Definition/Introduction
Bioavailability refers to the extent a substance or drug becomes completely available to its intended biological destination(s). More accurately, bioavailability is a measure of the rate and fraction of the initial dose of a drug that successfully reaches either; the site of action or the bodily fluid domain from which the drug's intended targets have unimpeded access.[1][2][3]
For most purposes, bioavailability is defined as the fraction of the active form of a drug that reaches systemic circulation unaltered. This definition assumes 100% of the active drug that enters systemic circulation will successfully reach the target site.[4] However, one should understand that this definition excludes drugs that do not require access to systemic circulation for function (eg, certain topical drugs). The bioavailability of these drugs is measured by different parameters discussed elsewhere.[2]
Bioavailability is an integral part of the pharmacokinetics paradigm. Pharmacokinetics is the study of drug movement through the body and is often represented by the acronym ABCD which stands for administration, bioavailability, clearance, and distribution. Administration refers to the route and dosing of a drug. Clearance is the active form of a drug being removed from the systemic circulation. Distribution measures how widely a drug can travel to fluid compartments of the body; this definition assumes distribution follows absorption if taken orally.[5]
The route of administration (ROA) and the drug dose can significantly impact both the rate and extent of bioavailability. The dose of a drug is indirectly proportional to its bioavailability (Equation 5). A drug with relatively low bioavailability requires a larger dose to reach the minimum effective concentration threshold. The various routes of administration each contain a unique capability to facilitate a certain plasma drug concentration for a certain length of time. In many cases, altering the route of administration calls for an alteration of the dosage. For example, an oral drug requires passage through the gastrointestinal (GI) system, subjecting it to intestinal absorption and hepatic first-pass metabolism.[4] On the contrary, an intravenous (IV) drug is assumed to be immediately delivered to the systemic circulation because it is not subject to absorption or first-pass metabolism to determine adequate dosage.
Drug clearance can be thought of as the metabolic and excretory factors on the rate and extent an active drug leaves the systemic circulation. Clearance is measured by the drug elimination rate divided by the plasma drug concentration. The drug elimination rate is classically categorized into a binary system. A drug is eliminated either by first-order or zero-order kinetics. In zero-order kinetics, a constant amount of a drug is eliminated over time regardless of plasma concentration. However, zero-order kinetics implies absorption and elimination can become saturated, potentially leading to toxicity. In first-order kinetics, a constant fraction of the drug is eliminated over time via the intrinsic half-life of the drug.
Further, first-order drug elimination is exponentially proportional to plasma concentration (unlike zero-order kinetics). This implies that drug elimination will exponentially increase when the drug has a higher plasma concentration. Therefore, providers should know which category of elimination the drugs they prescribe follow, as this will affect drug clearance and bioavailability. For drugs following first-order kinetics, accumulation can occur if doses are delivered too frequently. This could result in unintended supratherapeutic consequences and side effects.[6]
Together, bioavailability and clearance can be used to determine the steady-state concentration of a drug.[5] Steady-state concentration is the time frame in which the concentration of a drug in the plasma is constant. This occurs when the rate of a drug reaching systemic circulation is equal to the rate a drug is removed from systemic circulation.[6] Thus, disparities in factors affecting the respective medications' bioavailability are important to consider when assessing therapeutic efficacy. Factors that alter drug clearance will reliably alter bioavailability and steady-state concentration. Such is the case in renal diseases that inhibit the kidneys' ability to eliminate drugs in the urine. Any degree of failure to eliminate a drug may augment its bioavailability by maintaining a larger drug plasma concentration than would normally be expected over time.
In contrast with bioavailability which measures the rate and extent an active drug reaches the plasma of systemic circulation, distribution is a measure of the rate and extent a drug is delivered to the various compartments of the body; total body water, intracellular volume, extracellular volume, plasma volume, and blood volume. Drugs capable of venturing into multiple fluid compartments are considered in a multi-compartment distribution model. Drugs that are thought to immediately distribute to their target domains, and do not normally distribute to peripheral compartments, are considered part of the single-compartment model. In the single-compartment model, any reduction in plasma drug concentration is assumed to have resulted from drug elimination.[7]
The multi-compartment model is useful for tracking drug flow throughout the fluid compartments. In both models, distribution is referred to as the volume of distribution (Vd) since volume is a convenient metric to compartmentalize the distribution of solutes, including drugs. The volume of distribution can be an important indicator of changes in bioavailability. The volume of distribution can be determined instantaneously by the proportion of the total amount of a drug in the body compared to the plasma concentration of the drug at a given time.[7] (Equation 1):
Equation 1: Vd = total amount of drug in the body ÷ plasma drug concentration
Extrapolating from the equation, a drug with a larger Vd will have a larger distribution outside the central compartment (plasma systemic circulation). One must consider how the relative breadth of a drug's volume of distribution might affect the drug's potential bioavailability. To illustrate, a drug that readily flows across multiple compartments may not be ideal if the intention is to maximize the plasma drug concentration.
Foundational to how bioavailability is classically defined is that an intravenously administered active drug delivered directly into systemic circulation yields a bioavailability of 100%. The bioavailability (F) of a medication delivered via other routes of administration can be determined by the mass of the drug delivered to the plasma divided by the total mass of the drug administered (Equation 2):
Equation 2: F = mass of the drug delivered to the plasma ÷ total mass of the drug administered
In pharmacologic contexts, an area under the curve graph (AUC) plots the plasma concentration of a drug on the y-axis versus the time following drug administration on the x-axis (example shown in Figure 1).[8] The area under the curve is directly proportional to drug absorption. Recall that the bioavailability of any drug delivered intravenously is theoretically 100%, or 1. This allows for convenient calculation of the bioavailability of drugs not delivered intravenously. By dividing the area under the curve of a medication delivered orally, for example, by the area under the curve for the same dose of that same drug delivered intravenously, one may successfully calculate the bioavailability of the oral medication.[9]
Bioavailability can be derived from an area under the curve (AUC) graph (Equation 3), which can be observed in the associated Figure 1.[4] For clinical purposes, a conceptual understanding of an AUC graph is crucial.
Equation 3: F = AUC for X route of administration ÷ AUC for IV administration
Thus, bioavailability is measured on a continuous range from 0 to 1 but can be represented as a percentage.[4] As a memory device, "F" can be thought of as a "fraction" because bioavailability is a non-IV drug's AUC divided into its IV version.
Issues of Concern
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Issues of Concern
Limitations of current theoretical models of bioavailability do exist. Calculating bioavailability using AUC data assumes a constant drug clearance and a uniformly distributed concentration of the drug once it reaches the plasma. In all other cases, AUC data is unreliable.[10]
Oral drugs, unlike drugs with other ROAs (eg, IV medications), must undergo intestinal absorption and hepatic first-pass metabolism.[4] Many structural and physiological gastrointestinal (GI) alterations, such as GI surgery or chronic inflammatory intestinal conditions, affect this absorption, typically by reducing bioavailability.[11] Genetic polymorphisms of intestinal transporters that facilitate absorption (eg, P-glycoprotein 1) also affect drug bioavailability.[12] Verapamil, a calcium channel blocker that inhibits P-glycoprotein, has been shown to augment the plasma concentration of immunosuppressive drugs that utilize P-glycoprotein in their elimination, such as cyclosporine and tacrolimus, increasing the risk for toxicity.[13]
Following drug absorption into the intestines, drugs are delivered to the liver via the portal system. The liver is the site of first-pass metabolism. The bioavailability of a drug will be reduced proportionally to the fraction of the initial dose converted to inactive metabolites by liver enzymes.[14] Notably, hepatic cytochrome P450 metabolism can significantly alter drug bioavailability.[15]
Cytochrome P450 enzymes can be inhibited or induced by a concurrent drug, supplement, or food metabolism. Even non-prescription drugs have been demonstrated to interact with cytochrome P450 enzymes. The herbal drug St. John’s wort used most commonly for depression, has been shown to increase cytochrome P450 activity, reducing the plasma concentration, and therefore bioavailability, of other drugs (eg, warfarin) that are also metabolized by cytochrome P450 enzymes.[16]
These observed interactions have implications for providers inquiring about dietary supplements, over-the-counter (OTC) medications, and herbal drugs their patients take. Depending on interactions between the unique, temporal portfolio of substances and the target drug, bioavailability may be reduced or enhanced. Consideration of these interactions is critical in the prevention of undesirable clinical outcomes.
Clinical Significance
Both intrinsic and extrinsic variables can influence the bioavailability of a drug. Intrinsically, a drug's bioavailability can be affected by the drug's required metabolic steps to activation, the specificity of its target receptors, the patient's unique physiology (including phenotypic polymorphisms), the route of administration of the drug, and the site of drug absorption. Extrinsic variables affecting drug bioavailability include interactions with concurrent food or substance metabolic processes and drug interactions with medications.[17]
A common clinical experience where an appreciation for bioavailability becomes important is the interactions between warfarin and albumin in the plasma. Warfarin has a propensity for binding with human serum albumin. Albumin-warfarin binding inactivates warfarin and reduces the fraction of active, free warfarin in the systemic circulation. When albumin sequesters warfarin in the blood, both the clearance and bioavailability of warfarin are reduced.[18]
As previously mentioned, bioavailability refers to the fraction of active drug concentration in the plasma; albumin-bound warfarin is not an active form of warfarin. This phenomenon implies monitoring serum albumin and dietary choices (especially regarding protein) when evaluating an adequate warfarin dose to prevent toxicity or inefficacy.
The use of nitroglycerin in angina relief is another common clinical example that illustrates how differences in routes of administration with disparate bioavailabilities can affect clinical outcomes. Nitroglycerin delivered orally will be affected by the first-pass metabolism in the liver, reducing the rate and extent of the drug that reaches its target sites. In consequence, the therapeutic effect of oral nitroglycerin should be slower in development and more sustained. For these reasons, oral nitroglycerin is commonly prescribed to patients with coronary artery disease for prophylactic use.[19]
In contrast, oral administration of nitroglycerin might not be the most appropriate route of administration for the immediate relief of angina. Sublingual nitroglycerin diffuses immediately into the bloodstream, bypassing intestinal absorption and first-pass metabolism. Consequently, the drug's therapeutic effects are manifested as early as 2 minutes yet last only 10 to 15 minutes.[20] This is the rationale for using sublingual nitroglycerin to provide more immediate relief of anginal chest pain. Intravenous nitroglycerin can be used when the sublingual administration fails.[19]
Fortunately, many AUC and bioavailability data have been previously calculated in pharmacokinetic studies, which are available to providers. The clinical pearl is to use these numbers to make pharmacologic decisions such as dosage and schedule.
Factors such as dose (D), route of administration, bioavailability (F), and total clearance (CT) will affect AUC.[4] Accommodations for changes in these variables can be made with relatively simple calculations (Equation 4).[4]
Equation 4: AUC = (F x D) ÷ CT
Understanding bioavailability is important for the clinician to determine the most appropriate dose, route, schedule, and route of administration; a drug should be delivered in specific clinical scenarios. Bioavailability is integral in evaluating an appropriate loading and maintenance dose. The loading dose is part of the initiation of treatment and is typically higher than the maintenance dose. The intention is to frontload an adequate plasma drug concentration that the maintenance dose will subsequently maintain. If the volume of distribution (Vd), desired steady-state concentration (Css), and bioavailability (F) are known, one can calculate the loading dose (LD) using Equation 5. If the desired steady-state concentration (Css), dosing interval (DI), bioavailability (F), and clearance (CL) are known, the maintenance dose (MD) can be determined (Equation 6).[7]
Equation 5: LD = (Css x Vd) ÷ F
Equation 6: MD = (Css x CL x DI) ÷ F
Prescribing doses most appropriate for the clinical scenario is critical to patient outcomes. For example, an antimicrobial dose that cannot maintain a plasma concentration within the therapeutic window could fail to treat a patient's infection and contribute to antimicrobial resistance. Conversely, prescribing a dose too high could result in toxicities idiosyncratic to that drug. When considering the administration of a drug, the clinician must appreciate the relevance of bioavailability in each medication and patient interaction.
Nursing, Allied Health, and Interprofessional Team Interventions
As the diversity of drug delivery systems continues to expand to accommodate patient needs, the risk of prescribing errors will increase. Nuanced differences between medication labels or doses of the same drug with different routes of administration can lead to human error. In the most objective sense, the physiologic manifestation of these errors can be traced to aberrant changes in basic pharmacokinetic principles. A focus on bioavailability can serve as a logical hinge to consider drug-patient and drug-drug interactions.
The first step toward mitigating medical errors associated with bioavailability is a shared knowledge of the basic principles of bioavailability between members of the interprofessional healthcare team caring for the patient. Including this knowledge as part of staff training can be beneficial. Nurses with a solid understanding of bioavailability can use the patient’s history, vitals, and medication list more effectively and become another layer of protection against adverse drug events (ADEs). This knowledge may provide additional confidence to nurses administering drugs to patients.
With more opportunities for medical errors, the onus falls on creating and maintaining a system that mitigates these mistakes as best as possible. Open communication among interprofessional healthcare team members can significantly reduce the risk of prescription errors. Regularly utilizing pharmacists in pharmaceutical decision support has been shown to reduce the risk of adverse drug events.[21] [Level 3] Pharmacists can perform a medication reconciliation before drug administration. Pharmacist review is another safeguard against adverse drug events because their more detailed pharmaceutical expertise is well-equipped for surveying a wider scope of potential drug interactions.
After drug administration, scheduled checkpoints for patient monitoring can provide a safety net for unanticipated pharmacologic responses. One important checkpoint is obtaining laboratory values at regular intervals; this is a safety measure to ensure a prescribed drug is maintained within the therapeutic range. In addition to ensuring drug efficacy through adequate plasma drug concentration, this checkpoint can serve as a method to prevent drug accumulation and toxicity. Between these intervals, checking patient vitals more frequently in patients with complex illnesses and medication lists is useful for monitoring acute physiologic reactions secondary to changes in drug bioavailability.
Ultimately, obtaining and recording a complete medical history is critical in forming the breadth of medical background required to make safer prescription decisions. A previous study showed that 61% of patients (n=304) have one or more prescriptions not recorded.[22] [Level 3] This inadequacy reveals a significant vulnerability to patients in the context of potential adverse drug events. Further, nonprescription drugs, herbal supplements, and food products can alter drug bioavailability; such is the essence of special dietary monitoring of patients taking warfarin.[18] While augmented interprofessional healthcare teams can mitigate bioavailability-associated adverse patient outcomes, the systems are limited to the degree of medical and lifestyle knowledge about the patient and the drugs they take.
Media
(Click Image to Enlarge)
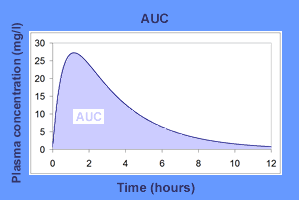
Area under the curve (AUC) graph example Contributed by Thierry Buclin, MD and Marc Sohrmann, PhD
References
Currie GM. Pharmacology, Part 2: Introduction to Pharmacokinetics. Journal of nuclear medicine technology. 2018 Sep:46(3):221-230. doi: 10.2967/jnmt.117.199638. Epub 2018 May 3 [PubMed PMID: 29724803]
Herkenne C, Alberti I, Naik A, Kalia YN, Mathy FX, Préat V, Guy RH. In vivo methods for the assessment of topical drug bioavailability. Pharmaceutical research. 2008 Jan:25(1):87-103 [PubMed PMID: 17985216]
Chow SC. Bioavailability and Bioequivalence in Drug Development. Wiley interdisciplinary reviews. Computational statistics. 2014:6(4):304-312 [PubMed PMID: 25215170]
Olivares-Morales A, Hatley OJ, Turner D, Galetin A, Aarons L, Rostami-Hodjegan A. The use of ROC analysis for the qualitative prediction of human oral bioavailability from animal data. Pharmaceutical research. 2014 Mar:31(3):720-30. doi: 10.1007/s11095-013-1193-2. Epub 2013 Sep 27 [PubMed PMID: 24072264]
Level 3 (low-level) evidenceDoogue MP, Polasek TM. The ABCD of clinical pharmacokinetics. Therapeutic advances in drug safety. 2013 Feb:4(1):5-7. doi: 10.1177/2042098612469335. Epub [PubMed PMID: 25083246]
Level 3 (low-level) evidenceWadhwa RR, Cascella M. Steady State Concentration. StatPearls. 2023 Jan:(): [PubMed PMID: 31985925]
Mansoor A, Mahabadi N. Volume of Distribution. StatPearls. 2023 Jan:(): [PubMed PMID: 31424864]
Kale P. Pharmacokinetics and bioavailability of single dose ibuprofen and pseudoephedrine alone or in combination: a randomized three-period, cross-over trial in healthy Indian volunteers. Frontiers in pharmacology. 2014:5():98. doi: 10.3389/fphar.2014.00098. Epub 2014 May 9 [PubMed PMID: 24847268]
Level 1 (high-level) evidenceScheff JD, Almon RR, Dubois DC, Jusko WJ, Androulakis IP. Assessment of pharmacologic area under the curve when baselines are variable. Pharmaceutical research. 2011 May:28(5):1081-9. doi: 10.1007/s11095-010-0363-8. Epub 2011 Jan 14 [PubMed PMID: 21234658]
Rescigno A. Area under the curve and bioavailability. Pharmacological research. 2000 Dec:42(6):539-40 [PubMed PMID: 11058405]
Level 3 (low-level) evidenceHatton GB, Madla CM, Rabbie SC, Basit AW. Gut reaction: impact of systemic diseases on gastrointestinal physiology and drug absorption. Drug discovery today. 2019 Feb:24(2):417-427. doi: 10.1016/j.drudis.2018.11.009. Epub 2018 Nov 16 [PubMed PMID: 30453059]
Dietrich CG, Geier A, Oude Elferink RP. ABC of oral bioavailability: transporters as gatekeepers in the gut. Gut. 2003 Dec:52(12):1788-95 [PubMed PMID: 14633964]
Yigitaslan S, Erol K, Cengelli C. The Effect of P-Glycoprotein Inhibition and Activation on the Absorption and Serum Levels of Cyclosporine and Tacrolimus in Rats. Advances in clinical and experimental medicine : official organ Wroclaw Medical University. 2016 Mar-Apr:25(2):237-42. doi: 10.17219/acem/35254. Epub [PubMed PMID: 27627555]
Level 3 (low-level) evidenceBecker DE. Drug therapy in dental practice: general principles. Part 1 - Pharmacokinetic considerations. Anesthesia progress. 2006 Winter:53(4):140-5; quiz 146 [PubMed PMID: 17177593]
Xie F, Ding X, Zhang QY. An update on the role of intestinal cytochrome P450 enzymes in drug disposition. Acta pharmaceutica Sinica. B. 2016 Sep:6(5):374-383 [PubMed PMID: 27709006]
Komoroski BJ, Zhang S, Cai H, Hutzler JM, Frye R, Tracy TS, Strom SC, Lehmann T, Ang CY, Cui YY, Venkataramanan R. Induction and inhibition of cytochromes P450 by the St. John's wort constituent hyperforin in human hepatocyte cultures. Drug metabolism and disposition: the biological fate of chemicals. 2004 May:32(5):512-8 [PubMed PMID: 15100173]
Martinez MN, Amidon GL. A mechanistic approach to understanding the factors affecting drug absorption: a review of fundamentals. Journal of clinical pharmacology. 2002 Jun:42(6):620-43 [PubMed PMID: 12043951]
Level 3 (low-level) evidenceFender AC, Dobrev D. Bound to bleed: How altered albumin binding may dictate warfarin treatment outcome. International journal of cardiology. Heart & vasculature. 2019 Mar:22():214-215. doi: 10.1016/j.ijcha.2019.02.007. Epub 2019 Mar 25 [PubMed PMID: 30963099]
Kim KH, Kerndt CC, Adnan G, Schaller DJ. Nitroglycerin. StatPearls. 2023 Jan:(): [PubMed PMID: 29494004]
Boden WE, Padala SK, Cabral KP, Buschmann IR, Sidhu MS. Role of short-acting nitroglycerin in the management of ischemic heart disease. Drug design, development and therapy. 2015:9():4793-805. doi: 10.2147/DDDT.S79116. Epub 2015 Aug 19 [PubMed PMID: 26316714]
Lesar TS. Prescribing errors involving medication dosage forms. Journal of general internal medicine. 2002 Aug:17(8):579-87 [PubMed PMID: 12213138]
Lau HS, Florax C, Porsius AJ, De Boer A. The completeness of medication histories in hospital medical records of patients admitted to general internal medicine wards. British journal of clinical pharmacology. 2000 Jun:49(6):597-603 [PubMed PMID: 10848724]