Introduction
An aortopulmonary (AP) septal defect, also known as an AP window, is one of the rarest congenital heart defects accounting for less than 0.5% of forms of congenital heart disease.[1] This defect can occur in isolation or concurrently with other forms of congenital heart disease, such as a ventricular septal defect, interruption of the aortic arch, tetralogy of Fallot, and, rarely, coronary artery anomalies.[2][3][4][5][6] By definition, the AP window is a direct "side to side" connection of the ascending aorta to the main pulmonary artery but with the formation of a normal aortic valve and a right ventricular outflow tract, differentiating it from truncus arteriosus (see Video. Echocardiogram of Aortopulmonary Window).[7] Embryologically an AP window develops when there is incomplete septation of the great arteries.
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
Aortopulmonary window defects account for significantly less than 0.5% of all congenital heart defects.[1] They are associated approximately 50% of the time with other congenital heart defects such as other conotruncal defects (eg, tetralogy of Fallot, interrupted aortic arch, D-transposition of the great arteries), coarctation of the aorta, ventricular septal defect, coronary artery anomalies, and tricuspid atresia.[2][3][4][5][6][7][8][9] While this defect would seem to be similar to other conotruncal malformations embryologically, there is a surprisingly low association with DiGeorge syndrome.[10][11] This suggests this is not related to an abnormality in the cardiac neural crest.[11]
Epidemiology
As discussed above, an AP window is a rare congenital heart defect occurring in less than 0.5% of all congenital heart defects.[1] Genetic associations such as VACTERL and Bohring-Opitz have been identified; however, no specific maternal exposures have been found to be an associative cause.[8][12]
Pathophysiology
An AP window occurs during embryonic life when there is incomplete septation of the common arterial trunk, allowing an abnormal connection between the ascending aorta and the main pulmonary artery. In an isolated AP window, the two separate semilunar valves (aortic and pulmonary) are typically formed normally.[13] The location of the window defect is between the semilunar valves and the branch pulmonary arteries. Three types of AP window are identified: type I (proximal), occurring between the posterior wall of the ascending aorta and lateral wall of the main pulmonary artery; type II (distal), occurring between the posterior wall of the ascending aorta and the anterior wall of the origin of the right pulmonary artery; and type III, which is a combination of type I and II. Type I is the most common type of AP window.[13] The size of the connection is variable but is usually large, unrestrictive, and hemodynamically significant. In less than 10% of the cases, the AP window is small and pressure-restrictive.[14]
History and Physical
While it is conceivable to diagnose an isolated AP window in utero by fetal echocardiography, the more typical presentation is in the neonatal period or early infancy.[15][16][17] The typical signs and symptoms associated with an AP window are pulmonary overcirculation occurring as the pulmonary vascular resistance falls over the first few weeks of life. This usually results in a large left-to-right shunt, and the symptoms would include diaphoresis (especially with feeding), tachypnea, tachycardia, poor weight gain, and increased respiratory symptoms with viral infections. The precordium is often hyperdynamic, and a mitral valve rumble can be appreciated. The pulses are often bounding as the systemic diastolic blood pressure is decreased secondary to aortic flow reversal in diastole. Rarely, there is a continuous murmur noted as the connection between the aorta, and the pulmonary artery is usually large, not allowing for a significant pressure gradient (restriction) to develop.
As noted above, an AP window can be associated with other types of congenital heart defects, and the associated defect can alter the presentation. In the tetralogy of Fallot, a pulmonary ejection murmur and pulmonary valve click can be noted. When the AP window occurs with an interrupted aortic arch, the neonate can present with shock as the ductus arteriosus constricts. Occasionally the AP window can be restrictive and present with less significant symptoms of overcirculation. In this scenario, a continuous heart murmur could be noted. Rarely, an AP window diagnosis is not made until later in childhood or even adult life. This could present with features of Eisenmenger syndrome, including cyanosis and clubbing.[18][19][20] However, a late presentation of the AP window still requires a detailed hemodynamic evaluation as some patients could still be candidates for repair if there is acceptable pulmonary vascular resistance or an appropriate response to vasodilator testing.[21][22]
Evaluation
In children with an AP window, a chest X-ray will show cardiomegaly and increased pulmonary vascular markings. An electrocardiogram will demonstrate tachycardia and increased right and left-sided voltages. The diagnosis of an AP window is made by echocardiography after suspicion of a large left-to-right shunt. Because the connection is usually without significant restriction, color Doppler echocardiography will not detect a high-velocity color jet. When the suspicion is high, the 2D images are usually sufficient to measure the AP window communication (see Video. Aortopulmonary Window). If echocardiogram imaging is insufficient, a CT scan can potentially delineate the AP connection (see Image. Aortopulmonary Window, Computed Tomography Scan).
Echocardiography should completely evaluate the remaining cardiac structures, including evaluating other cardiac diseases such as tetralogy of Fallot, and interruption of the aortic arch, and care should be taken to identify the coronary arteries. Unless the diagnosis is made late or cannot be made with less invasive means, cardiac catheterization adds little to the diagnostic management. When the diagnosis is made after infancy or even into adult life, especially if cyanosis is noted, catheterization could be utilized to evaluate pulmonary vascular resistance as Eisenmenger syndrome can develop. If there is pulmonary vascular disease, reactivity testing should be performed during catheterization to determine the advisability of closure.[22]
Treatment / Management
In general, the treatment for a large AP window is surgical patch closure, and the results are generally good when the repair occurs early in infancy.[23][24] Smaller defects can be surgically closed primarily with double ligation or suture closure.[24][25] Logically, associated congenital heart disease makes surgical repair more difficult and worsens outcomes.[23][26] Catheterization device closure with double disk closure devices (atrial septal defect and patent ductus arteriosus devices) has been described in case reports in defects that are typically restrictive, allowing for closure later in childhood.[27][28] (B2)
Anticongestive medications such as diuretics (eg, furosemide and chlorothiazide) and digoxin can provide temporary symptomatic improvement but should not significantly alter the course of the disease. Afterload reduction can be considered with angiotensin-converting enzyme inhibition. Medical therapy should be approached with caution, as there can be abnormal renal perfusion. Surgery should be considered at the time of diagnosis as there will likely be little growth with this physiology, and there is a risk of developing irreversible pulmonary hypertension over time. Furthermore, the AP window does not restrict or get hemodynamically less significant over time. In general, surgery involves separating the great arteries with either suture division or patch closure of the aorta and pulmonary artery, with low surgical mortality.
Catheterization can be considered when a defect is small enough to allow for device closure without causing stenosis of the great arteries or interference with the semilunar valves. If other cardiac defects are present, repair or palliation of the other defects should occur at the same operation. Postoperative aorta, rarely, and pulmonary artery stenosis in the main and branch pulmonary arteries, more commonly, can occur. If this stenosis is hemodynamically important, future cardiac catheterization with balloon angioplasty or stent implantation can be employed. Furthermore, in the setting of interrupted aortic arch repair, transcatheter aortic interventions for residual arch obstruction could be necessary.[26](B2)
Differential Diagnosis
- Truncus arteriosus is the most common lesion confused with an AP window but has a truncal valve instead of two semilunar valves. The early pathophysiology is similar.
- Patent ductus arteriosus (PDA) has similar physiology. A "window" type PDA is a connection between the proximal descending aorta and the left pulmonary artery near the bifurcation. The AP window is a connection between the ascending aorta and the main pulmonary artery.
- A large ventricular septal defect has similar physiology but should be easily distinguished from an AP window.
Prognosis
The prognosis for an isolated AP window is good when treated early with surgical closure and avoidance of the development of pulmonary vascular disease.[24] Late diagnosis can be associated with the development of pulmonary hypertension/Eisenmenger syndrome, which carries a poor prognosis. An associated AP window with other congenital heart lesions increases the complexity of the surgery.[23][26]
Complications
Complications of missed diagnosis include the development of Eisenmenger syndrome.[29] The repair can be associated with residual defects, including branch pulmonary artery stenosis and residual AP window that might need to be addressed in the future.[30] Caution should be used in balloon angioplasty as an AP window can theoretically recur with aggressive angioplasty. Recurrent laryngeal nerve injury should be suspected if hoarseness or choking with feeds is noted post-repair.[31]
Consultations
Treatment of AP window requires an interprofessional team approach. Generally, consultation should occur with cardiology, cardiac surgery, and cardiac intensive care to optimize outcomes.
Pearls and Other Issues
The diagnosis of an AP window is usually made by echocardiography. It cannot be emphasized enough that a high clinical suspicion of a large left-to-right shunt and bounding pulses should prompt an echocardiogram focusing on an evaluation for an AP window. Late repair of AP window is a risk for the persistence of substantial pulmonary hypertension, and preoperative catheterization is recommended in advance of late AP window repair. There should be a low threshold for postoperative cardiac catheterization for evaluating pulmonary vascular resistance. In general, early repair of an AP window results in an excellent long-term prognosis.
Enhancing Healthcare Team Outcomes
The diagnosis and management of AP window require an interprofessional team that includes a pediatrician, cardiologist, cardiac surgeon, intensivist, and radiologist. The disorder is typically managed with surgery, but the key is that the child does not have any other associated congenital heart defects. Once the surgery is performed, the infant is usually monitored by cardiac or critical care nurses. If this is an isolated defect, the outcomes are good and are typically good, even when associated with other congenital heart defects. Pharmacists should review the dosing of medications and provide teaching to families of patients. Specialty care nurses help coordinate care and update the team on changes in patient status. Indefinite long-term follow-up with cardiology is advised after successful repair to screen for late abnormalities such as branch pulmonary artery stenosis.
Media
(Click Video to Play)
Aortopulmonary Window. This 2-dimensional image of the aortopulmonary window is shown from the subcostal view.
Contributed by P Shivaram, MD
(Click Video to Play)
Echocardiogram of Aortopulmonary Window. Echocardiogram and color display of AP window demonstrating the flow between the aorta and pulmonary artery.
Contributed by P Shivaram, MD
(Click Image to Enlarge)
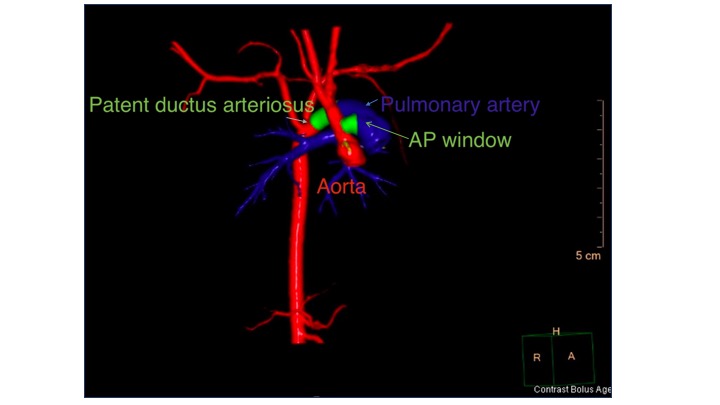
Aortopulmonary Window, Computed Tomography Scan. This 3-dimensional reconstruction demonstrates an aortopulmonary window.
Contributed by M Law, MD
References
Samánek M, Vorísková M. Congenital heart disease among 815,569 children born between 1980 and 1990 and their 15-year survival: a prospective Bohemia survival study. Pediatric cardiology. 1999 Nov-Dec:20(6):411-7 [PubMed PMID: 10556387]
Prabhu S, Keshav M, Ramachandra P, Raj V, John C, Karl TR. Tetralogy of Fallot with pulmonary atresia and aortopulmonary window may mimic common arterial trunk. Cardiology in the young. 2022 Mar:32(3):410-414. doi: 10.1017/S1047951121002298. Epub 2021 Jun 17 [PubMed PMID: 34134806]
Sondhi S, Negi PC, Sharma R, Mehta A. Aortopulmonary window with pumonary atresia with ventricular septal defect with D-transposition of great arteries: extremely rare anomaly. BMJ case reports. 2018 Jun 13:2018():. pii: bcr-2018-224401. doi: 10.1136/bcr-2018-224401. Epub 2018 Jun 13 [PubMed PMID: 29898907]
Level 3 (low-level) evidenceBin-Moallim M, Hamadah HK, Alhabshan F, Alghamdi AA, Kabbani MS. Aortopulmonary window: Types, associated cardiovascular anomalies, and surgical outcome. Retrospective analysis of a single center experience. Journal of the Saudi Heart Association. 2020:32(2):127-133. doi: 10.37616/2212-5043.20. Epub 2020 May 10 [PubMed PMID: 33154906]
Level 2 (mid-level) evidenceAlakhfash AA, Tagelden M, Almesned A, Alqwaiee A. Aortopulmonary window with anomalous right coronary artery from the pulmonary artery. Case report and literature review. Cardiology in the young. 2020 Jan:30(1):47-49. doi: 10.1017/S1047951119002543. Epub 2019 Dec 19 [PubMed PMID: 31854282]
Level 3 (low-level) evidenceGrünenfelder J, Zünd G, Vogt PR, Turina MI. Aortopulmonary window with anomalous origin of the right coronary artery. The Annals of thoracic surgery. 1999 Jan:67(1):233-5 [PubMed PMID: 10086558]
Level 3 (low-level) evidenceRajagopal R, Sinha M, Pandey NN, Bhambri K, Kumar S. Tetralogy of fallot with pulmonary atresia and aorto-pulmonary window: Or is it truncus arteriosus? Journal of cardiovascular computed tomography. 2020 Sep-Oct:14(5):e20-e21. doi: 10.1016/j.jcct.2018.10.022. Epub 2018 Oct 26 [PubMed PMID: 30385324]
Trowitzsch E, Schneider M, Urban A, Asfour B. Congenital pulmonary sling, aorto-pulmonary window and pulmonary vein obstruction as a diagnostic and therapeutic challenge in an infant with VACTERL association. Clinical research in cardiology : official journal of the German Cardiac Society. 2006 Jun:95(6):338-43 [PubMed PMID: 16598388]
Level 3 (low-level) evidenceMilovanovic V, Stefanovic I, Ilic S. Tricuspid atresia associated with aortopulmonary window: diagnostic and therapeutic dilemmas. Cardiology in the young. 2017 Apr:27(3):580-583. doi: 10.1017/S1047951116001499. Epub 2016 Sep 29 [PubMed PMID: 27680574]
Van Mierop LH, Kutsche LM. Cardiovascular anomalies in DiGeorge syndrome and importance of neural crest as a possible pathogenetic factor. The American journal of cardiology. 1986 Jul 1:58(1):133-7 [PubMed PMID: 3728313]
Kutsche LM, Van Mierop LH. Anatomy and pathogenesis of aorticopulmonary septal defect. The American journal of cardiology. 1987 Feb 15:59(5):443-7 [PubMed PMID: 3812313]
Verma B, Abhinay A, Singh A, Kumar M. Double outlet right ventricle and aortopulmonary window in a neonate with Bohring-Opitz (Oberklaid-Danks) syndrome: First case report. Journal of family medicine and primary care. 2019 Mar:8(3):1279-1281. doi: 10.4103/jfmpc.jfmpc_74_19. Epub [PubMed PMID: 31041292]
Level 3 (low-level) evidenceJacobs JP, Quintessenza JA, Gaynor JW, Burke RP, Mavroudis C. Congenital Heart Surgery Nomenclature and Database Project: aortopulmonary window. The Annals of thoracic surgery. 2000 Apr:69(4 Suppl):S44-9 [PubMed PMID: 10798415]
Awasthy N, Jawid SA. Aortopulmonary Window with Crisscross Pulmonary Arteries: Anatomically Type 1, Physiologically Type 2. Journal of cardiovascular echography. 2017 Oct-Dec:27(4):143-144. doi: 10.4103/jcecho.jcecho_12_17. Epub [PubMed PMID: 29142813]
Tongprasert F, Sittiwangkul R, Jatavan P, Tongsong T. Prenatal Diagnosis of Aortopulmonary Window: A Case Series and Literature Review. Journal of ultrasound in medicine : official journal of the American Institute of Ultrasound in Medicine. 2017 Aug:36(8):1733-1738. doi: 10.7863/ultra.16.08025. Epub 2017 Apr 10 [PubMed PMID: 28393388]
Level 2 (mid-level) evidenceCollinet P, Chatelet-Cheront C, Houze de l'Aulnoit D, Rey C. Prenatal diagnosis of an aorto-pulmonary window by fetal echocardiography. Fetal diagnosis and therapy. 2002 Sep-Oct:17(5):302-7 [PubMed PMID: 12169817]
Level 3 (low-level) evidenceDemir IH, Erdem A, Sarıtaş T, Demir F, Erol N, Yücel IK, Aydemir NA, Celebi A. Diagnosis, treatment and outcomes of patients with aortopulmonary window. Balkan medical journal. 2013 Jun:30(2):191-6. doi: 10.5152/balkanmedj.2013.6995. Epub 2013 Jun 1 [PubMed PMID: 25207099]
Chen J, Guo J, Cao S. Unexplained pulmonary hypertension: an overlooked aortopulmonary window. European heart journal. 2020 Mar 14:41(11):1217. doi: 10.1093/eurheartj/ehaa021. Epub [PubMed PMID: 32034904]
Myers PO, Lador F, Hachulla AL, Bouchardy J, Noble S, Licker M, Pache JC, Kalimanovaska-Ostric D, Djukic M, Kalangos A, Beghetti M. Unrestrictive Aortopulmonary Window: Extreme Presentation as Non-Eisenmenger in a 30-Year-Old Patient. Circulation. 2016 May 10:133(19):1907-10. doi: 10.1161/CIRCULATIONAHA.115.020819. Epub [PubMed PMID: 27166350]
Dev M, Sharma M, Rana N. Large Unrepaired Aortopulmonary Window Presenting in Adulthood. Current cardiology reviews. 2020:16(1):73-76. doi: 10.2174/1573403X15666190513105231. Epub [PubMed PMID: 31092183]
Talwar S, Siddharth B, Gupta SK, Choudhary SK, Kothari SS, Juneja R, Saxena A, Airan B. Aortopulmonary window: results of repair beyond infancy. Interactive cardiovascular and thoracic surgery. 2017 Nov 1:25(5):740-744. doi: 10.1093/icvts/ivx158. Epub [PubMed PMID: 28633352]
Zografos PM, Protopapas EM, Hakim NI, Alexopoulos C, Sarris GE. Remarkably Still Repairable Large Aortopulmonary Window in an Adult Patient. World journal for pediatric & congenital heart surgery. 2020 Jan:11(1):117-119. doi: 10.1177/2150135119878703. Epub 2019 Nov 22 [PubMed PMID: 31755375]
Gangana CS, Malheiros AF, Alves EV, de Azevedo MA, Bernardes RM, Simões LC. Aortopulmonary window--impact of associated lesions on surgical results. Arquivos brasileiros de cardiologia. 2007 Apr:88(4):402-7 [PubMed PMID: 17546268]
Level 2 (mid-level) evidenceGowda D, Gajjar T, Rao JN, Chavali P, Sirohi A, Pandarinathan N, Desai N. Surgical management of aortopulmonary window: 24 years of experience and lessons learned. Interactive cardiovascular and thoracic surgery. 2017 Aug 1:25(2):302-309. doi: 10.1093/icvts/ivx099. Epub [PubMed PMID: 28475712]
Talwar S, Agarwal P, Choudhary SK, Kothari SS, Juneja R, Saxena A, Airan B. Aortopulmonary window: Morphology, diagnosis, and long-term results. Journal of cardiac surgery. 2017 Feb:32(2):138-144. doi: 10.1111/jocs.12936. Epub 2017 Jan 30 [PubMed PMID: 28139013]
Konstantinov IE, Karamlou T, Williams WG, Quaegebeur JM, del Nido PJ, Spray TL, Caldarone CA, Blackstone EH, McCrindle BW, Congenital Heart Surgeons Society. Surgical management of aortopulmonary window associated with interrupted aortic arch: a Congenital Heart Surgeons Society study. The Journal of thoracic and cardiovascular surgery. 2006 May:131(5):1136-1141.e2 [PubMed PMID: 16678601]
Level 2 (mid-level) evidenceUçar T, Karagözlü S, Ramoğlu MG, Tutar E. Transcatheter closure of aortopulmonary window with Amplatzer duct occluder II: additional size. Cardiology in the young. 2020 Mar:30(3):424-426. doi: 10.1017/S1047951119003342. Epub 2020 Jan 13 [PubMed PMID: 31928548]
Trehan V, Nigam A, Tyagi S. Percutaneous closure of nonrestrictive aortopulmonary window in three infants. Catheterization and cardiovascular interventions : official journal of the Society for Cardiac Angiography & Interventions. 2008 Feb 15:71(3):405-11. doi: 10.1002/ccd.21366. Epub [PubMed PMID: 18288731]
Level 3 (low-level) evidenceEl Dick J, El-Rassi I, Tayeh C, Bitar F, Arabi M. Aortopulmonary window in adults: A rare entity leading to Eisenmenger syndrome. Echocardiography (Mount Kisco, N.Y.). 2019 Jun:36(6):1173-1178. doi: 10.1111/echo.14368. Epub 2019 May 22 [PubMed PMID: 31116466]
Barnes ME, Mitchell ME, Tweddell JS. Aortopulmonary window. Seminars in thoracic and cardiovascular surgery. Pediatric cardiac surgery annual. 2011:14(1):67-74. doi: 10.1053/j.pcsu.2011.01.017. Epub [PubMed PMID: 21444051]
Condon LM, Katkov H, Singh A, Helseth HK. Cardiovocal syndrome in infancy. Pediatrics. 1985 Jul:76(1):22-5 [PubMed PMID: 4011354]
Level 3 (low-level) evidence