
Introduction
Adrenal hypoplasia is a condition in which there is underdevelopment or hypotrophy of the adrenal cortex due to several clinical conditions. It can divide into two categories: primary adrenal hypoplasia and secondary adrenal hypoplasia. Primary adrenal hypoplasia is caused by the adrenal hypoplasia congenita (AHC) or intrinsic defect during adrenal gland development. Adrenal hypoplasia congenita involves hypofunctional and hypertrophic adrenal glands, which are caused by defects during differentiation of the fetal adrenal glands. It occurs in many forms. Secondary adrenal hypoplasia results from pituitary or hypothalamic dysfunction. These conditions cause a decreased synthesis and secretion of adrenocorticotropic hormone (ACTH), leading to inadequate adrenal development and adrenal dysfunction because of the reduced adrenal stimulus.[1][2][3]
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
Adrenal hypoplasia can be due to adrenal hypoplasia congenita (AHC). Affected patients present with adrenocortical insufficiency in infancy and childhood. Research has identified four forms of AHC.
- The X-linked form is the result of a mutation or deletion of the nuclear receptor protein DAX-1 gene located on the X-chromosome. NROB1 gene encodes for this DAX-1 protein, which is thought to be important in the development and function of the hypothalamic-pituitary-adrenal axis. There have also been suggestions that this chromosomal aberrancy could be a part of a contiguous chromosomal deletion of a large segment on the X-chromosome carrying the NROB1 gene. Such a large deletion can additionally result in Duchenne muscular dystrophy and glycerol kinase deficiency.
- The autosomal recessive form involves a gene that codes for steroidogenic factor 1 (SF-1). This gene is on chromosome 9q33. Another discovered form of the disease is associated with metaphysical dysplasia, intrauterine growth retardation, and genital abnormalities.
- IMAGE syndrome, SERKAL syndrome (rare autosomal form). Few other rare syndromes and mutations can lead to adrenal hypoplasia and aplasia.
- Autosomal recessive ACTH resistance syndrome, such as triple-A syndrome and familial glucocorticoid deficiency present with ACTH insensitivity.
Secondary adrenal hypoplasia can arise from pituitary or hypothalamic dysfunction. These include transcription factor defects in pituitary development, pituitary adenomas, POMC defect, and convertase 1 (PCSK1) enzyme defect. Other causes of secondary adrenal hypoplasia are isolated ACTH deficiency, diseases of pituitary development, and central nervous system developmental anomalies. Adrenal hypoplasia can occur due to chronic exogenous glucocorticoid treatment. The resultant reduced ACTH action leads to adrenal hypofunction and hypoplasia due to a low stimulus.[1][4][5][6][7]
Epidemiology
Congenital adrenal hypoplasia or AHC is very rare. Clinical experience has suggested it to be more uncommon than congenital adrenal hyperplasia (which has an incidence of approximately 1 per 10000 to 15000 births worldwide). Patients are more commonly male as one of the mentioned forms is X-linked. The estimated AHC frequency is 1 in 12500 live births. Triple-A syndrome or Allgrove syndrome has a prevalence of 1 per 1000000 individuals. Triple-A syndrome accounts for approximately 1% of all AHC cases and is more common in the middle east, Puerto Rico, and south Europe.[1]
Histopathology
The anterior pituitary contains special cells having distinct hormonal functions. Somatotrophs produce growth hormones and are acidophilic on light microscopy. Mammosomatotrophs are similar to somatotrophs. ACTH-producing cells are called corticotrophs, which are basophilic cells on staining. These basophils contain complex lysosomes and large vacuoles. The secretory granules vary in shape and size, and their membranes have indentations and evaginations. Adrenocortical hypofunction results in increased demand for ACTH, causing an increase in sparsely granulated basophils. Moreover, the tissue section of adrenals shows an absence of fasciculata or reticularis cells, as well as disorganization of glomerulosa cells in adrenal hypoplasia.[1][8]
History and Physical
Adrenal Hypoplasia Clinical Presentation
Adrenal Insufficiency
The hallmark of adrenal hypoplasia is adrenal insufficiency, which is a decrease in adrenal gland function. Patients often present in infancy or childhood, but there are reports of adult-onset cases. Females are mostly carriers and are generally unaffected. Undiagnosed patients usually present with adrenal crisis, but adrenal crisis can also occur in patients who are taking glucocorticoid replacement therapy. Patients usually present early in infancy or childhood with the salt-wasting crisis. Non-specific symptoms of adrenal insufficiency like feeling unwell, nausea, vomiting, lethargy, fatigue, anorexia, feeding difficulties, poor weight gain, and abdominal pain are present early in life or slightly later. The patient may have a fever, convulsions, and altered consciousness. These varied symptoms can cause misdiagnosis in these children. There is hypoglycemia, hyperkalemia, hyponatremia, and metabolic acidosis. Patients have associated mineralocorticoid deficiency and can present with hypotension, hypovolaemia, dehydration, and shock.
Hypogonadotropic Hypogonadism
Apart from adrenal insufficiency, affected males can lack male sex hormones, which can lead to hypogonadotropic hypogonadism, underdeveloped reproductive tissues, cryptorchidism, and infertility (azoospermia). Patients may have central precocious puberty.
- Short height can occur due to associated growth hormone deficiency.
- Adrenal hypoplasia can rarely present as respiratory symptoms, such as chronic respiratory distress.
- Triple-A syndrome causes esophageal achalasia, alacrima (absence of tears), and various types of central, peripheral, and autonomic neurological defects.
- Other unusual presentations include excessive pigmentation, hearing loss, and monosomy.
- Various other rare syndromes and mutations that can cause adrenal hypoplasia/aplasia can present with cardiac defects, central nervous system malformations, pulmonary hypoplasia, polydactyly, and other anomalies.[1][5][7][9][10][11]
Evaluation
Lab Findings
- In X-linked AHC with adrenal insufficiency, lab findings show electrolyte disturbances, including hyponatremia, hyperkalemia, hypoglycemia, and metabolic acidosis.
- Adrenal functions show a high ACTH level and low or baseline cortisol level.
- There is increased plasma renin activity, decreased aldosterone level, and no response to high-dose Cosyntropin stimulation.
- The 17-OH-progesterone level is normal or low.
- Androgen secretion is also impaired, and there are low androstenedione and low testosterone levels.
- Growth hormone deficiency may be present. Gonadotropin-releasing hormone (GnRH) test may show impaired gonadotropin secretion.
Genetic Screening
Precise genetic screening is advisable when adrenal hypoplasia is suspected. Identify the carrier status of women in the family.[12]
Imaging Studies
- Adrenal imaging, including CT and MRI, are necessary. Imaging may show small size adrenal glands. An ultrasound of adrenals may demonstrate small for age adrenal glands.
- A detailed gastroenterological and neurological evaluation should take place.
- Rarely, the patient may present with respiratory distress or other respiratory symptoms. Adrenal hypoplasia should be taken into account if a child is having respiratory symptoms along with electrolyte disturbances, and hypoglycemia. In AHC, due to DAX1 mutation, elevated 11-deoxycortisol levels have been noticed if checked very early in life.
- Schirmer test is a recommended procedure in Triple-A syndrome.[1][5][7][10][12]
Treatment / Management
It is essential to differentiate between adrenal insufficiency due to primary adrenal hypoplasia or secondary adrenal hypoplasia because treatment gets tailored according to the deficient hormones. In primary adrenal hypoplasia, all the adrenal gland hormones that are synthesized by adrenals, i.e., cortisol, aldosterone, and sex steroids, are deficient. In secondary adrenal hypoplasia, aldosterone replacement is not necessary since it is under the regulation of the renin-angiotensin-aldosterone system. Only ACTH-controlled hormones, i.e., cortisol and sex steroids, are decreased.
The fundamentals of the treatment of an adrenal crisis are similar in children and adult patients. Children warrant close monitoring. X-linked AHC gets treated with glucocorticoid, mineralocorticoid, and salt supplementation starting at two weeks of life. Symptoms usually resolve by two months of age, and ACTH and plasma renin activity return to normal. Management involves life-long glucocorticoid replacement therapy. According to guidelines, all adrenal hypoplasia patients who are receiving treatment should keep steroid emergency cards. Hydrocortisone is the preferred steroid as it has mineralocorticoid activity. However, if ordering a cosyntropin stimulation test to confirm the diagnosis, dexamethasone can be given before the test without any interference of results.
Growth hormone therapy (GnRH analogs) should start in patients with growth hormone deficiency.
Testosterone replacement is required when there is delayed puberty during adolescence to ensure normal development of secondary sexual characteristics and bone mineralization.
Emergency treatment of an adrenal crisis in adrenal hypoplasia should focus on correcting metabolic and electrolytic disturbances, including the correction of hypovolaemia and hypoglycemia. If the patient is hypotensive, aggressive fluid resuscitation, as well as, glucocorticoid treatment may be needed.[5][11][12](B3)
Differential Diagnosis
Obstructive uropathy, pyelonephritis, and tubulointerstitial disorders can mimic adrenal hypoplasia due to adrenal resistance.
Prognosis
Adrenal insufficiency in adrenal hypoplasia is a potentially life-threatening condition. There is reduced life expectancy and high morbidity, even if the patient receives optimal management. It is imperative to find the etiology of adrenal hypoplasia to figure out associated inheritance and comorbidities. The majority of the patients (35%) having adrenal insufficiency dies from cardiovascular disease, while 15% die from infections.[9][11]
Complications
- Life-threatening adrenal crisis
- Delayed or arrested puberty
- Infertility
- Hypoglycemia
- Short final height
- Myopathy (in associated Duchenne muscular dystrophy and glycerol kinase deficiency)
Consultations
Endocrinology consult is recommended for expert advice.
Deterrence and Patient Education
Adrenal hypoplasia patients should wear an alert bracelet or similar identification. Patient education is central to the management of adrenal hypoplasia to prevent adrenal crisis. A regular visit to an endocrinologist is required after diagnosis and especially after an episode of adrenal crisis. Follow-up at 6 to 12 months is the recommendation. Adult patients need to be more independent and be able to take care of the dose adjustments according to their needs. An additional dose of hydrocortisone is necessary during periods of stress, such as psychological stress or prolonged physical exercise.[11]
Enhancing Healthcare Team Outcomes
Patient or parents/family education is critical in the management of adrenal hypoplasia irrespective of the cause. They should be aware of the clinical picture of any impending crisis. Patients and caregivers should seek immediate medical attention if symptoms develop. Prevention is imperative, and patients and their families should be taught parenteral glucocorticoid self-administration to prevent an emerging crisis. The pharmacist should educate the patient and family about the importance of medication compliance. Prior to any procedure, the parent should ask for a booster dose of the corticosteroid. They should be informed about a low threshold to administer hydrocortisone emergency injection. The objective is to achieve that patients can acquire competencies to protect themselves from the life-threatening situations associated with the disease.[11]
The pharmacist should also make the parents aware of the potential adverse effects of long-term corticosteroids. Regular screening for bone density is another recommendation. Nursing staff should work closely with the family to answer questions, verify compliance with therapeutic measures, and inform the treating clinician on any concerns or issues. All patients should be urged to wear an ID bracelet indicating their medical condition. All clinicians who manage these patients should communicate with each other to ensure that the patient receives the current standard of care treatment. This type of interprofessional team approach is crucial to the successful management of the patient with adrenal hypoplasia. [Level 5]
Media
(Click Image to Enlarge)
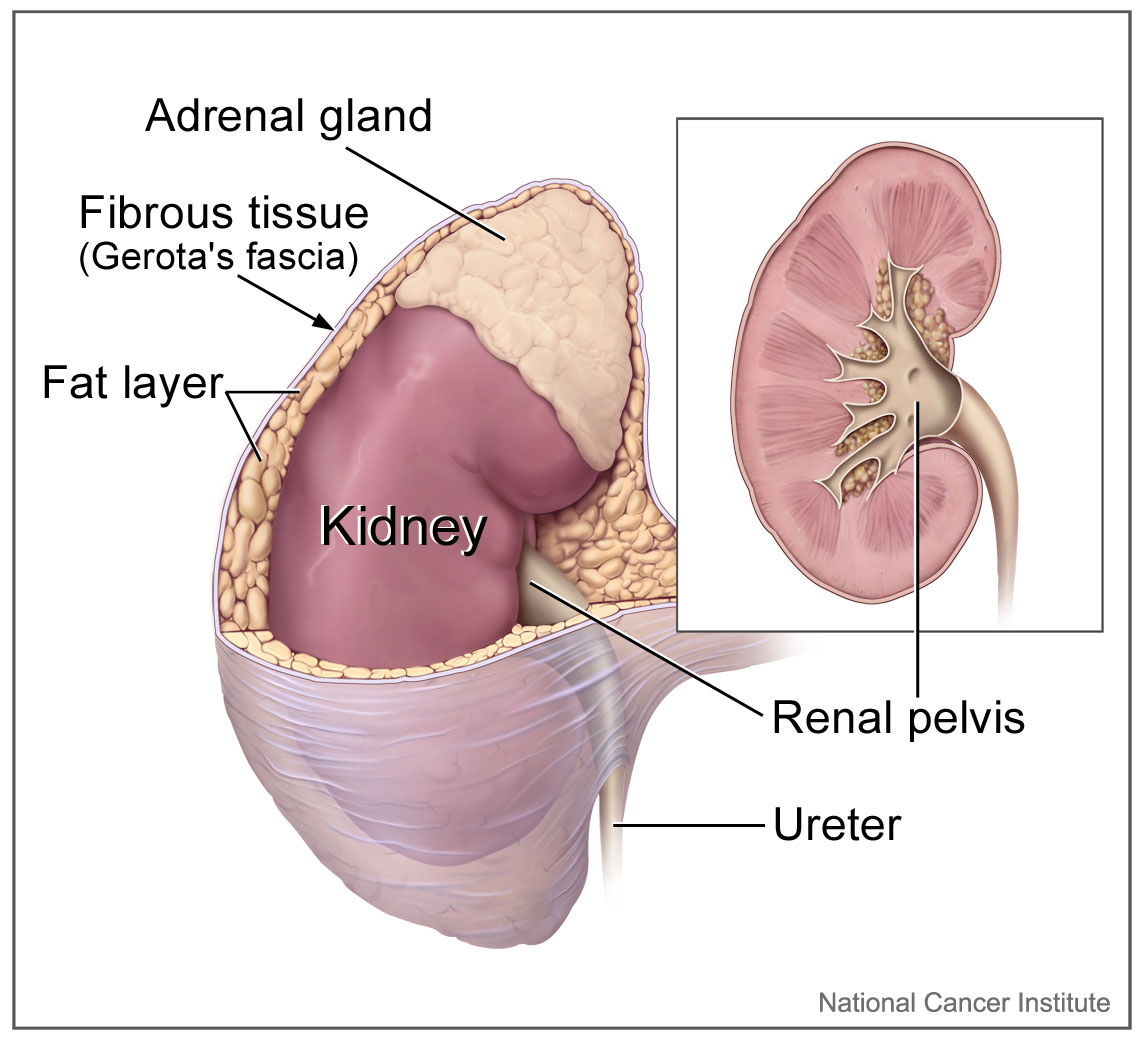
Kidney Gross Anatomy. Kidney anatomy includes the surrounding fibrous tissue and fat layer, renal pelvis, ureter, and the adrenal gland, with a close-up view of the renal pelvis.
National Cancer Institute
References
Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, de Herder WW, Dhatariya K, Dungan K, Hofland J, Kalra S, Kaltsas G, Kapoor N, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrère B, Levy M, McGee EA, McLachlan R, New M, Purnell J, Sahay R, Shah AS, Singer F, Sperling MA, Stratakis CA, Trence DL, Wilson DP, Kyritsi EM, Sertedaki A, Charmandari E, Chrousos GP. Familial or Sporadic Adrenal Hypoplasia Syndromes. Endotext. 2000:(): [PubMed PMID: 25905355]
Lotfi CFP, Kremer JL, Dos Santos Passaia B, Cavalcante IP. The human adrenal cortex: growth control and disorders. Clinics (Sao Paulo, Brazil). 2018 Sep 6:73(suppl 1):e473s. doi: 10.6061/clinics/2018/e473s. Epub 2018 Sep 6 [PubMed PMID: 30208164]
Aljabri KS, Bokhari SA, Assiri FY, Alshareef MA, Khan PM. The epidemiology of pituitary adenomas in a community-based hospital: a retrospective single center study in Saudi Arabia. Annals of Saudi medicine. 2016 Sep-Oct:36(5):341-345 [PubMed PMID: 27710986]
Level 2 (mid-level) evidenceRojek A, Krawczynski MR, Jamsheer A, Sowinska-Seidler A, Iwaniszewska B, Malunowicz E, Niedziela M. X-Linked Adrenal Hypoplasia Congenita in a Boy due to a Novel Deletion of the Entire NR0B1 (DAX1) and MAGEB1-4 Genes. International journal of endocrinology. 2016:2016():5178953. doi: 10.1155/2016/5178953. Epub 2016 Aug 30 [PubMed PMID: 27656210]
Chung ST, Chi CH, Haymond MW, Jeha GS. Infantile Growth Hormone Deficiency and X- Linked Adrenal Hypoplasia Congenita. Jacobs journal of pediatrics. 2015 Nov:1(1):. pii: 003. Epub 2015 May 4 [PubMed PMID: 27110597]
Francke U, Harper JF, Darras BT, Cowan JM, McCabe ER, Kohlschütter A, Seltzer WK, Saito F, Goto J, Harpey JP. Congenital adrenal hypoplasia, myopathy, and glycerol kinase deficiency: molecular genetic evidence for deletions. American journal of human genetics. 1987 Mar:40(3):212-27 [PubMed PMID: 2883886]
Schwarz K, Thwaites R, Minford A, Day C, Butler G. Congenital adrenal hypoplasia presenting as a chronic respiratory condition. Archives of disease in childhood. 2003 Mar:88(3):261-2 [PubMed PMID: 12598398]
Level 3 (low-level) evidenceFeingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, de Herder WW, Dhatariya K, Dungan K, Hofland J, Kalra S, Kaltsas G, Kapoor N, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrère B, Levy M, McGee EA, McLachlan R, New M, Purnell J, Sahay R, Shah AS, Singer F, Sperling MA, Stratakis CA, Trence DL, Wilson DP, Asa SL. Pituitary Histopathology in Man: Normal and Abnormal. Endotext. 2000:(): [PubMed PMID: 25905234]
Kyriakakis N, Shonibare T, Kyaw-Tun J, Lynch J, Lagos CF, Achermann JC, Murray RD. Late-onset X-linked adrenal hypoplasia (DAX-1, NR0B1): two new adult-onset cases from a single center. Pituitary. 2017 Oct:20(5):585-593. doi: 10.1007/s11102-017-0822-x. Epub [PubMed PMID: 28741070]
Level 3 (low-level) evidenceShima H, Yatsuga S, Nakamura A, Sano S, Sasaki T, Katsumata N, Suzuki E, Hata K, Nakabayashi K, Momozawa Y, Kubo M, Okamura K, Kure S, Matsubara Y, Ogata T, Narumi S, Fukami M. NR0B1 Frameshift Mutation in a Boy with Idiopathic Central Precocious Puberty. Sexual development : genetics, molecular biology, evolution, endocrinology, embryology, and pathology of sex determination and differentiation. 2016:10(4):205-209 [PubMed PMID: 27648561]
Dineen R, Thompson CJ, Sherlock M. Adrenal crisis: prevention and management in adult patients. Therapeutic advances in endocrinology and metabolism. 2019:10():2042018819848218. doi: 10.1177/2042018819848218. Epub 2019 Jun 13 [PubMed PMID: 31223468]
Level 3 (low-level) evidenceLoureiro M, Reis F, Robalo B, Pereira C, Sampaio L. Adrenal Hypoplasia Congenita: A Rare Cause of Primary Adrenal Insufficiency and Hypogonadotropic Hypogonadism. Pediatric reports. 2015 Sep 28:7(3):5936. doi: 10.4081/pr.2015.5936. Epub 2015 Sep 28 [PubMed PMID: 26500747]