Introduction
Renal cystic disease (RCD) refers to a group of pathologic conditions associated with the development of renal cysts. These conditions may present in children and adults with extrarenal symptoms and have genetic or non-inherited etiologies. The renal cysts caused by these conditions are some of the most common renal abnormalities and can vary from being clinically insignificant to resulting in end-stage renal disease. The most common genetic RCD in adults is autosomal dominant polycystic kidney disease (ADPKD), a type of hepatorenal fibrocystic disease (HRFCD); the most common acquired kidney cyst is a simple renal cyst.[1][2][3]
Renal cystic disease is typically differentiated and diagnosed by clinical features, such as patient age, symptoms, renal function, and cyst characteristics (eg, size, shape, location, and number).[4][5] Diagnostic studies consist of renal imaging, laboratory tests, and genetic testing. Management typically involves supportive therapy, surveillance and treatment of complications, and, in some patients, transplantation.[1]
Most renal cystic disorders are not well-known or understood by the general population. Therefore, patients and their families must receive adequate counseling and education to understand the particular disorder diagnosed as various rare cystic renal syndromes may have genetic implications. Healthcare professionals should enhance their competence in evaluating, managing, and educating patients about renal cystic disease. Furthermore, clinicians should be empowered to drive positive changes, enhance patient safety, and improve overall care quality, enabling them to provide evidence-based and patient-centered care.[4]
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
RCD comprises various underlying conditions and is often classified as inherited or non-inherited (developmental dysplasia, de novo mutations, or systemic disorders). Advancements in research have led to a better understanding of the pathophysiologic causes of RCD. As a result, certain conditions can be categorized as dysplasias (encompassing abnormalities in renal structure) or ciliopathies (which involve mutations affecting renal tubular cilia). RCD usually has genetic etiologies; aside from ADPKD, most types first present in children or teenagers. The RCD most often observed in adults is ADPKD.[6]
Inherited Renal Cystic Disease
Polycystic kidney disease includes the 3 forms below.
- ADPKD is one of the most common causes of adult-onset, end-stage kidney disease caused by PKD1 and PKD2 mutations, which code for polycystin-1 and polycystin-2, respectively.[7] PKD1 is the more prevalent mutation, identified in approximately 85% of patients. It is associated with many extrarenal manifestations and has a 5% to 10% prevalence in the dialysis population.[3][6]
- Autosomal recessive polycystic kidney disease (ARPKD) is a rare ciliopathy caused by PKHD1 mutations that affects the fibrocystin-polyductin complex, a protein typically found in the primary cilia and basal bodies of renal tubular cells, leading to hepatorenal fibrocystic abnormalities. It has a high perinatal mortality due to pulmonary hypoplasia and is associated with liver developmental abnormalities and hypertension.[6]
- Glomerulocystic kidney disease is primarily found in neonates and infants with a family history of PKD, but not all cases are associated with mutations in PKD1 or PKD2. This condition is associated with cyst formation in the glomerulus and increased Bowman's space due to gene mutations in renal tubular primary cilia or the centrosome. About 10% of cases are associated with mutations in HFN1β, and this phenotype results in hypoplastic kidneys with defects in the renal calyces and collecting systems. Of note, HFN1β is thought to regulate transcription of PKD2 and PKHD1.[5][8]
Nephronophthisis: Nephronophthisis is a rare, autosomal recessive disorder that affects individuals at any age but is more frequently seen in children and young adults, leading to end-stage renal failure before age 30. Nephronophthisis, the most common genetic cause of end-stage renal failure in the first 3 decades of life, results from mutations in various genes (eg, NPH1 and NPH5).[9] The variants of this condition are further categorized based on the age of onset of ESRD. The most common variant is juvenile nephronophthisis, with an incidence rate of 9:8.3 million births. About 10% to 20% of cases are associated with extrarenal symptoms, including genetic syndromes like Senior-Løken syndrome (associated with retinitis pigmentosa) or Joubert syndrome (associated with cerebellar vermis aplasia). Cysts are usually found attached to the collecting tubules rather than dissociated as in PKD.[5][9][10][11]
Autosomal dominant tubulointerstitial kidney disease (ADTKD): ADTKD, formerly known as medullary cystic kidney disease, is a group of rare, autosomal dominant genetic disorders that have been recently reclassified due to mutations in multiple different genes. This classification is evolving but can be identified by ADTDK followed by the specific gene mutation (eg, ADTKD-UMOD). These mutations lead to misfolded proteins causing cytotoxicity, which leads to interstitial and tubular fibrosis. The most common mutation is in the UMOD gene. ADTKD presents as chronic kidney disease that slowly progresses to ESRD between the ages of 40 and 70. Extrarenal manifestations depend on the specific gene mutation and can include diabetes, gout, or neurodevelopmental delay. The kidneys are usually normal-sized initially but can shrink as disease progresses.[12][13]
Cysts associated with tumor syndromes: These cysts, such as tuberous sclerosis and Von Hippel-Lindau disease, result from autosomal dominant gene mutations that exhibit variable penetrance. These are described in further detail in the history and physical section.[5]
Noninherited Cystic Kidney Disease
Noninherited RCDs can be due to developmental renal dysplasia, de novo genetic mutations, or acquired cysts from systemic disease. However, due to improved genetic analysis techniques, some etiologies in this group previously thought to be noninherited, eg, multicystic dysplastic kidney, have been found to have a genetic component likely. Most of these etiologies are considered to be largely a result of renal developmental malformations.[5][14] Noninherited cystic kidney diseases include those discussed below.
Acquired cystic disease: This occurs in patients requiring dialysis and manifests as atrophic kidneys with microcysts. It is strongly associated with length of dialysis, and close to 100% of patients who are dialysis-dependent for longer than 10 years have this condition. The exact etiology is unknown but thought to be related to uremic toxins, kidney fibrosis, and calcium oxalate crystal deposition. Although usually asymptomatic, screening is recommended because these cysts are a significant risk of bleeding and renal cell carcinoma.[15][16]
Simple renal cysts: Cysts that typically develop after the age of 20 years and then increase in frequency as patients age. These are the most common RCDs, and autopsy studies show that 50% of those older than 50 years will have at least one renal cyst. Simple cysts vary in size but, on average, are approximately 1 cm. The Bosniak classification system classifies cysts to stratify the risk of malignancy. As opposed to simple cysts, complex cysts may contain calcifications, septae, and loculations. The most common etiology of simple cysts is hypertension. The etiology is thought to be hypertrophy from localized ischemia or obstruction. Unless very large, simple cysts are usually asymptomatic and do not affect renal function.[5][15]
Multicystic dysplastic kidney: This condition occurs during fetal development when the nephrons and collecting ducts do not develop in one or both kidneys. The affected kidney has many noncommunicating cysts surrounded by nonfunctional renal parenchyma. About 70% of cases are unilateral; bilateral disease requires close observation for oliguria. About 30% of cases are associated with contralateral urinary tract complications, usually urinary reflux. Fortunately, this condition is usually benign and resolves itself by a median time of 10 years old.[17] There is a genetic component in some patients, and about 10% of cases are associated with mutations in the HFN1β gene.[5][8][18]
Medullary sponge kidney: This is characterized by cystic dilatation of the papillary collecting ducts, medullary cysts, nephrocalcinosis, and nephrolithiasis. Traditionally, this etiology has been classified as a spontaneous malformation; however, it may also have a genetic component, considering that it is associated with other developmental disorders and shows familial clustering. About 10% of patients will progress to chronic kidney disease (CKD).[19][20][21]
Rare miscellaneous abnormalities: These include isolated anomalies resulting in renal cysts, such as pyelocaliceal diverticula, unilateral renal cystic disease, and multilocular renal cystic nephroma.[5][15][22]
Epidemiology
The incidence of RCD depends on the underlying etiology.
Polycystic Kidney Disease
- ADPKD is the most common RCD overall, with a prevalence of around 1 per 2500 persons, affecting 12.5 million worldwide, mostly adults.[6][23][24] ADPKD is also one of the most common causes of adult-onset end-stage kidney disease, with 50% of affected individuals progressing to renal failure by age 70 years.[6] ADPKD affects all racial and ethnic groups. Because of its delayed and indolent presentation, as many as 50% of affected individuals may go undiagnosed.[25] About 10% to 15% of affected individuals will not have a positive family history of the disease.[26][27]
- ARPKD is a rare ciliopathy with an incidence of 1 per 20,000 infants and a perinatal mortality rate of 30% due to complications of pulmonary hypoplasia. In contrast to ADPK, the kidney is enlarged at birth, but around age 3 years, the kidney can start to shrink due to atrophy. ARPKD is primarily diagnosed in infancy, childhood, or adolescence and is the cause of 5% of end-stage renal disease (ESRD) in children and adults.[6]
Nephronophthisis: This rare, autosomal recessive genetic disorder, with the most common variant being juvenile nephronophthisis (JNPHP), has an incidence of 98.3 cases per million births. About 10% to 20% of children with JNPHP present with chronic renal failure, and between 1% and 5% of all affected patients ultimately need dialysis or transplantation. JNPHP is the most common cause of genetic ESRD in children.[5][9][10][28][29]
Autosomal-dominant tubulointerstitial kidney disease: The estimated population prevalence of this condition is 9 in 1 million births, and it tends to inevitably lead to renal failure later in life, typically between the ages of 30 and 60 years, with an average of 45 years.[30][23][31][32][33]
Cysts related to tumor syndromes: These conditions consist of autosomal-dominant gene mutations with an estimated incidence ranging from 1 in 6,000 to 1 in 10,000 births.[5] Von Hippel-Lindau syndrome has a reported incidence of 1 per 50,000 individuals.[15]
Acquired cystic disease: The occurrence of renal cysts is secondary to dialysis. The incidence increases in parallel to the number of years on dialysis and is more common in males.[5][34]
Simple renal cysts: Typically, these cysts develop in adults, usually after the age of 20 years, and then increase in frequency, occurring in 7% to 10% of the general populace. Incidence is associated with male sex, hypertension, and advanced age.[5]
Multicystic dysplastic kidney: Unilateral variants have a higher incidence, estimated to occur in 1 per 4300 births; bilateral kidney involvement has a reported incidence of 1 per 10,000 births.[18]
Medullary sponge kidney: The precise incidence of this etiology is unknown; however, between 3% and 20% of individuals with renal stones are estimated to have medullary sponge kidney.[19][20]
Rare miscellaneous abnormalities: These cystic renal anomalies rarely occur, often with only a few cases reported worldwide. For instance, pyelocaliceal diverticula are identified in up to 0.6% of patients who have had an intravenous pyelogram performed.[5][22]
Pathophysiology
Due to the various etiologies underlying the development of RCDs, several abnormalities in renal function can result. It is believed that the primary physiologic abnormality in RCD is the loss of functional nephrons to cysts; this can be due to genetic mutations of renal tubular cilia (ie, ciliopathies), developmental anomalies in renal structure (ie, dysplasia), or acquired due to systemic factors.
Primary cilia usually line renal tubules and project into the tubule lumen, contributing significantly to the kidney's normal physiologic function. Generally, kidney cysts develop from renal tubule segments and detach from the parent tubule after growing a few millimeters. The fluid inside the early developing cysts is initially glomerular filtrate, but as the tubules dilate and separate from the nephron, the cystic fluid is actively secreted. Therefore, ciliopathies (eg, polycystic kidney disease, nephronophthisis, and ADTKD) are believed to be associated with abnormal proteins and gene mutations that result in renal tubular ciliary mechanosensors becoming unable to detect luminal flow rates.[35][36][37] This ciliary malfunction leads to the overproduction of epidermal growth factors, epithelial cell proliferation, increased fluid secretion, tubular ectasia, and renal cyst formation.[35][36][37][38]
For instance, ADPKD develops due to mutations in the genes PKD1 and PKD2. These mutations cause abnormal tubular ciliary activity, dysregulation of tubular epithelial adhesion, cell differentiation, and ion channel dysfunction, which causes active fluid secretion into cysts.[35][36][37][39] Renal cysts in ADPKD can hemorrhage and cause hematuria or become infected. Patients are at higher than average risk of pyelonephritis, and 25% will develop nephrolithiasis. Estrogen levels play a role in hepatic cyst development, leading to a higher prevalence of liver cysts in women compared to men. Multiple pregnancies or other estrogen exposure can further increase this likelihood. Many hepatic cysts are common, but liver failure is rare. Vascular sclerosis and interstitial fibrosis may also develop. Failure of normal ductal plate remodeling causes the persistence of embryologic bile ducts, which become dilated and form hepatic cysts.
ARPKD is due to mutations in the PKHD1 gene and presents in childhood. Severe cases result in perinatal death, most often due to pulmonary hypoplasia, and patients who survive until adulthood have some degree of liver fibrosis. Imaging shows small cystic lesions due to renal tubular dilation rather than the large non-communicating cysts seen in ADPKD.[6][15]
Autosomal dominant tubulointerstitial kidney disease is also characterized by progressive interstitial fibrosis and tubular damage due to ciliary dysfunction. Patients usually present with ESRD within 10 years of when their affected parent had ESRD. At least 6 separate genes are known to be involved, and the most common are UMOD, MUC1, and REN. Other physiologic abnormalities, including polydipsia and polyuria, may also result.[33][40]
Nephronophthisis is an autosomal recessive genetic disorder caused by mutations in the NPH gene. There are 26 known mutations, and many more are unidentified.
Simple cysts are thin-walled sacs filled with serous fluid lined with cuboidal or flattened epithelial cells. It is thought their genesis is a hypertrophic response to ischemia or obstruction. They project outside the renal parenchyma into the pelvic area and are called parapelvic renal cysts.[15]
Hypertension related to RCD can be a prominent feature from as young as the neonatal period. It results from the cystic distortion of the intrarenal architecture, which causes activation of the renin-angiotensin-aldosterone system and increased sympathetic nervous system activation.[41][42] The polyuria caused by renal tubule concentrating defects can also activate the V1 vasopressin receptors, causing vasoconstriction. It is also thought that the polycystin proteins (which are deficient in PKD) help decrease vascular tone.[41]
Histopathology
The primary histological abnormalities in RCD are cystic dilatations within the nephron's various components, including the glomerulus, tubule, and collecting ducts. On microscopic examination, the cystic dilatation of these structures can be 2 to 3 times larger than normal size.[5][43]
History and Physical
Often, RCD is suspected due to clinical features but must be confirmed with diagnostic studies. Clinical findings, for the most part, are nonspecific and, when present, vary based on the underlying etiology and may range from asymptomatic to ESRD. However, a few conditions may be somewhat differentiated by extrarenal symptoms that are associated with them. Additionally, obtaining a comprehensive family history can significantly inform the diagnosis. The following are the clinical features of some common CKD etiologies.[5]
Autosomal-dominant polycystic kidney disease: Patients typically do not show symptoms until they are aged 30 to 50; however, children with ADPK have higher rates of proteinuria and hypertension than children without ADPKD.[6] Clinical presentation in adults typically includes flank pain, intermittent hematuria, cyst hemorrhage, hypertension, and chronic renal failure. Furthermore, up to 25% of ADPKD patients will develop kidney stones. Chronic flank or back pain is frequently noted secondary to comorbid conditions and complications, including the following: cyst rupture, infection, or hemorrhage; kidney enlargement; urolithiasis; or urinary tract infection.[6][44] Additionally, ADPKD commonly has several extrarenal clinical features. Hepatic cysts are the most common, followed by hepatomegaly, pancreatic cysts, diverticulosis, valvular cardiac disease, cerebral aneurysms, and seminal vesicle cysts. Clinical features can be similar to other RCDs. Therefore, a positive family history can assist in diagnosis, as 75% to 90% of these patients have a parent who shares the same diagnosis.[45]
Autosomal recessive polycystic kidney disease: Clinical features typically appear antenatally and in newborns but may also present in early adulthood. Patients have concurrent liver disease with developmental biliary defects resulting in dilated bile ducts, sometimes with cyst formation. Clinical expression of hepatic disease is variable, with the most common complications being portal hypertension, hypersplenism, esophageal varices, and cholangitis. One estimate of long-term survivors is that 7% of patients will need liver transplantation due to portal hypertension or recurrent cholangitis.[15][46] In two-thirds of affected newborns, significant hypertension develops within the first few months after birth. Due to the early onset of the disease, perinatal clinical features are commonly seen, including oligohydramnios or anhydramnios; low Apgar scores; Potter facies with limb defects; stomach compression and feeding difficulty secondary to enlarged kidneys; and pulmonary hypoplasia with respiratory distress (a common cause of mortality).[47][48][49] Other clinical manifestations may include left ventricular hypertrophy, neurocognitive impairment, polyuria, polydipsia, recurrent urinary tract infections, and ESRD.[5][50][51]
Autosomal-dominant tubulointerstitial kidney disease: Clinical presentation depends on the specific gene mutation, but polydipsia and polyuria are common. Blood pressure is usually normal, and renal failure progresses slowly with a median age of 45 for ESRD. The most common gene abnormality occurs with the UMOD gene and is associated with hyperuricemia and gout.[13][15]
Nephronophthisis: This condition is characterized by symptoms of ESRD by the age of 30 years with normal blood pressure. Extrarenal lesions present in 10% to 20% of patients, including retinitis pigmentosa, cerebellar vermis hypoplasia, gaze palsy, and hepatic fibrosis. JNPHP presents with growth retardation, polyuria and polydipsia, skeletal dysplasia, anemia, and progressive renal failure.[15]
Tuberous sclerosis: This condition is due to mutations in the genes TSC1 and TSC2, which result in hamartoma formation throughout the body. Patients can present with several characteristic findings, including angiofibroma, facial nevi, cardiac rhabdomyoma, epilepsy, and mental retardation. Renal findings include angiomyolipomas, renal cysts, and renal cell carcinomas. One study showed that about 71% of patients had renal lesions; the most common was angiomyolipomas (61%), and 28% had renal cysts.[15][52]
Von Hippel-Lindau syndrome: This syndrome is caused by defects in the VHL gene, which produces a tumor suppressor. In addition to cystic disease of the kidneys, which is typically asymptomatic, characteristic cystic lesions can be present in other organs, such as the pancreas and epidermis. The diagnosis of this condition varies based on family history. In patients with a positive family history of this condition, the presence of a von Hippel-Lindau–associated lesion—retinal angiomas, spinal or cerebellar hemangioblastomas, endolymphatic sac tumors, renal cell carcinoma, pheochromocytoma, pancreatic cysts, neuroendocrine tumors, or epididymal cystadenomas—meets the criteria for clinical diagnosis. In patients without a family history, clinical diagnosis is confirmed by the presence of either (1) 2 hemangioblastomas or retinal angiomas or (2) at least 1 hemangioblastoma or retinal angioma, in addition to at least 1 other von Hippel-Lindau–associated lesion. A positive result for a VHL gene mutation is also diagnostic if the family history is negative.[15][53][54]
Acquired renal cystic disease: In patients on dialysis, this condition usually progresses without symptoms; however, it can present with a palpable renal mass, gross hematuria, flank pain, urinary tract infection, or renal colic.[55][56]
Multicystic dysplastic kidney: Most commonly, patients with this condition are identified during antenatal fetal ultrasound imaging. Patients with bilateral kidney involvement may have clinical findings of oliguria and amenorrhea. Genital tract anomalies, eg, Mayer-Rokitansky, are also common. Children with this condition are more likely to have ureteral reflex and UTIs. In most children with this condition, the cysts will spontaneously regress and have no residual symptoms.[3][15][57]
Simple renal cysts: Patients are typically asymptomatic with no family history of kidney dysfunction. They can cause pain, hematuria, or infection, especially if the cysts are large.[3][15][3]
Evaluation
In patients presenting with nonspecific clinical features of RCD and unknown or negative family history, clinicians should perform diagnostic studies to assist in determining an accurate diagnosis. Generally, imaging studies are the primary diagnostic tool used to characterize cystic kidneys; genetic testing may be considered to assess other potential inherited etiologies. Additional laboratory studies may be used to monitor for complications and exclude differential diagnoses.[15]
Diagnostic Imaging Studies
Ultrasound of the kidneys is the preferred modality to evaluate RCD. Indications for ultrasound imaging include individuals with clinical symptoms of RCD (eg, flank pain, significant hypertension, or recurrent UTI), renal cysts incidentally identified on prenatal ultrasounds, or with a family history of polycystic kidney disease who are at least aged 18 years. In patients with a positive family history, ultrasound imaging is used as a screening modality to diagnose RCD.[58] Ultrasound is often also used to monitor disease progression. Characteristic diagnostic findings for RCD on kidney ultrasound depend on the etiology, including the following:
Autosomal-dominant polycystic kidney disease
- Enlarged echogenic kidneys, often with a "lumpy" contour
- Multiple, large, bilateral renal cysts; the following is generally used for diagnosis:
- Age 15 to 39 years: ≥3 cysts in one or both kidneys
- Age 40 to 59 years: ≥2 cysts in both kidneys
- Age >60 years: ≥4 cysts in both kidneys [59]
- Cyst number and size increase with patient age [6]
Autosomal recessive polycystic kidney disease
- Enlarged kidneys, normal contour, echogenic with poor cortico-medullary junction differentiation
- Dilated collecting ducts
- Multiple small bilateral cysts, 5 to 7 mm on average
- Compression of the renal cortex with hypoechoic halos
- No evidence of renal cysts in parents of infants with RCD [3]
Nephronophthisis
- Hyperechoic normal or small kidneys with normal to small-sized cysts; in the infantile variant, kidneys are enlarged [3]
- Renal cysts primarily within the corticomedullary junction [6]
Autosomal-dominant tubulointerstitial kidney disease
- Bilateral atrophic or normal-sized kidneys with microcysts
- Antenatal fetal anatomy ultrasound may show associated fetal anomalies (eg, cleft palate, spina bifida, and polydactyly) [15]
Medullary cystic kidney disease
- Unconnected multiple cysts of various sizes
- Decreased amount of renal parenchyma
- Usually unilateral, but occasionally bilateral
- Genital anomalies may also be visualized [60]
Acquired renal cystic disease
- Bilateral small cysts primarily in the cortical area of the kidney
- Small or atrophic kidneys [59]
Simple renal cysts
- Sharply-defined cysts with thin, smooth walls without internal debris or cystic communication
- Normal-sized kidneys
- Typically unilateral [5][15]
Renal CT and MRI may be used to evaluate RCD further, particularly if findings are nonspecific on ultrasound imaging.[60] These modalities can better visualize cyst properties or disease progression for some conditions, such as tuberous sclerosis and simple and complex renal cysts.[5] Additionally, the liver and spleen should be imaged with CT or MRI if initial ultrasound imaging is inadequate to evaluate associated extrarenal pathologies (eg, biliary duct enlargement, esophageal varices, and hepatosplenomegaly).[3][61]
The Bosniak classification may be used in interpreting findings on CT imaging, with and without contrast, by classifying cystic lesions.[62] Cyst characteristics are categorized into 5 different groups according to their malignant potential. This classification aids clinicians in determining the best management approach with the following recommendations for each respective category.[5][56][63]
- Class I: A benign simple cyst with a thin wall without septa, calcifications, or solid components. Further evaluation is not required. Malignant potential is estimated at < 1%.
- Class II: There are two categories: (1) A benign simple cyst with a few thin walls and septa with fine calcifications but no enhancement. (2) Homogenous high-attenuating masses ≤3 cm that have sharp margins and do not enhance. Follow-up is usually unnecessary but may be suggested 6 or 12 months later in selected patients to confirm the diagnosis.
- Class IIF: An indeterminate cyst designation between categories II and III with two types: (1) Cysts with multiple thin septa with smooth, minimally thickened walls that may have nodular calcification. There may be the impression of some mild enhancement. (2) High attenuation masses >3cm but with no enhancement. The estimated malignant potential is 5%. Follow-up imaging with CT or ultrasound is recommended, usually at 6 months and then annually for 5 years.
- Class III: The cysts demonstrate a multilocular lesion with thickened walls or septa, irregular calcifications, or measurable vascular enhancement >10 Hounsfield units. Close follow-up or surgical excision with partial or total nephrectomy is recommended. These cysts have an estimated malignancy rate of 55%.
- Class IV: These cysts are typically large nodular lesions with enhancing solid or necrotic components. These cysts have an extremely high malignant potential of almost 100%. Partial or total nephrectomy surgery is recommended.
Diagnostic imaging is also recommended for some etiologies to monitor RCD progression and extrarenal manifestations. For instance, annual abdominal ultrasound examinations are recommended in patients diagnosed with ARPKD to identify early portal hypertension and to monitor kidney size in infants with severe or rapidly progressive disease.[64] Individuals diagnosed with ADPKD who work in high-risk occupations (eg, airline pilots), are scheduled to have major surgery, experience central nervous system symptoms (eg, new-onset severe headache or loss of consciousness), or have a family history of intracranial aneurysms should have imaging studies to screen for intracranial aneurysm.[58]
Genetic Testing
Genetic testing for known mutations may be considered to help establish a diagnosis in patients with suspected hereditary etiologies of RCD. However, clinicians should understand that genetic testing is not definitive, as current testing may not identify newer mutations. For instance, though there are 26 known genetic mutations of the NPHP genes that cause nephronophthisis, recent genetic analysis only detects 9 to 12 mutations.[3]
Prenatal genetic testing is recommended for pregnancies where clinicians have identified fetuses with RCD. Indications for which clinicians should offer prenatal genetic testing include fetuses with 1 or more unilateral cysts if there are extrarenal manifestations present, fetuses with bilateral CKD or enlarged kidneys regardless of oligohydramnios or extrarenal malformations, or if ADPKD or ARPKD are suspected.[3]
Laboratory Studies
Laboratory studies are performed to assess kidney function or identify extra renal manifestations of RCD, such as liver disease. In patients with ADPKD, albuminuria, proteinuria, and a decreased glomerular filtration rate (GFR) are frequently identified.[3] Furthermore, urinary cultures are often positive for Escherichia coli, Staphylococcus aureus, Enterococcus, Lactobacillus, or anaerobic bacteria. However, negative urine culture results are possible when infected renal cysts are not in contact with the urinary space.[58]
Characteristic findings of ARPKD in laboratory studies include hyponatremia and dilute urine osmolality.[5] Also, clinicians may perform bilirubin, hepatic enzyme, and complete blood cell counts to help identify associated portal hypertension and splenic dysfunction. Thrombocytopenia due to splenic activity and portal hypertension can also be observed.[65]
Depending on the genetic mutation, ADTKD can present with hyperuricemia, hyperkalemia, anemia, acidosis, elevated liver enzymes, neutropenia, or hypogammaglobulinemia. Nephronophthisis can show hyponatremia due to impaired sodium reabsorption.[15]
Treatment / Management
The management of RCDs is primarily supportive with treatment and monitoring of any sequelae, eg, hypertension, infection, and pain. Hypertension is an almost universal finding in polycystic kidney disease, and several options exist to control high blood pressure. Preferred agents include angiotensin receptor blockers and angiotensin-converting enzyme inhibitors.[41] Calcium channel blockers may also maintain a blood pressure of <130/80 mm Hg. About two-thirds of hypertensive patients will require more than 1 agent; however, optimal management of hypertension may slow the decline in renal function with this disease.[66] The following are other management recommendations that are targeted to the underlying etiologies.
Autosomal Dominant Polycystic Kidney Disease
In ADPKD, recommendations include drinking at least 2 to 3 liters of water daily to naturally suppress vasopressin, which can help reduce cyst formation and growth.[67][68][69] Additional measures include restricting sodium to ≤2 g of salt daily, regularly exercising, maintaining a normal weight, and managing hypertension.[67][70][71](B3)
Tolvaptan is a vasopressin receptor antagonist at the V2 receptor that is effective for ADPKD and has been shown to slow the growth and development of renal cysts.[72][73][74][75] Tolvaptan blocks vasopressin signaling, which would otherwise increase intracellular cyclic adenosine monophosphate that stimulates the growth and proliferation of renal cysts; it is the only US Food and Drug Administration (FDA)-approved medication for ADPKD.[72][76] However, tolvaptan appears to be most useful in younger patients with ADPKD and early or rapidly progressive renal disease; it is less effective in patients older than 55.[75][77] (B2)
Clinicians should be aware that tolvaptan is a potentially dangerous drug that can cause significant liver damage or even acute liver failure requiring transplantation. Renal function tests should be monitored regularly. A recommended surveillance schedule is before therapy, 2 and 4 weeks after starting treatment, followed by every month for 18 months, and then quarterly. Elevated liver enzymes are most likely to occur during the first 18 months of use but will generally resolve after treatment discontinuation.[77] The drug also causes significant diuresis with polyuria and severe thirst. Adverse effects result in drug discontinuation in about 8% of patients.
Cyst infections can develop in ADPKD, which can be treated with a course of antibiotics that penetrate cysts well, such as ciprofloxacin, clindamycin, sulfamethoxazole-trimethoprim, erythromycin, chloramphenicol, or tetracycline. For chronic pain, which frequently occurs in patients with ADPKD, alternative pain management strategies (eg, antidepressants and avoidance of activities that can trigger pain) and referral to a pain management specialist should be considered. Nonsteroidal anti-inflammatory drugs (NSAIDs) should be avoided due to the risk of toxicity.[58]
Autosomal Recessive Polycystic Kidney Disease
ARPKD has no curative treatment other than transplantation; therefore, there is only symptomatic management, including supportive respiratory therapy for pulmonary insufficiency, dialysis or kidney transplantation for end-stage renal failure, salt restriction, antihypertensives for hypertension, and loop diuretics for edema.[78] The associated hepatobiliary disease may require the administration of synthetic bile acids, portosystemic vascular shunting, or a liver transplant. Several ongoing trials are evaluating its usefulness in pediatric ARPKD.[79] Adverse effects and cost limit its use. Tolvaptan is the only drug specifically FDA-approved for ADPKD, but the repurposing of other medications that might be effective in polycystic kidney disease is being investigated.[74](B3)
Tuberous Sclerosis Syndrome
Patients with tuberous sclerosis-associated RCD should be monitored for angiomyolipoma lesions that often develop. The 2012 International Tuberous Sclerosis Consensus Group recommends initiating first-line treatment with mTOR inhibitor therapy for asymptomatic lesions >3 cm. Another study showed evidence that the drug everolimus effectively reduced the volume of angiomyolipoma lesions.[5] Embolization is necessary for angiomyolipoma lesions that lead to acute hemorrhage; for asymptomatic lesions >3 cm, embolization or nephron-sparing resection may also be considered.[5](B3)
Von Hippel-Lindau Syndrome
Treatment of RCD associated with this condition is based on the management recommendations specified in the Bosniak classification. Follow-up and imaging at 3, 6, and 12 months after diagnosis, and then yearly, is recommended for patients in the IIF class. Patients in class III should have close follow-up and possibly surgical excision due to the risk of malignancy; those in class IV should have renal cysts surgically excised.[5][15](B3)
Nephronophthisis and Autosomal-Dominant Tubulointerstitial Kidney Disease
Nephronophthisis and ADTKD are managed supportively to treat sequelae symptoms; currently, there are no pharmacologic treatments. Salt supplementation should be used when there is severe salt wasting. In addition, diuretics, sodium restriction, NSAIDs, and renin-angiotensin-suppressing medications should be avoided. In patients with ESRD, dialysis and kidney transplantation should be considered. Allopurinol or febuxostat are used to treat the hyperuricemia associated with ADTKD.[15]
Acquired Cystic Renal Disease
Patients with this condition are at significantly increased risk of hemorrhage and renal cell carcinoma, so initial and periodic screening is recommended in dialysis patients. Although transplant diminishes the size of the cysts, it does not eliminate the risk of renal cell carcinoma, and transplanted patients should also have a yearly ultrasound of the native kidney as part of follow-up.[80]
Simple Renal Cysts
Cysts that are Bosniak classes I and II generally do not require follow-up. Renal cysts generally produce no symptoms, but if they become symptomatic, needle aspiration, sclerotherapy, or cyst unroofing may be needed. Laparoscopic techniques can be performed for complex cysts.[81] Surgical excision should be considered for patients in class III and is recommended for those in Bosniak class IV.[5](B3)
Investigational Therapies
Targeted therapies for genetic renal cystic diseases are being actively investigated, and several are undergoing extensive clinical trials.[82][83] More investigation is required before these agents can be recommended for clinical use.(A1)
- Various drugs such as methylprednisone, urinary alkalinization, lovastatin, epidermal growth factor tyrosine kinase receptor inhibitors, paclitaxel, bardoxolone methyl, peroxisome proliferator-activated receptor agonists, mitogen-activated protein kinase inhibitors, and cyclin-dependent kinase inhibitors are undergoing animal studies to assess clinical utility.[45][47][48][82][84][85][86] (A1)
- Drugs targeting mTOR signaling pathways, such as rapamycin, by checking cellular proliferation, are under phase 2 and 3 clinical trials.[82][87][88][89] (A1)
- Statins have shown some benefits in lowering serum lipid levels in ADPKD patients, reducing proteinuria, and slowing the decline in GFR, but their effect on disease progression is still uncertain.[90]
- Combined somatostatin and tolvaptan block the effect of cyclic adenosine monophosphate and inhibit fluid secretion and cell proliferation.[82][91][92] (A1)
- Triptolide, which affects calcium signaling, also exhibits antiproliferative effects in ADPKD.[93] (B2)
- Salsalate, an NSAID, and metformin, either together or separately, may help slow the progression of ADPKD.[94][95][96]
- Amiloride and caffeine restriction have shown some benefits in limiting cyst growth in animals but have not demonstrated similar efficacy in humans.[97][98] (B3)
- Bosutinib, an oral tyrosine kinase inhibitor, was studied for possible use in ADPKD. Bosutinib reduced the renal growth rate but did not affect the decline in GFR.[99]
Differential Diagnosis
Excluding differential diagnoses is essential to arrive at the most accurate diagnosis. Accurate diagnosis enables clinicians to employ the most suitable, currently available, and targeted treatment strategies. A complete history and physical examination for secondary findings, imaging modalities, and genetic testing can help differentiate the various etiologies of renal cysts. Some conditions that can appear as RCDs include angiomyolipomas, cystic renal neoplasms, extrarenal pelvis, hemangiomas, hydronephrosis, nephrolithiasis, neuroblastoma, parapelvic renal cysts, renal abscess, renal dysplasia, lipomas, and many rare genetic disorders.[6][15]
Prognosis
Generally, the prognosis in cystic renal disease depends on the rate of declining renal function and extrarenal manifestations. Patients who progress to ESRD at a younger age typically have a poorer prognosis.
For instance, ADPKD will typically progress to some degree of renal insufficiency in patients aged > 30 years. Ultimately, 35% to 45% of patients with ADPKD will develop end-stage renal failure by the age of 60 years, 50% by the age of 70 years, and up to 75% by the age of 75 years. The GFR in ADPKD declines by about 5 mL/min/year in patients older than 40. The most reliable predictor of disease progression to end-stage renal failure appears to be kidney and cyst volume measurements, particularly a total renal volume of > 1500 mL.[100] The faster the total renal volume increases, the more rapidly renal function decreases. Other predictors of a more rapid progression and poorer prognosis include the following: [101][102][103][104][105][106]
- Black ethnicity or genetic heritage
- Diagnosis at an earlier age
- Diabetes
- Gross hematuria
- Hypertension
- Larger kidney and cyst size
- Male gender
- PKD1 genotype
- Rapidly enlarging kidneys or cyst dimensions
- Sickle cell trait
- Worsening proteinuria
ADPKD does not increase the incidence of developing renal cancer, but if such a malignancy were to develop, it is more likely to be bilateral than in the general population. Renal cancer rarely causes death in such cases. Mortality is most commonly due to valvular heart disease, disseminated infection, hypertensive nephropathy, renal failure, or cerebral vascular accident due to a ruptured cerebral aneurysm from the combination of cerebral aneurysms and hypertension. Hepatic cysts are common but rarely clinically significant.[58]
In ARPKD, hypoplastic and immature lungs are the most significant neonatal clinical problems, often requiring mechanical ventilation. Portal hypertension may develop, resulting in hypersplenism and thrombocytopenia. Frequent causes of death aside from renal failure include pulmonary insufficiency and hepatobiliary disease. Liver and kidney transplantation at a younger age is associated with longer survival rates.[3]
Complications
ADPKD has several complications, including hypertension, renal failure, cyst infection, severe abdominal and flank pain, and subarachnoid hemorrhage.[15] The most common complication of ARPKD is respiratory distress due to pulmonary hypoplasia; other complications include portal hypertension, variceal bleeding, and renal insufficiency. Nephronophthisis can be present with both hepatic and other extrarenal complications (eg, hepatic fibrosis, biliary duct enlargement, growth retardation, retinitis pigmentosa, cerebellar vermis hypoplasia, gaze palsy, hepatic fibrosis, and skeletal abnormalities).[15] ADTKD complications depend on the gene mutation and can include hyperuricemia, gout, hyponatremia, and polyuria.[1][40]
Consultations
A multidisciplinary care team is required to manage patients with RCD. This involves perinatologists, neonatologists, hepatologists, nephrologists, and geneticists to organize care for the patient from the perinatal period to adulthood. Prenatal consultation with neonatologists, maternal-fetal medicine specialists, and genetic specialists is often warranted if a kidney anomaly is identified on prenatal ultrasonography. At this consultation, topics such as ultrasonographic findings, family history, prognosis factors, and the risk of recurrence should be discussed. The patient should also receive various prenatal and postnatal genetic testing options.[3]
Deterrence and Patient Education
Genetic counseling is essential for genetic renal cystic diseases such as ADPKD and ARPKD. Furthermore, patient education on lifestyle modification can help mitigate these complications, and suggestions include regular medical care, salt restriction, hypertension control, a healthy BMI, hydration, and regular exercise.[107]
Pearls and Other Issues
Key facts pertinent when managing polycystic disease are as follows:
- Screening family members for polycystic kidney disease has been an ongoing controversy. Genetic testing can be conclusive but requires appropriate genetic counseling, which may not be readily available. Now that tolvaptan is available and has proven to be effective in slowing the progression of the disease, considering renal ultrasonography in relatives of affected individuals seems reasonable if patients or family members desire it. However, screening asymptomatic family members without hypertension, renal failure, or proteinuria is not generally recommended.[108]
- Cyst volume and total kidney size appear to be the best prognostic factors in tracking polycystic kidney disease progression in ADPKD. Cysts are present throughout the renal cortex and medulla.[109][110]
- Angiotensin-converting enzyme inhibitors and blockers are preferred for managing hypertension in patients with polycystic kidney disease.
- Tolvaptan is a vasopressin V2 receptor antagonist that slows the development and growth of the cysts and thus is an effective treatment of ADPKD, especially if rapidly progressive.[72][76] It can have significant adverse effects of polyuria and severe thirst but should be considered for all appropriate ADPKD patients.
- ARKD is always associated with liver fibrosis, but hepatic symptoms are variable. When severe, hepatic symptoms include portal hypertension, splenomegaly, thrombocytopenia, and esophageal varices.
- Autosomal dominant tubulointerstitial disease is characterized by tubulointerstitial fibrosis and has been recently reclassified to be called ADTKD-(abnormal gene name). Urine sediment is bland, and renal failure is slowly progressive. Symptoms depend on the gene abnormality, and the most common mutation is in the UMOD gene, which codes for the uromodulin protein, and is associated with hyperuricemia and gout. Cysts are not found in the renal cortex.
- Multicystic dysplastic kidney disease can be diagnosed prenatally and involves renal cysts surrounded by undifferentiated renal parenchyma, along with an undeveloped renal collecting system and vasculature. It is usually unilateral but can result in urinary reflux and UTI in the unaffected kidney. It usually spontaneously regresses around age 10 years without treatment.[111]
- Nephronophthisis is a common cause of ESRD in those aged younger than 30 years. It is associated with impaired renal concentrating ability, polyuria, and polydipsia. The most common type is Juvenile nephronophthisis, where ESRD is reached at a median age of 13. Cysts are found primarily in the corticomedullary junction and are often attached to the cortical collecting ducts instead of dissociated.
- Medullary sponge kidney presents with many measurable abnormalities, such as distal renal tubular acidosis, hypocitraturia, hypercalciuria, and proteinuria. The cysts are generally small (microcysts) in the papilla and medulla. Ultrasound will show cystic dilation in the renal papilla (giving the impression of a "sponge" on autopsy studies) and multiple calcifications. It is strongly associated with nephrolithiasis.[111]
- Acquired renal cystic disease is associated with ESRD and is asymptomatic but requires monitoring for the increased incidence of renal cell carcinoma. The risk of RCC is decreased with transplant but not eliminated, so transplant ultrasounds should include the native kidneys as well.
Enhancing Healthcare Team Outcomes
Adult and pediatric nephrology clinicians should coordinate to ensure appropriate continuity of care for a patient with renal cystic disease who may initially present as a child but survives to become an adult.[6] RCD, especially those conditions diagnosed in patients of very young age, usually requires a care team with varied expertise (eg, perinatologists, neonatologists, hepatologists, nephrologists, and geneticists) to coordinate care for the patient.[3]
Lastly, all healthcare team members should support and promote general health and lifestyle measures, optimal control of blood pressure, increased fluid intake, and dietary sodium restriction. Given the long-term progression of these diseases, it is often helpful to include a multidisciplinary team of social workers, dieticians, pharmacists, and genetic counselors.
Media
(Click Image to Enlarge)
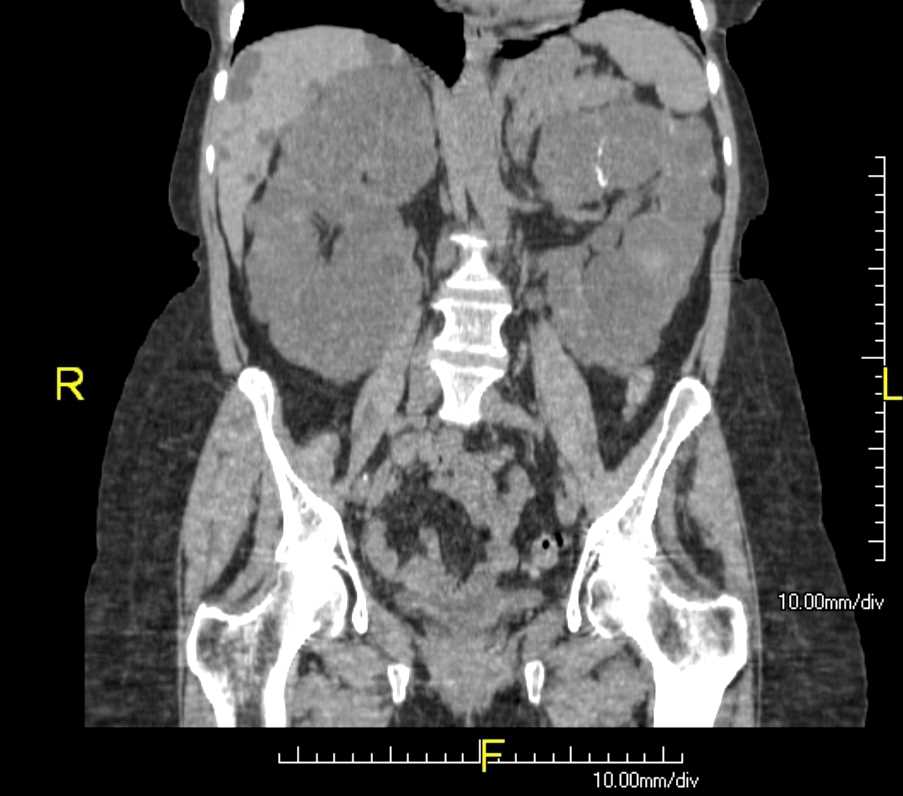
Polycystic Kidneys (ADPKD) and Liver Cysts. CT coronal view of abdomen.
Contributed by Scott Dulebohn, MD
(Click Image to Enlarge)
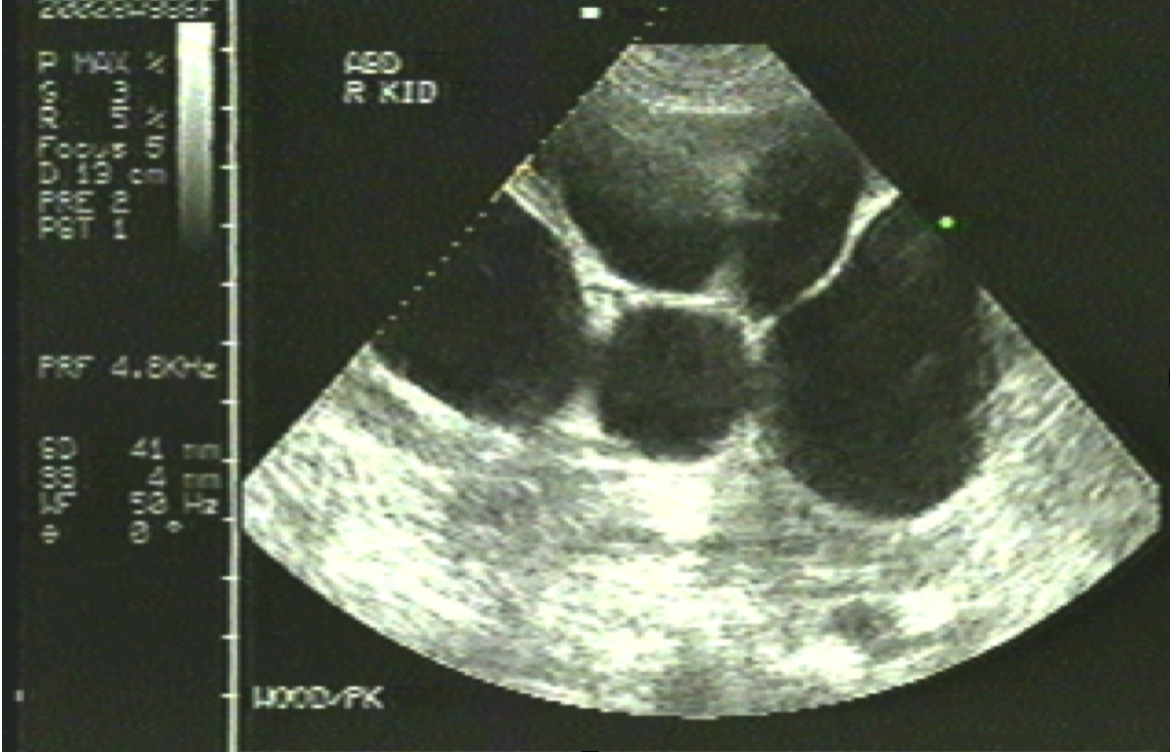
Polycystic Kidney
Contributed by Michael Lambert, MD
(Click Image to Enlarge)
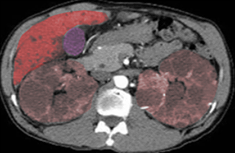
Autosomal Polycystic Kidney Disease
Image courtesy S Bhimji, MD
References
Bisceglia M, Galliani CA, Senger C, Stallone C, Sessa A. Renal cystic diseases: a review. Advances in anatomic pathology. 2006 Jan:13(1):26-56 [PubMed PMID: 16462154]
Level 3 (low-level) evidenceWei L, Xiao Y, Xiong X, Li L, Yang Y, Han Y, Zhao H, Yang M, Sun L. The Relationship Between Simple Renal Cysts and Renal Function in Patients With Type 2 Diabetes. Frontiers in physiology. 2020:11():616167. doi: 10.3389/fphys.2020.616167. Epub 2020 Dec 15 [PubMed PMID: 33384617]
Raina R, Chakraborty R, Sethi SK, Kumar D, Gibson K, Bergmann C. Diagnosis and Management of Renal Cystic Disease of the Newborn: Core Curriculum 2021. American journal of kidney diseases : the official journal of the National Kidney Foundation. 2021 Jul:78(1):125-141. doi: 10.1053/j.ajkd.2020.10.021. Epub 2021 Jan 6 [PubMed PMID: 33418012]
Kurschat CE, Müller RU, Franke M, Maintz D, Schermer B, Benzing T. An approach to cystic kidney diseases: the clinician's view. Nature reviews. Nephrology. 2014 Dec:10(12):687-99. doi: 10.1038/nrneph.2014.173. Epub 2014 Sep 30 [PubMed PMID: 25266212]
Cramer MT, Guay-Woodford LM. Cystic kidney disease: a primer. Advances in chronic kidney disease. 2015 Jul:22(4):297-305. doi: 10.1053/j.ackd.2015.04.001. Epub [PubMed PMID: 26088074]
Level 3 (low-level) evidenceMüller RU, Benzing T. Cystic Kidney Diseases From the Adult Nephrologist's Point of View. Frontiers in pediatrics. 2018:6():65. doi: 10.3389/fped.2018.00065. Epub 2018 Mar 22 [PubMed PMID: 29623269]
Kim B, King BF Jr, Vrtiska TJ, Irazabal MV, Torres VE, Harris PC. Inherited renal cystic diseases. Abdominal radiology (New York). 2016 Jun:41(6):1035-51. doi: 10.1007/s00261-016-0754-3. Epub [PubMed PMID: 27167233]
Shao A, Chan SC, Igarashi P. Role of transcription factor hepatocyte nuclear factor-1β in polycystic kidney disease. Cellular signalling. 2020 Jul:71():109568. doi: 10.1016/j.cellsig.2020.109568. Epub 2020 Feb 14 [PubMed PMID: 32068086]
Hildebrandt F, Zhou W. Nephronophthisis-associated ciliopathies. Journal of the American Society of Nephrology : JASN. 2007 Jun:18(6):1855-71 [PubMed PMID: 17513324]
Level 3 (low-level) evidenceSalomon R, Saunier S, Niaudet P. Nephronophthisis. Pediatric nephrology (Berlin, Germany). 2009 Dec:24(12):2333-44. doi: 10.1007/s00467-008-0840-z. Epub 2008 Jul 8 [PubMed PMID: 18607645]
Gupta S, Ozimek-Kulik JE, Phillips JK. Nephronophthisis-Pathobiology and Molecular Pathogenesis of a Rare Kidney Genetic Disease. Genes. 2021 Nov 5:12(11):. doi: 10.3390/genes12111762. Epub 2021 Nov 5 [PubMed PMID: 34828368]
Devuyst O, Olinger E, Weber S, Eckardt KU, Kmoch S, Rampoldi L, Bleyer AJ. Autosomal dominant tubulointerstitial kidney disease. Nature reviews. Disease primers. 2019 Sep 5:5(1):60. doi: 10.1038/s41572-019-0109-9. Epub 2019 Sep 5 [PubMed PMID: 31488840]
Econimo L, Schaeffer C, Zeni L, Cortinovis R, Alberici F, Rampoldi L, Scolari F, Izzi C. Autosomal Dominant Tubulointerstitial Kidney Disease: An Emerging Cause of Genetic CKD. Kidney international reports. 2022 Nov:7(11):2332-2344. doi: 10.1016/j.ekir.2022.08.012. Epub 2022 Aug 29 [PubMed PMID: 36531871]
Kalyoussef E, Hwang J, Prasad V, Barone J. Segmental multicystic dysplastic kidney in children. Urology. 2006 Nov:68(5):1121.e9-11 [PubMed PMID: 17095057]
Level 3 (low-level) evidenceSekine A, Hidaka S, Moriyama T, Shikida Y, Shimazu K, Ishikawa E, Uchiyama K, Kataoka H, Kawano H, Kurashige M, Sato M, Suwabe T, Nakatani S, Otsuka T, Kai H, Katayama K, Makabe S, Manabe S, Shimabukuro W, Nakanishi K, Nishio S, Hattanda F, Hanaoka K, Miura K, Hayashi H, Hoshino J, Tsuchiya K, Mochizuki T, Horie S, Narita I, Muto S. Cystic Kidney Diseases That Require a Differential Diagnosis from Autosomal Dominant Polycystic Kidney Disease (ADPKD). Journal of clinical medicine. 2022 Nov 3:11(21):. doi: 10.3390/jcm11216528. Epub 2022 Nov 3 [PubMed PMID: 36362756]
Scandling JD. Acquired cystic kidney disease and renal cell cancer after transplantation: time to rethink screening? Clinical journal of the American Society of Nephrology : CJASN. 2007 Jul:2(4):621-2 [PubMed PMID: 17699473]
Rabelo EA, Oliveira EA, Diniz JS, Silva JM, Filgueiras MT, Pezzuti IL, Tatsuo ES. Natural history of multicystic kidney conservatively managed: a prospective study. Pediatric nephrology (Berlin, Germany). 2004 Oct:19(10):1102-7 [PubMed PMID: 15258845]
Society for Maternal-Fetal Medicine (SMFM), Chetty S. Multicystic dysplastic kidney. American journal of obstetrics and gynecology. 2021 Nov:225(5):B21-B22. doi: 10.1016/j.ajog.2021.06.046. Epub 2021 Sep 8 [PubMed PMID: 34507790]
Pfau A, Knauf F. Update on Nephrolithiasis: Core Curriculum 2016. American journal of kidney diseases : the official journal of the National Kidney Foundation. 2016 Dec:68(6):973-985. doi: 10.1053/j.ajkd.2016.05.016. Epub 2016 Aug 3 [PubMed PMID: 27497526]
Imam TH, Patail H, Patail H. Medullary Sponge Kidney: Current Perspectives. International journal of nephrology and renovascular disease. 2019:12():213-218. doi: 10.2147/IJNRD.S169336. Epub 2019 Sep 26 [PubMed PMID: 31576161]
Level 3 (low-level) evidenceGranata S, Bruschi M, Candiano G, Catalano V, Ghiggeri GM, Stallone G, Zaza G. Proteomics Insights into Medullary Sponge Kidney Disease: Review of the Recent Results of an Italian Research Collaborative Network. Kidney & blood pressure research. 2022:47(12):683-692. doi: 10.1159/000527195. Epub 2022 Oct 20 [PubMed PMID: 36265463]
Waingankar N, Hayek S, Smith AD, Okeke Z. Calyceal diverticula: a comprehensive review. Reviews in urology. 2014:16(1):29-43 [PubMed PMID: 24791153]
Harris PC, Torres VE. Polycystic kidney disease. Annual review of medicine. 2009:60():321-37. doi: 10.1146/annurev.med.60.101707.125712. Epub [PubMed PMID: 18947299]
Level 3 (low-level) evidenceLiebau MC, Mekahli D, Perrone R, Soyfer B, Fedeles S. Polycystic Kidney Disease Drug Development: A Conference Report. Kidney medicine. 2023 Mar:5(3):100596. doi: 10.1016/j.xkme.2022.100596. Epub 2022 Dec 27 [PubMed PMID: 36698747]
Finnigan NA, Leslie SW. Polycystic Kidney Disease In Adults. StatPearls. 2023 Jan:(): [PubMed PMID: 29261941]
Chapman AB, Devuyst O, Eckardt KU, Gansevoort RT, Harris T, Horie S, Kasiske BL, Odland D, Pei Y, Perrone RD, Pirson Y, Schrier RW, Torra R, Torres VE, Watnick T, Wheeler DC, Conference Participants. Autosomal-dominant polycystic kidney disease (ADPKD): executive summary from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney international. 2015 Jul:88(1):17-27. doi: 10.1038/ki.2015.59. Epub 2015 Mar 18 [PubMed PMID: 25786098]
Reed B, McFann K, Kimberling WJ, Pei Y, Gabow PA, Christopher K, Petersen E, Kelleher C, Fain PR, Johnson A, Schrier RW. Presence of de novo mutations in autosomal dominant polycystic kidney disease patients without family history. American journal of kidney diseases : the official journal of the National Kidney Foundation. 2008 Dec:52(6):1042-50. doi: 10.1053/j.ajkd.2008.05.015. Epub 2008 Jul 21 [PubMed PMID: 18640754]
Guay-Woodford LM. Renal cystic diseases: diverse phenotypes converge on the cilium/centrosome complex. Pediatric nephrology (Berlin, Germany). 2006 Oct:21(10):1369-76 [PubMed PMID: 16823577]
Saunier S, Salomon R, Antignac C. Nephronophthisis. Current opinion in genetics & development. 2005 Jun:15(3):324-31 [PubMed PMID: 15917209]
Level 3 (low-level) evidenceTorres VE, Harris PC. Autosomal dominant polycystic kidney disease: the last 3 years. Kidney international. 2009 Jul:76(2):149-68. doi: 10.1038/ki.2009.128. Epub 2009 May 20 [PubMed PMID: 19455193]
Xue C, Mei CL. Polycystic Kidney Disease and Renal Fibrosis. Advances in experimental medicine and biology. 2019:1165():81-100. doi: 10.1007/978-981-13-8871-2_5. Epub [PubMed PMID: 31399962]
Level 3 (low-level) evidenceGast C, Marinaki A, Arenas-Hernandez M, Campbell S, Seaby EG, Pengelly RJ, Gale DP, Connor TM, Bunyan DJ, Hodaňová K, Živná M, Kmoch S, Ennis S, Venkat-Raman G. Autosomal dominant tubulointerstitial kidney disease-UMOD is the most frequent non polycystic genetic kidney disease. BMC nephrology. 2018 Oct 30:19(1):301. doi: 10.1186/s12882-018-1107-y. Epub 2018 Oct 30 [PubMed PMID: 30376835]
Shamam YM, Hashmi MF. Autosomal Dominant Tubulointerstitial Kidney Disease. StatPearls. 2024 Jan:(): [PubMed PMID: 33760469]
Ishikawa I. Uremic acquired renal cystic disease. Natural history and complications. Nephron. 1991:58(3):257-67 [PubMed PMID: 1896090]
Dell KM. The role of cilia in the pathogenesis of cystic kidney disease. Current opinion in pediatrics. 2015 Apr:27(2):212-8. doi: 10.1097/MOP.0000000000000187. Epub [PubMed PMID: 25575298]
Level 3 (low-level) evidenceHildebrandt F, Attanasio M, Otto E. Nephronophthisis: disease mechanisms of a ciliopathy. Journal of the American Society of Nephrology : JASN. 2009 Jan:20(1):23-35. doi: 10.1681/ASN.2008050456. Epub 2008 Dec 31 [PubMed PMID: 19118152]
Fliegauf M, Benzing T, Omran H. When cilia go bad: cilia defects and ciliopathies. Nature reviews. Molecular cell biology. 2007 Nov:8(11):880-93 [PubMed PMID: 17955020]
Level 3 (low-level) evidenceWilson PD, Goilav B. Cystic disease of the kidney. Annual review of pathology. 2007:2():341-68 [PubMed PMID: 18039103]
Level 3 (low-level) evidenceMcConnachie DJ, Stow JL, Mallett AJ. Ciliopathies and the Kidney: A Review. American journal of kidney diseases : the official journal of the National Kidney Foundation. 2021 Mar:77(3):410-419. doi: 10.1053/j.ajkd.2020.08.012. Epub 2020 Oct 9 [PubMed PMID: 33039432]
Živná M, Kidd KO, Barešová V, Hůlková H, Kmoch S, Bleyer AJ Sr. Autosomal dominant tubulointerstitial kidney disease: A review. American journal of medical genetics. Part C, Seminars in medical genetics. 2022 Sep:190(3):309-324. doi: 10.1002/ajmg.c.32008. Epub 2022 Oct 17 [PubMed PMID: 36250282]
Li X, Wüthrich RP, Kistler AD, Rodriguez D, Kapoor S, Mei C. Blood Pressure Control for Polycystic Kidney Disease. Polycystic Kidney Disease. 2015 Nov:(): [PubMed PMID: 27512778]
Chapman AB, Johnson A, Gabow PA, Schrier RW. The renin-angiotensin-aldosterone system and autosomal dominant polycystic kidney disease. The New England journal of medicine. 1990 Oct 18:323(16):1091-6 [PubMed PMID: 2215576]
Torres VE, Harris PC. Mechanisms of Disease: autosomal dominant and recessive polycystic kidney diseases. Nature clinical practice. Nephrology. 2006 Jan:2(1):40-55; quiz 55 [PubMed PMID: 16932388]
Level 3 (low-level) evidenceReiterová J, Tesař V. Autosomal Dominant Polycystic Kidney Disease: From Pathophysiology of Cystogenesis to Advances in the Treatment. International journal of molecular sciences. 2022 Mar 19:23(6):. doi: 10.3390/ijms23063317. Epub 2022 Mar 19 [PubMed PMID: 35328738]
Level 3 (low-level) evidenceTorres VE, Harris PC, Pirson Y. Autosomal dominant polycystic kidney disease. Lancet (London, England). 2007 Apr 14:369(9569):1287-1301. doi: 10.1016/S0140-6736(07)60601-1. Epub [PubMed PMID: 17434405]
Level 3 (low-level) evidenceHartung EA, Guay-Woodford LM. Autosomal recessive polycystic kidney disease: a hepatorenal fibrocystic disorder with pleiotropic effects. Pediatrics. 2014 Sep:134(3):e833-45. doi: 10.1542/peds.2013-3646. Epub 2014 Aug 11 [PubMed PMID: 25113295]
Level 3 (low-level) evidenceGuay-Woodford LM, Desmond RA. Autosomal recessive polycystic kidney disease: the clinical experience in North America. Pediatrics. 2003 May:111(5 Pt 1):1072-80 [PubMed PMID: 12728091]
Level 3 (low-level) evidenceBergmann C, Senderek J, Windelen E, Küpper F, Middeldorf I, Schneider F, Dornia C, Rudnik-Schöneborn S, Konrad M, Schmitt CP, Seeman T, Neuhaus TJ, Vester U, Kirfel J, Büttner R, Zerres K, APN (Arbeitsgemeinschaft für Pädiatrische Nephrologie). Clinical consequences of PKHD1 mutations in 164 patients with autosomal-recessive polycystic kidney disease (ARPKD). Kidney international. 2005 Mar:67(3):829-48 [PubMed PMID: 15698423]
Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, Burgmaier K, Gimpel C, Schaefer F, Liebau M. Autosomal Recessive Polycystic Kidney Disease – PKHD1. GeneReviews(®). 1993:(): [PubMed PMID: 20301501]
Chinali M, Lucchetti L, Ricotta A, Esposito C, D'Anna C, Rinelli G, Emma F, Massella L. Cardiac Abnormalities in Children with Autosomal Recessive Polycystic Kidney Disease. Cardiorenal medicine. 2019:9(3):180-189. doi: 10.1159/000496473. Epub 2019 Mar 7 [PubMed PMID: 30844805]
Hartung EA, Matheson M, Lande MB, Dell KM, Guay-Woodford LM, Gerson AC, Warady BA, Hooper SR, Furth SL. Neurocognition in children with autosomal recessive polycystic kidney disease in the CKiD cohort study. Pediatric nephrology (Berlin, Germany). 2014 Oct:29(10):1957-65. doi: 10.1007/s00467-014-2816-5. Epub 2014 May 15 [PubMed PMID: 24828609]
Level 2 (mid-level) evidenceWataya-Kaneda M, Tanaka M, Hamasaki T, Katayama I. Trends in the prevalence of tuberous sclerosis complex manifestations: an epidemiological study of 166 Japanese patients. PloS one. 2013:8(5):e63910. doi: 10.1371/journal.pone.0063910. Epub 2013 May 17 [PubMed PMID: 23691114]
Level 2 (mid-level) evidenceMikhail MI, Singh AK. Von Hippel-Lindau Syndrome. StatPearls. 2024 Jan:(): [PubMed PMID: 29083737]
Aufforth RD, Ramakant P, Sadowski SM, Mehta A, Trebska-McGowan K, Nilubol N, Pacak K, Kebebew E. Pheochromocytoma Screening Initiation and Frequency in von Hippel-Lindau Syndrome. The Journal of clinical endocrinology and metabolism. 2015 Dec:100(12):4498-504. doi: 10.1210/jc.2015-3045. Epub 2015 Oct 9 [PubMed PMID: 26451910]
Garfield K, Leslie SW. Simple Renal Cyst. StatPearls. 2024 Jan:(): [PubMed PMID: 29763075]
Sigmon DF, Shikhman R, Nielson JL. Renal Cyst. StatPearls. 2024 Jan:(): [PubMed PMID: 29261909]
Erlich T, Lipsky AM, Braga LH. A meta-analysis of the incidence and fate of contralateral vesicoureteral reflux in unilateral multicystic dysplastic kidney. Journal of pediatric urology. 2019 Feb:15(1):77.e1-77.e7. doi: 10.1016/j.jpurol.2018.10.023. Epub 2018 Nov 3 [PubMed PMID: 30482499]
Level 1 (high-level) evidenceSrivastava A, Patel N. Autosomal dominant polycystic kidney disease. American family physician. 2014 Sep 1:90(5):303-7 [PubMed PMID: 25251090]
Gaur P, Gedroyc W, Hill P. ADPKD-what the radiologist should know. The British journal of radiology. 2019 Jun:92(1098):20190078. doi: 10.1259/bjr.20190078. Epub 2019 Apr 30 [PubMed PMID: 31039325]
Avni FE, Garel C, Cassart M, D'Haene N, Hall M, Riccabona M. Imaging and classification of congenital cystic renal diseases. AJR. American journal of roentgenology. 2012 May:198(5):1004-13. doi: 10.2214/AJR.11.8083. Epub [PubMed PMID: 22528889]
Rajanna DK, Reddy A, Srinivas NS, Aneja A. Autosomal recessive polycystic kidney disease: antenatal diagnosis and histopathological correlation. Journal of clinical imaging science. 2013:3():13. doi: 10.4103/2156-7514.109733. Epub 2013 Mar 29 [PubMed PMID: 23814685]
Israel GM, Bosniak MA. An update of the Bosniak renal cyst classification system. Urology. 2005 Sep:66(3):484-8 [PubMed PMID: 16140062]
Silverman SG, Pedrosa I, Ellis JH, Hindman NM, Schieda N, Smith AD, Remer EM, Shinagare AB, Curci NE, Raman SS, Wells SA, Kaffenberger SD, Wang ZJ, Chandarana H, Davenport MS. Bosniak Classification of Cystic Renal Masses, Version 2019: An Update Proposal and Needs Assessment. Radiology. 2019 Aug:292(2):475-488. doi: 10.1148/radiol.2019182646. Epub 2019 Jun 18 [PubMed PMID: 31210616]
Gimpel C, Avni EF, Breysem L, Burgmaier K, Caroli A, Cetiner M, Haffner D, Hartung EA, Franke D, König J, Liebau MC, Mekahli D, Ong ACM, Pape L, Titieni A, Torra R, Winyard PJD, Schaefer F. Imaging of Kidney Cysts and Cystic Kidney Diseases in Children: An International Working Group Consensus Statement. Radiology. 2019 Mar:290(3):769-782. doi: 10.1148/radiol.2018181243. Epub 2019 Jan 1 [PubMed PMID: 30599104]
Level 3 (low-level) evidenceGunay-Aygun M, Font-Montgomery E, Lukose L, Tuchman M, Graf J, Bryant JC, Kleta R, Garcia A, Edwards H, Piwnica-Worms K, Adams D, Bernardini I, Fischer RE, Krasnewich D, Oden N, Ling A, Quezado Z, Zak C, Daryanani KT, Turkbey B, Choyke P, Guay-Woodford LM, Gahl WA. Correlation of kidney function, volume and imaging findings, and PKHD1 mutations in 73 patients with autosomal recessive polycystic kidney disease. Clinical journal of the American Society of Nephrology : CJASN. 2010 Jun:5(6):972-84. doi: 10.2215/CJN.07141009. Epub 2010 Apr 22 [PubMed PMID: 20413436]
Irazabal MV, Abebe KZ, Bae KT, Perrone RD, Chapman AB, Schrier RW, Yu AS, Braun WE, Steinman TI, Harris PC, Flessner MF, Torres VE, HALT Investigators. Prognostic enrichment design in clinical trials for autosomal dominant polycystic kidney disease: the HALT-PKD clinical trial. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 2017 Nov 1:32(11):1857-1865. doi: 10.1093/ndt/gfw294. Epub [PubMed PMID: 27484667]
Torres VE. Salt, water, and vasopressin in polycystic kidney disease. Kidney international. 2020 Oct:98(4):831-834. doi: 10.1016/j.kint.2020.06.001. Epub [PubMed PMID: 32998813]
Torres VE, Bankir L, Grantham JJ. A case for water in the treatment of polycystic kidney disease. Clinical journal of the American Society of Nephrology : CJASN. 2009 Jun:4(6):1140-50. doi: 10.2215/CJN.00790209. Epub 2009 May 14 [PubMed PMID: 19443627]
Level 3 (low-level) evidenceBarash I, Ponda MP, Goldfarb DS, Skolnik EY. A pilot clinical study to evaluate changes in urine osmolality and urine cAMP in response to acute and chronic water loading in autosomal dominant polycystic kidney disease. Clinical journal of the American Society of Nephrology : CJASN. 2010 Apr:5(4):693-7. doi: 10.2215/CJN.04180609. Epub 2010 Feb 18 [PubMed PMID: 20167686]
Level 3 (low-level) evidenceKramers BJ, Koorevaar IW, Drenth JPH, de Fijter JW, Neto AG, Peters DJM, Vart P, Wetzels JF, Zietse R, Gansevoort RT, Meijer E. Salt, but not protein intake, is associated with accelerated disease progression in autosomal dominant polycystic kidney disease. Kidney international. 2020 Oct:98(4):989-998. doi: 10.1016/j.kint.2020.04.053. Epub 2020 Jun 10 [PubMed PMID: 32534051]
Torres VE, Abebe KZ, Schrier RW, Perrone RD, Chapman AB, Yu AS, Braun WE, Steinman TI, Brosnahan G, Hogan MC, Rahbari FF, Grantham JJ, Bae KT, Moore CG, Flessner MF. Dietary salt restriction is beneficial to the management of autosomal dominant polycystic kidney disease. Kidney international. 2017 Feb:91(2):493-500. doi: 10.1016/j.kint.2016.10.018. Epub 2016 Dec 16 [PubMed PMID: 27993381]
Torres VE. Pro: Tolvaptan delays the progression of autosomal dominant polycystic kidney disease. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 2019 Jan 1:34(1):30-34. doi: 10.1093/ndt/gfy297. Epub [PubMed PMID: 30312438]
Masuda H, Shimizu N, Sekine K, Okato A, Hou K, Suyama T, Araki K, Kojima S, Naya Y. Efficacy and Safety of Tolvaptan for Patients With Autosomal Dominant Polycystic Kidney Disease in Real-world Practice: A Single Institution Retrospective Study. In vivo (Athens, Greece). 2023 Mar-Apr:37(2):801-805. doi: 10.21873/invivo.13144. Epub [PubMed PMID: 36881088]
Level 2 (mid-level) evidenceZhou JX, Torres VE. Drug repurposing in autosomal dominant polycystic kidney disease. Kidney international. 2023 May:103(5):859-871. doi: 10.1016/j.kint.2023.02.010. Epub 2023 Mar 2 [PubMed PMID: 36870435]
Raina R, Houry A, Rath P, Mangat G, Pandher D, Islam M, Khattab AG, Kalout JK, Bagga S. Clinical Utility and Tolerability of Tolvaptan in the Treatment of Autosomal Dominant Polycystic Kidney Disease (ADPKD). Drug, healthcare and patient safety. 2022:14():147-159. doi: 10.2147/DHPS.S338050. Epub 2022 Sep 8 [PubMed PMID: 36105663]
Müller RU, Messchendorp AL, Birn H, Capasso G, Cornec-Le Gall E, Devuyst O, van Eerde A, Guirchoun P, Harris T, Hoorn EJ, Knoers NVAM, Korst U, Mekahli D, Le Meur Y, Nijenhuis T, Ong ACM, Sayer JA, Schaefer F, Servais A, Tesar V, Torra R, Walsh SB, Gansevoort RT. An update on the use of tolvaptan for autosomal dominant polycystic kidney disease: consensus statement on behalf of the ERA Working Group on Inherited Kidney Disorders, the European Rare Kidney Disease Reference Network and Polycystic Kidney Disease International. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 2022 Apr 25:37(5):825-839. doi: 10.1093/ndt/gfab312. Epub [PubMed PMID: 35134221]
Level 3 (low-level) evidenceChebib FT, Perrone RD, Chapman AB, Dahl NK, Harris PC, Mrug M, Mustafa RA, Rastogi A, Watnick T, Yu ASL, Torres VE. A Practical Guide for Treatment of Rapidly Progressive ADPKD with Tolvaptan. Journal of the American Society of Nephrology : JASN. 2018 Oct:29(10):2458-2470. doi: 10.1681/ASN.2018060590. Epub 2018 Sep 18 [PubMed PMID: 30228150]
Guay-Woodford LM, Bissler JJ, Braun MC, Bockenhauer D, Cadnapaphornchai MA, Dell KM, Kerecuk L, Liebau MC, Alonso-Peclet MH, Shneider B, Emre S, Heller T, Kamath BM, Murray KF, Moise K, Eichenwald EE, Evans J, Keller RL, Wilkins-Haug L, Bergmann C, Gunay-Aygun M, Hooper SR, Hardy KK, Hartung EA, Streisand R, Perrone R, Moxey-Mims M. Consensus expert recommendations for the diagnosis and management of autosomal recessive polycystic kidney disease: report of an international conference. The Journal of pediatrics. 2014 Sep:165(3):611-7. doi: 10.1016/j.jpeds.2014.06.015. Epub 2014 Jul 9 [PubMed PMID: 25015577]
Level 3 (low-level) evidenceMekahli D, Liebau MC, Cadnapaphornchai MA, Goldstein SL, Greenbaum LA, Litwin M, Seeman T, Schaefer F, Guay-Woodford LM. Design of two ongoing clinical trials of tolvaptan in the treatment of pediatric patients with autosomal recessive polycystic kidney disease. BMC nephrology. 2023 Feb 13:24(1):33. doi: 10.1186/s12882-023-03072-x. Epub 2023 Feb 13 [PubMed PMID: 36782137]
Schwarz A, Vatandaslar S, Merkel S, Haller H. Renal cell carcinoma in transplant recipients with acquired cystic kidney disease. Clinical journal of the American Society of Nephrology : CJASN. 2007 Jul:2(4):750-6 [PubMed PMID: 17699492]
Emre H, Turgay A, Ali A, Murat B, Ozgür Y, Cankon G. 'Stepped procedure' in laparoscopic cyst decortication during the learning period of laparoscopic surgery: Detailed evaluation of initial experiences. Journal of minimal access surgery. 2010 Apr:6(2):37-41. doi: 10.4103/0972-9941.65162. Epub [PubMed PMID: 20814509]
Tsukamoto S, Urate S, Yamada T, Azushima K, Yamaji T, Kinguchi S, Uneda K, Kanaoka T, Wakui H, Tamura K. Comparative Efficacy of Pharmacological Treatments for Adults With Autosomal Dominant Polycystic Kidney Disease: A Systematic Review and Network Meta-Analysis of Randomized Controlled Trials. Frontiers in pharmacology. 2022:13():885457. doi: 10.3389/fphar.2022.885457. Epub 2022 May 18 [PubMed PMID: 35662736]
Level 1 (high-level) evidenceRichards T, Modarage K, Malik SA, Goggolidou P. The cellular pathways and potential therapeutics of Polycystic Kidney Disease. Biochemical Society transactions. 2021 Jun 30:49(3):1171-1188. doi: 10.1042/BST20200757. Epub [PubMed PMID: 34156429]
Woo DD, Miao SY, Pelayo JC, Woolf AS. Taxol inhibits progression of congenital polycystic kidney disease. Nature. 1994 Apr 21:368(6473):750-3 [PubMed PMID: 7908721]
Level 3 (low-level) evidenceGile RD, Cowley BD Jr, Gattone VH 2nd, O'Donnell MP, Swan SK, Grantham JJ. Effect of lovastatin on the development of polycystic kidney disease in the Han:SPRD rat. American journal of kidney diseases : the official journal of the National Kidney Foundation. 1995 Sep:26(3):501-7 [PubMed PMID: 7645559]
Level 3 (low-level) evidenceOmori S, Hida M, Fujita H, Takahashi H, Tanimura S, Kohno M, Awazu M. Extracellular signal-regulated kinase inhibition slows disease progression in mice with polycystic kidney disease. Journal of the American Society of Nephrology : JASN. 2006 Jun:17(6):1604-14 [PubMed PMID: 16641154]
Level 3 (low-level) evidenceJamal MH, Nunes ACF, Vaziri ND, Ramchandran R, Bacallao RL, Nauli AM, Nauli SM. Rapamycin treatment correlates changes in primary cilia expression with cell cycle regulation in epithelial cells. Biochemical pharmacology. 2020 Aug:178():114056. doi: 10.1016/j.bcp.2020.114056. Epub 2020 May 26 [PubMed PMID: 32470549]
Wahl PR, Serra AL, Le Hir M, Molle KD, Hall MN, Wüthrich RP. Inhibition of mTOR with sirolimus slows disease progression in Han:SPRD rats with autosomal dominant polycystic kidney disease (ADPKD). Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 2006 Mar:21(3):598-604 [PubMed PMID: 16221708]
Level 3 (low-level) evidenceTao Y, Kim J, Schrier RW, Edelstein CL. Rapamycin markedly slows disease progression in a rat model of polycystic kidney disease. Journal of the American Society of Nephrology : JASN. 2005 Jan:16(1):46-51 [PubMed PMID: 15563559]
Level 3 (low-level) evidenceXue C, Zhang LM, Zhou C, Mei CL, Yu SQ. Effect of Statins on Renal Function and Total Kidney Volume in Autosomal Dominant Polycystic Kidney Disease. Kidney diseases (Basel, Switzerland). 2020 Nov:6(6):407-413. doi: 10.1159/000509087. Epub 2020 Jul 29 [PubMed PMID: 33313061]
Hogan MC, Masyuk TV. Concurrent Targeting of Vasopressin Receptor 2 and Somatostatin Receptors in Autosomal Dominant Polycystic Kidney Disease: A Promising Approach for Autosomal Dominant Polycystic Kidney Disease Treatment? Clinical journal of the American Society of Nephrology : CJASN. 2023 Feb 1:18(2):154-156. doi: 10.2215/CJN.0000000000000055. Epub 2023 Jan 16 [PubMed PMID: 36754002]
Capuano I, Buonanno P, Riccio E, Rizzo M, Pisani A. Tolvaptan vs. somatostatin in the treatment of ADPKD: A review of the literature. Clinical nephrology. 2022 Mar:97(3):131-140. doi: 10.5414/CN110510. Epub [PubMed PMID: 34846296]
Jing Y, Wu M, Zhang D, Chen D, Yang M, Mei S, He L, Gu J, Qi N, Fu L, Li L, Mei C. Triptolide delays disease progression in an adult rat model of polycystic kidney disease through the JAK2-STAT3 pathway. American journal of physiology. Renal physiology. 2018 Sep 1:315(3):F479-F486. doi: 10.1152/ajprenal.00329.2017. Epub 2018 Mar 7 [PubMed PMID: 29513074]
Level 2 (mid-level) evidenceSeliger SL, Watnick T, Althouse AD, Perrone RD, Abebe KZ, Hallows KR, Miskulin DC, Bae KT. Baseline Characteristics and Patient-Reported Outcomes of ADPKD Patients in the Multicenter TAME-PKD Clinical Trial. Kidney360. 2020 Dec 31:1(12):1363-1372. doi: 10.34067/KID.0004002020. Epub [PubMed PMID: 33768205]
Pastor-Soler NM, Li H, Pham J, Rivera D, Ho PY, Mancino V, Saitta B, Hallows KR. Metformin improves relevant disease parameters in an autosomal dominant polycystic kidney disease mouse model. American journal of physiology. Renal physiology. 2022 Jan 1:322(1):F27-F41. doi: 10.1152/ajprenal.00298.2021. Epub 2021 Nov 22 [PubMed PMID: 34806449]
Leonhard WN, Song X, Kanhai AA, Iliuta IA, Bozovic A, Steinberg GR, Peters DJM, Pei Y. Salsalate, but not metformin or canagliflozin, slows kidney cyst growth in an adult-onset mouse model of polycystic kidney disease. EBioMedicine. 2019 Sep:47():436-445. doi: 10.1016/j.ebiom.2019.08.041. Epub 2019 Aug 28 [PubMed PMID: 31473186]
Belibi FA, Wallace DP, Yamaguchi T, Christensen M, Reif G, Grantham JJ. The effect of caffeine on renal epithelial cells from patients with autosomal dominant polycystic kidney disease. Journal of the American Society of Nephrology : JASN. 2002 Nov:13(11):2723-9 [PubMed PMID: 12397042]
Grantham JJ, Uchic M, Cragoe EJ Jr, Kornhaus J, Grantham JA, Donoso V, Mangoo-Karim R, Evan A, McAteer J. Chemical modification of cell proliferation and fluid secretion in renal cysts. Kidney international. 1989 Jun:35(6):1379-89 [PubMed PMID: 2770116]
Level 3 (low-level) evidenceTesar V, Ciechanowski K, Pei Y, Barash I, Shannon M, Li R, Williams JH, Levisetti M, Arkin S, Serra A. Bosutinib versus Placebo for Autosomal Dominant Polycystic Kidney Disease. Journal of the American Society of Nephrology : JASN. 2017 Nov:28(11):3404-3413. doi: 10.1681/ASN.2016111232. Epub 2017 Aug 24 [PubMed PMID: 28838955]
Grantham JJ, Torres VE, Chapman AB, Guay-Woodford LM, Bae KT, King BF Jr, Wetzel LH, Baumgarten DA, Kenney PJ, Harris PC, Klahr S, Bennett WM, Hirschman GN, Meyers CM, Zhang X, Zhu F, Miller JP, CRISP Investigators. Volume progression in polycystic kidney disease. The New England journal of medicine. 2006 May 18:354(20):2122-30 [PubMed PMID: 16707749]
Ars E, Bernis C, Fraga G, Martínez V, Martins J, Ortiz A, Rodríguez-Pérez JC, Sans L, Torra R, Spanish Working Group on Inherited Kidney Disease. Spanish guidelines for the management of autosomal dominant polycystic kidney disease. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 2014 Sep:29 Suppl 4():iv95-105. doi: 10.1093/ndt/gfu186. Epub [PubMed PMID: 25165191]
Cornec-Le Gall E, Audrézet MP, Chen JM, Hourmant M, Morin MP, Perrichot R, Charasse C, Whebe B, Renaudineau E, Jousset P, Guillodo MP, Grall-Jezequel A, Saliou P, Férec C, Le Meur Y. Type of PKD1 mutation influences renal outcome in ADPKD. Journal of the American Society of Nephrology : JASN. 2013 May:24(6):1006-13. doi: 10.1681/ASN.2012070650. Epub 2013 Feb 21 [PubMed PMID: 23431072]
Level 2 (mid-level) evidenceMurphy EL, Dai F, Blount KL, Droher ML, Liberti L, Crews DC, Dahl NK. Revisiting racial differences in ESRD due to ADPKD in the United States. BMC nephrology. 2019 Feb 14:20(1):55. doi: 10.1186/s12882-019-1241-1. Epub 2019 Feb 14 [PubMed PMID: 30764782]
Woon C, Bielinski-Bradbury A, O'Reilly K, Robinson P. A systematic review of the predictors of disease progression in patients with autosomal dominant polycystic kidney disease. BMC nephrology. 2015 Aug 15:16():140. doi: 10.1186/s12882-015-0114-5. Epub 2015 Aug 15 [PubMed PMID: 26275819]
Level 1 (high-level) evidenceReed B, Helal I, McFann K, Wang W, Yan XD, Schrier RW. The impact of type II diabetes mellitus in patients with autosomal dominant polycystic kidney disease. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 2012 Jul:27(7):2862-5. doi: 10.1093/ndt/gfr744. Epub 2011 Dec 29 [PubMed PMID: 22207329]
Niemczyk M, Gradzik M, Niemczyk S, Bujko M, Gołębiowski M, Pączek L. Intracranial aneurysms in autosomal dominant polycystic kidney disease. AJNR. American journal of neuroradiology. 2013 Aug:34(8):1556-9. doi: 10.3174/ajnr.A3456. Epub 2013 Feb 28 [PubMed PMID: 23449651]
Gimpel C, Bergmann C, Bockenhauer D, Breysem L, Cadnapaphornchai MA, Cetiner M, Dudley J, Emma F, Konrad M, Harris T, Harris PC, König J, Liebau MC, Marlais M, Mekahli D, Metcalfe AM, Oh J, Perrone RD, Sinha MD, Titieni A, Torra R, Weber S, Winyard PJD, Schaefer F. International consensus statement on the diagnosis and management of autosomal dominant polycystic kidney disease in children and young people. Nature reviews. Nephrology. 2019 Nov:15(11):713-726. doi: 10.1038/s41581-019-0155-2. Epub [PubMed PMID: 31118499]
Level 3 (low-level) evidenceCornec-Le Gall E, Alam A, Perrone RD. Autosomal dominant polycystic kidney disease. Lancet (London, England). 2019 Mar 2:393(10174):919-935. doi: 10.1016/S0140-6736(18)32782-X. Epub 2019 Feb 25 [PubMed PMID: 30819518]
Odedra D, Sabongui S, Khalili K, Schieda N, Pei Y, Krishna S. Autosomal Dominant Polycystic Kidney Disease: Role of Imaging in Diagnosis and Management. Radiographics : a review publication of the Radiological Society of North America, Inc. 2023 Jan:43(1):e220126. doi: 10.1148/rg.220126. Epub [PubMed PMID: 36459494]
Bae KT, Tao C, Feldman R, Yu ASL, Torres VE, Perrone RD, Chapman AB, Brosnahan G, Steinman TI, Braun WE, Mrug M, Bennett WM, Harris PC, Srivastava A, Landsittel DP, Abebe KZ, CRISP and HALT PKD Consortium. Volume Progression and Imaging Classification of Polycystic Liver in Early Autosomal Dominant Polycystic Kidney Disease. Clinical journal of the American Society of Nephrology : CJASN. 2022 Mar:17(3):374-384. doi: 10.2215/CJN.08660621. Epub 2022 Feb 25 [PubMed PMID: 35217526]
Pisani I, Giacosa R, Giuliotti S, Moretto D, Regolisti G, Cantarelli C, Vaglio A, Fiaccadori E, Manenti L. Ultrasound to address medullary sponge kidney: a retrospective study. BMC nephrology. 2020 Oct 12:21(1):430. doi: 10.1186/s12882-020-02084-1. Epub 2020 Oct 12 [PubMed PMID: 33046028]
Level 2 (mid-level) evidence