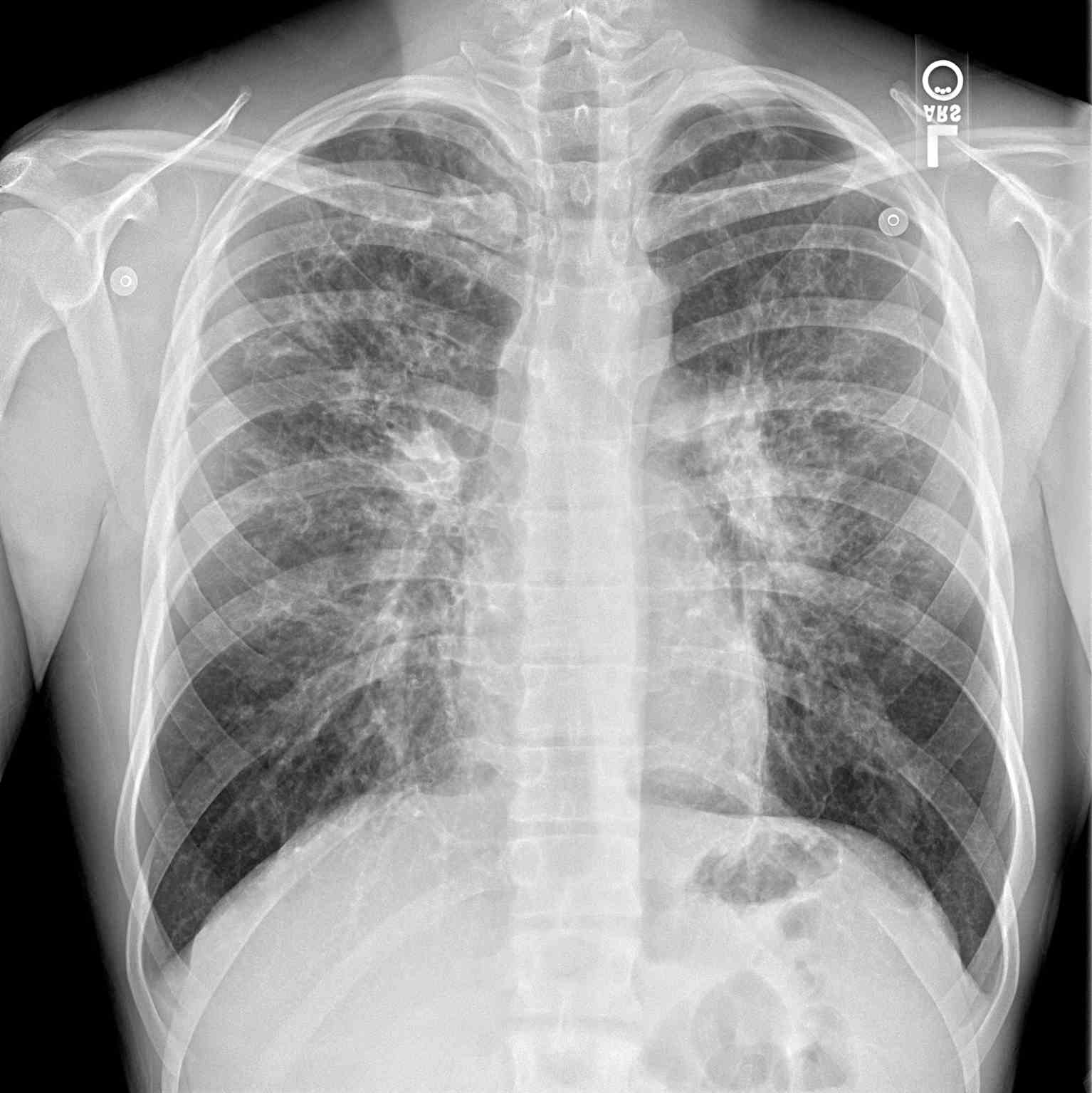
Periapical Chest Radiograph, Cystic Fibrosis
Contributed by S Dulebohn, MD
References
Lyamin AV, Ismatullin DD, Zhestkov AV, Kondratenko OV. [The laboratory diagnostic in patients with mucoviscidosis: A review.]. Klinicheskaia laboratornaia diagnostika. 2018:63(5):315-320. doi: 10.18821/0869-2084-2018-63-5-315-320. Epub [PubMed PMID: 30689329]
Poncin W, Lebecque P. [Lung clearance index in cystic fibrosis]. Revue des maladies respiratoires. 2019 Mar:36(3):377-395. doi: 10.1016/j.rmr.2018.03.007. Epub 2019 Jan 25 [PubMed PMID: 30686561]
Bianconi I, D'Arcangelo S, Esposito A, Benedet M, Piffer E, Dinnella G, Gualdi P, Schinella M, Baldo E, Donati C, Jousson O. Persistence and Microevolution of Pseudomonas aeruginosa in the Cystic Fibrosis Lung: A Single-Patient Longitudinal Genomic Study. Frontiers in microbiology. 2018:9():3242. doi: 10.3389/fmicb.2018.03242. Epub 2019 Jan 11 [PubMed PMID: 30692969]
Eschenhagen P, Schwarz C. [Patients with cystic fibrosis become adults : Treatment hopes and disappointments]. Der Internist. 2019 Jan:60(1):98-108. doi: 10.1007/s00108-018-0536-9. Epub [PubMed PMID: 30627755]
Davis PB. Cystic fibrosis since 1938. American journal of respiratory and critical care medicine. 2006 Mar 1:173(5):475-82 [PubMed PMID: 16126935]
Welsh MJ, Smith AE. Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell. 1993 Jul 2:73(7):1251-4 [PubMed PMID: 7686820]
Awatade NT, Wong SL, Hewson CK, Fawcett LK, Kicic A, Jaffe A, Waters SA. Human Primary Epithelial Cell Models: Promising Tools in the Era of Cystic Fibrosis Personalized Medicine. Frontiers in pharmacology. 2018:9():1429. doi: 10.3389/fphar.2018.01429. Epub 2018 Dec 7 [PubMed PMID: 30581387]
Hamosh A, FitzSimmons SC, Macek M Jr, Knowles MR, Rosenstein BJ, Cutting GR. Comparison of the clinical manifestations of cystic fibrosis in black and white patients. The Journal of pediatrics. 1998 Feb:132(2):255-9 [PubMed PMID: 9506637]
Level 2 (mid-level) evidenceGuo J, Garratt A, Hill A. Worldwide rates of diagnosis and effective treatment for cystic fibrosis. Journal of cystic fibrosis : official journal of the European Cystic Fibrosis Society. 2022 May:21(3):456-462. doi: 10.1016/j.jcf.2022.01.009. Epub 2022 Feb 4 [PubMed PMID: 35125294]
Pallin M. Cystic fibrosis vigilance in Arab countries: The role of genetic epidemiology. Respirology (Carlton, Vic.). 2019 Feb:24(2):93-94. doi: 10.1111/resp.13461. Epub 2018 Dec 13 [PubMed PMID: 30548951]
Fanen P, Wohlhuter-Haddad A, Hinzpeter A. Genetics of cystic fibrosis: CFTR mutation classifications toward genotype-based CF therapies. The international journal of biochemistry & cell biology. 2014 Jul:52():94-102. doi: 10.1016/j.biocel.2014.02.023. Epub 2014 Mar 12 [PubMed PMID: 24631642]
Pilewski JM, Frizzell RA. Role of CFTR in airway disease. Physiological reviews. 1999 Jan:79(1 Suppl):S215-55 [PubMed PMID: 9922383]
Panos RJ, Mortenson RL, Niccoli SA, King TE Jr. Clinical deterioration in patients with idiopathic pulmonary fibrosis: causes and assessment. The American journal of medicine. 1990 Apr:88(4):396-404 [PubMed PMID: 2183601]
Kelly T, Buxbaum J. Gastrointestinal Manifestations of Cystic Fibrosis. Digestive diseases and sciences. 2015 Jul:60(7):1903-13. doi: 10.1007/s10620-015-3546-7. Epub 2015 Feb 4 [PubMed PMID: 25648641]
Bush A, Floto RA. Pathophysiology, causes and genetics of paediatric and adult bronchiectasis. Respirology (Carlton, Vic.). 2019 Nov:24(11):1053-1062. doi: 10.1111/resp.13509. Epub 2019 Feb 25 [PubMed PMID: 30801930]
García-Clemente M, Enríquez-Rodríguez AI, Iscar-Urrutia M, Escobar-Mallada B, Arias-Guillén M, López-González FJ, Madrid-Carbajal C, Pérez-Martínez L, Gonzalez-Budiño T. Severe asthma and bronchiectasis. The Journal of asthma : official journal of the Association for the Care of Asthma. 2020 May:57(5):505-509. doi: 10.1080/02770903.2019.1579832. Epub 2019 Feb 20 [PubMed PMID: 30784336]
Blasi F, Elborn JS, Palange P. Adults with cystic fibrosis and pulmonologists: new training needed to recruit future specialists. The European respiratory journal. 2019 Jan:53(1):. pii: 1802209. doi: 10.1183/13993003.02209-2018. Epub 2019 Jan 17 [PubMed PMID: 30655450]
Cystic Fibrosis Foundation, Borowitz D, Robinson KA, Rosenfeld M, Davis SD, Sabadosa KA, Spear SL, Michel SH, Parad RB, White TB, Farrell PM, Marshall BC, Accurso FJ. Cystic Fibrosis Foundation evidence-based guidelines for management of infants with cystic fibrosis. The Journal of pediatrics. 2009 Dec:155(6 Suppl):S73-93. doi: 10.1016/j.jpeds.2009.09.001. Epub [PubMed PMID: 19914445]
Level 1 (high-level) evidenceAccurso FJ, Sontag MK, Wagener JS. Complications associated with symptomatic diagnosis in infants with cystic fibrosis. The Journal of pediatrics. 2005 Sep:147(3 Suppl):S37-41 [PubMed PMID: 16202780]
Pillarisetti N, Williamson E, Linnane B, Skoric B, Robertson CF, Robinson P, Massie J, Hall GL, Sly P, Stick S, Ranganathan S, Australian Respiratory Early Surveillance Team for Cystic Fibrosis (AREST CF). Infection, inflammation, and lung function decline in infants with cystic fibrosis. American journal of respiratory and critical care medicine. 2011 Jul 1:184(1):75-81. doi: 10.1164/rccm.201011-1892OC. Epub 2011 Apr 14 [PubMed PMID: 21493738]
Penketh AR, Wise A, Mearns MB, Hodson ME, Batten JC. Cystic fibrosis in adolescents and adults. Thorax. 1987 Jul:42(7):526-32 [PubMed PMID: 3438896]
Bayfield KJ, Douglas TA, Rosenow T, Davies JC, Elborn SJ, Mall M, Paproki A, Ratjen F, Sly PD, Smyth AR, Stick S, Wainwright CE, Robinson PD. Time to get serious about the detection and monitoring of early lung disease in cystic fibrosis. Thorax. 2021 Dec:76(12):1255-1265. doi: 10.1136/thoraxjnl-2020-216085. Epub 2021 Apr 29 [PubMed PMID: 33927017]
Wucherpfennig L, Wuennemann F, Eichinger M, Schmitt N, Seitz A, Baumann I, Stahl M, Graeber SY, Chung J, Schenk JP, Alrajab A, Kauczor HU, Mall MA, Sommerburg O, Wielpütz MO. Longitudinal Magnetic Resonance Imaging Detects Onset and Progression of Chronic Rhinosinusitis from Infancy to School Age in Cystic Fibrosis. Annals of the American Thoracic Society. 2023 May:20(5):687-697. doi: 10.1513/AnnalsATS.202209-763OC. Epub [PubMed PMID: 36548543]
De Boeck K, Weren M, Proesmans M, Kerem E. Pancreatitis among patients with cystic fibrosis: correlation with pancreatic status and genotype. Pediatrics. 2005 Apr:115(4):e463-9 [PubMed PMID: 15772171]
Sellers ZM, Assis DN, Paranjape SM, Sathe M, Bodewes F, Bowen M, Cipolli M, Debray D, Green N, Hughan KS, Hunt WR, Leey J, Ling SC, Morelli G, Peckham D, Pettit RS, Philbrick A, Stoll J, Vavrina K, Allen S, Goodwin T, Hempstead SE, Narkewicz MR. Cystic fibrosis screening, evaluation, and management of hepatobiliary disease consensus recommendations. Hepatology (Baltimore, Md.). 2024 May 1:79(5):1220-1238. doi: 10.1097/HEP.0000000000000646. Epub 2023 Oct 26 [PubMed PMID: 37934656]
Level 3 (low-level) evidenceAris RM, Merkel PA, Bachrach LK, Borowitz DS, Boyle MP, Elkin SL, Guise TA, Hardin DS, Haworth CS, Holick MF, Joseph PM, O'Brien K, Tullis E, Watts NB, White TB. Guide to bone health and disease in cystic fibrosis. The Journal of clinical endocrinology and metabolism. 2005 Mar:90(3):1888-96 [PubMed PMID: 15613415]
von Drygalski A, Biller J. Anemia in cystic fibrosis: incidence, mechanisms, and association with pulmonary function and vitamin deficiency. Nutrition in clinical practice : official publication of the American Society for Parenteral and Enteral Nutrition. 2008 Oct-Nov:23(5):557-63. doi: 10.1177/0884533608323426. Epub [PubMed PMID: 18849562]
Alexopoulos A, Chouliaras G, Kakourou T, Dakoutrou M, Nasi L, Petrocheilou A, Siahanidou S, Kanaka-Gantenbein C, Chrousos G, Loukou I, Michos A. Aquagenic wrinkling of the palms after brief immersion to water test as a screening tool for cystic fibrosis diagnosis. Journal of the European Academy of Dermatology and Venereology : JEADV. 2021 Aug:35(8):1717-1724. doi: 10.1111/jdv.17312. Epub 2021 Jun 9 [PubMed PMID: 33914973]
Crone J, Huber WD, Eichler I, Granditsch G. Acrodermatitis enteropathica-like eruption as the presenting sign of cystic fibrosis--case report and review of the literature. European journal of pediatrics. 2002 Sep:161(9):475-8 [PubMed PMID: 12200605]
Level 3 (low-level) evidenceDodge JA. Male fertility in cystic fibrosis. Lancet (London, England). 1995 Sep 2:346(8975):587-8 [PubMed PMID: 7650999]
Berg P, Jeppesen M, Leipziger J. Cystic fibrosis in the kidney: new lessons from impaired renal HCO3- excretion. Current opinion in nephrology and hypertension. 2021 Jul 1:30(4):437-443. doi: 10.1097/MNH.0000000000000725. Epub [PubMed PMID: 34027905]
Level 3 (low-level) evidenceFrost F, Nazareth D, Fauchier L, Wat D, Shelley J, Austin P, Walshaw MJ, Lip GYH. Prevalence, risk factors and outcomes of cardiac disease in cystic fibrosis: a multinational retrospective cohort study. The European respiratory journal. 2023 Oct:62(4):. doi: 10.1183/13993003.00174-2023. Epub 2023 Oct 26 [PubMed PMID: 37474158]
Level 2 (mid-level) evidenceRajapaksha IG, Angus PW, Herath CB. Current therapies and novel approaches for biliary diseases. World journal of gastrointestinal pathophysiology. 2019 Jan 5:10(1):1-10. doi: 10.4291/wjgp.v10.i1.1. Epub [PubMed PMID: 30622832]
Mandalia A, Wamsteker EJ, DiMagno MJ. Recent advances in understanding and managing acute pancreatitis. F1000Research. 2018:7():. pii: F1000 Faculty Rev-959. doi: 10.12688/f1000research.14244.2. Epub 2018 Jun 28 [PubMed PMID: 30026919]
Level 3 (low-level) evidenceFenker DE, McDaniel CT, Panmanee W, Panos RJ, Sorscher EJ, Sabusap C, Clancy JP, Hassett DJ. A Comparison between Two Pathophysiologically Different yet Microbiologically Similar Lung Diseases: Cystic Fibrosis and Chronic Obstructive Pulmonary Disease. International journal of respiratory and pulmonary medicine. 2018:5(2):. pii: 098. doi: 10.23937/2378-3516/1410098. Epub 2018 Nov 29 [PubMed PMID: 30627668]
Kiedrowski MR, Bomberger JM. Viral-Bacterial Co-infections in the Cystic Fibrosis Respiratory Tract. Frontiers in immunology. 2018:9():3067. doi: 10.3389/fimmu.2018.03067. Epub 2018 Dec 20 [PubMed PMID: 30619379]
Stern M, Picard C, Roux A. [Lung transplantation]. La Revue du praticien. 2018 Feb:68(2):189-194 [PubMed PMID: 30801151]
Goss CH. Acute Pulmonary Exacerbations in Cystic Fibrosis. Seminars in respiratory and critical care medicine. 2019 Dec:40(6):792-803. doi: 10.1055/s-0039-1697975. Epub 2019 Oct 28 [PubMed PMID: 31659730]
Declercq D, Van Meerhaeghe S, Marchand S, Van Braeckel E, van Daele S, De Baets F, Van Biervliet S. The nutritional status in CF: Being certain about the uncertainties. Clinical nutrition ESPEN. 2019 Feb:29():15-21. doi: 10.1016/j.clnesp.2018.10.009. Epub 2018 Nov 7 [PubMed PMID: 30661680]
Radovanovic D, Santus P, Blasi F, Sotgiu G, D'Arcangelo F, Simonetta E, Contarini M, Franceschi E, Goeminne PC, Chalmers JD, Aliberti S. A comprehensive approach to lung function in bronchiectasis. Respiratory medicine. 2018 Dec:145():120-129. doi: 10.1016/j.rmed.2018.10.031. Epub 2018 Nov 2 [PubMed PMID: 30509700]
Lapp V, Chase SK. How Do Youth with Cystic Fibrosis Perceive Their Readiness to Transition to Adult Healthcare Compared to Their Caregivers' Views? Journal of pediatric nursing. 2018 Nov-Dec:43():104-110. doi: 10.1016/j.pedn.2018.09.012. Epub 2018 Sep 28 [PubMed PMID: 30473151]
Pawlaczyk-Kamieńska T, Borysewicz-Lewicka M, Śniatała R, Batura-Gabryel H, Cofta S. Dental and periodontal manifestations in patients with cystic fibrosis - A systematic review. Journal of cystic fibrosis : official journal of the European Cystic Fibrosis Society. 2019 Nov:18(6):762-771. doi: 10.1016/j.jcf.2018.11.007. Epub 2018 Nov 23 [PubMed PMID: 30473190]
Level 1 (high-level) evidenceLangton Hewer SC, Smith S, Rowbotham NJ, Yule A, Smyth AR. Antibiotic strategies for eradicating Pseudomonas aeruginosa in people with cystic fibrosis. The Cochrane database of systematic reviews. 2023 Jun 2:6(6):CD004197. doi: 10.1002/14651858.CD004197.pub6. Epub 2023 Jun 2 [PubMed PMID: 37268599]
Level 1 (high-level) evidenceHewer SCL, Smyth AR, Brown M, Jones AP, Hickey H, Kenna D, Ashby D, Thompson A, Williamson PR, TORPEDO-CF study group. Intravenous versus oral antibiotics for eradication of Pseudomonas aeruginosa in cystic fibrosis (TORPEDO-CF): a randomised controlled trial. The Lancet. Respiratory medicine. 2020 Oct:8(10):975-986. doi: 10.1016/S2213-2600(20)30331-3. Epub [PubMed PMID: 33007285]
Level 1 (high-level) evidenceTerlizzi V, Masi E, Francalanci M, Taccetti G, Innocenti D. Hypertonic saline in people with cystic fibrosis: review of comparative studies and clinical practice. Italian journal of pediatrics. 2021 Aug 6:47(1):168. doi: 10.1186/s13052-021-01117-1. Epub 2021 Aug 6 [PubMed PMID: 34362426]
Level 2 (mid-level) evidenceDentice RL, Elkins MR, Middleton PG, Bishop JR, Wark PA, Dorahy DJ, Harmer CJ, Hu H, Bye PT. A randomised trial of hypertonic saline during hospitalisation for exacerbation of cystic fibrosis. Thorax. 2016 Feb:71(2):141-7. doi: 10.1136/thoraxjnl-2014-206716. Epub [PubMed PMID: 26769016]
Level 1 (high-level) evidenceBalfour-Lynn IM, Welch K, Smith S. Inhaled corticosteroids for cystic fibrosis. The Cochrane database of systematic reviews. 2019 Jul 4:7(7):CD001915. doi: 10.1002/14651858.CD001915.pub6. Epub 2019 Jul 4 [PubMed PMID: 31271656]
Level 1 (high-level) evidenceElphick HE, Mallory G. Oxygen therapy for cystic fibrosis. The Cochrane database of systematic reviews. 2013 Jul 25:2013(7):CD003884. doi: 10.1002/14651858.CD003884.pub4. Epub 2013 Jul 25 [PubMed PMID: 23888484]
Level 1 (high-level) evidenceMoran F, Bradley JM, Piper AJ. Non-invasive ventilation for cystic fibrosis. The Cochrane database of systematic reviews. 2013 Apr 30:(4):CD002769. doi: 10.1002/14651858.CD002769.pub4. Epub 2013 Apr 30 [PubMed PMID: 23633308]
Level 1 (high-level) evidenceSiuba M, Attaway A, Zein J, Wang X, Han X, Strausbaugh S, Jacono F, Dasenbrook EC. Mortality in Adults with Cystic Fibrosis Requiring Mechanical Ventilation. Cross-Sectional Analysis of Nationwide Events. Annals of the American Thoracic Society. 2019 Aug:16(8):1017-1023. doi: 10.1513/AnnalsATS.201804-268OC. Epub [PubMed PMID: 31026405]
Level 2 (mid-level) evidenceJones M, Moffatt F, Harvey A, Ryan JM. Interventions for improving adherence to airway clearance treatment and exercise in people with cystic fibrosis. The Cochrane database of systematic reviews. 2023 Jul 18:7(7):CD013610. doi: 10.1002/14651858.CD013610.pub2. Epub 2023 Jul 18 [PubMed PMID: 37462324]
Level 1 (high-level) evidenceHilton N, Solis-Moya A. Respiratory muscle training for cystic fibrosis. The Cochrane database of systematic reviews. 2018 May 24:5(5):CD006112. doi: 10.1002/14651858.CD006112.pub4. Epub 2018 May 24 [PubMed PMID: 29797578]
Level 1 (high-level) evidenceSermet-Gaudelus I. Ivacaftor treatment in patients with cystic fibrosis and the G551D-CFTR mutation. European respiratory review : an official journal of the European Respiratory Society. 2013 Mar 1:22(127):66-71. doi: 10.1183/09059180.00008512. Epub [PubMed PMID: 23457167]
Ramsey BW, Davies J, McElvaney NG, Tullis E, Bell SC, Dřevínek P, Griese M, McKone EF, Wainwright CE, Konstan MW, Moss R, Ratjen F, Sermet-Gaudelus I, Rowe SM, Dong Q, Rodriguez S, Yen K, Ordoñez C, Elborn JS, VX08-770-102 Study Group. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. The New England journal of medicine. 2011 Nov 3:365(18):1663-72. doi: 10.1056/NEJMoa1105185. Epub [PubMed PMID: 22047557]
Wainwright CE, Elborn JS, Ramsey BW, Marigowda G, Huang X, Cipolli M, Colombo C, Davies JC, De Boeck K, Flume PA, Konstan MW, McColley SA, McCoy K, McKone EF, Munck A, Ratjen F, Rowe SM, Waltz D, Boyle MP, TRAFFIC Study Group, TRANSPORT Study Group. Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. The New England journal of medicine. 2015 Jul 16:373(3):220-31. doi: 10.1056/NEJMoa1409547. Epub 2015 May 17 [PubMed PMID: 25981758]
Taylor-Cousar JL, Munck A, McKone EF, van der Ent CK, Moeller A, Simard C, Wang LT, Ingenito EP, McKee C, Lu Y, Lekstrom-Himes J, Elborn JS. Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del. The New England journal of medicine. 2017 Nov 23:377(21):2013-2023. doi: 10.1056/NEJMoa1709846. Epub 2017 Nov 3 [PubMed PMID: 29099344]
Solomon M, Mallory GB. Lung transplant referrals for individuals with cystic fibrosis: A pediatric perspective on the cystic fibrosis foundation consensus guidelines. Pediatric pulmonology. 2021 Feb:56(2):465-471. doi: 10.1002/ppul.25215. Epub 2020 Dec 23 [PubMed PMID: 33300243]
Level 3 (low-level) evidenceLeard LE, Holm AM, Valapour M, Glanville AR, Attawar S, Aversa M, Campos SV, Christon LM, Cypel M, Dellgren G, Hartwig MG, Kapnadak SG, Kolaitis NA, Kotloff RM, Patterson CM, Shlobin OA, Smith PJ, Solé A, Solomon M, Weill D, Wijsenbeek MS, Willemse BWM, Arcasoy SM, Ramos KJ. Consensus document for the selection of lung transplant candidates: An update from the International Society for Heart and Lung Transplantation. The Journal of heart and lung transplantation : the official publication of the International Society for Heart Transplantation. 2021 Nov:40(11):1349-1379. doi: 10.1016/j.healun.2021.07.005. Epub 2021 Jul 24 [PubMed PMID: 34419372]
Level 3 (low-level) evidenceGreaney C, Doyle A, Drummond N, King S, Hollander-Kraaijeveld F, Robinson K, Tierney A. What do people with cystic fibrosis eat? Diet quality, macronutrient and micronutrient intakes (compared to recommended guidelines) in adults with cystic fibrosis-A systematic review. Journal of cystic fibrosis : official journal of the European Cystic Fibrosis Society. 2023 Nov:22(6):1036-1047. doi: 10.1016/j.jcf.2023.08.004. Epub 2023 Aug 28 [PubMed PMID: 37648586]
Level 1 (high-level) evidence