 Surgical Approaches to Congenital Aortic Stenosis
Surgical Approaches to Congenital Aortic Stenosis
Introduction
Managing obstruction of the left ventricular outflow tract (LVOT) in neonates, infants, and young children with congenital aortic stenosis (AS) poses significant surgical challenges due to the complexity of the LVOT's anatomy, mechanical function, and the aortic root's performance. Surgical options for relieving AS in this population require careful consideration, especially when the hypoplastic aortic annulus is involved. A thorough preoperative assessment is essential to identify suitable candidates for timely aortic root or LVOT interventions. Various approaches, including valve-conserving procedures such as the Ross procedure, sometimes modified with the Konno procedure, allow children to grow before definitive interventions like aortic valve replacement become necessary. Determining the optimal timing for aortic root enlargement is crucial, as delaying surgery can lead to severe circulatory failure and ventricular hypertrophy.
In neonates and infants with congenital AS, multiple comorbidities such as borderline left ventricles (LVs), small mitral valves, and pulmonary hypertension complicate surgical decision-making. The Ross-Konno procedure, often combined with institution-specific modifications like endocardial fibroelastosis resection, remains a preferred approach when valve remodeling is not feasible, offering a wide-open and competent LVOT. However, alternative techniques such as the Konno-Rastan procedure, which involves replacing the aortic valve with a mechanical or bioprosthetic valve, may lead to late complications like prosthesis mismatch as the child grows or the need for lifelong anticoagulation in cases of mechanical prostheses.[1] A careful balance between timely intervention and long-term management of these complexities is essential for optimizing patient outcomes.
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
Cardiac valves comprise a highly organized extracellular matrix, including collagen, elastin, and proteoglycans. The proper development of these valves relies on specific structural proteins. Numerous transcription factors and promoter genes regulate the intricate process of cardiogenesis.[2] Variations in the genes that encode these structural proteins—such as elastin, fibrillin 1, and collagen type III alpha 1 chain—have been linked to congenital valve defects. Additionally, gene variants that encode proteins necessary for the precise localization of cells through cell-cell and cell-matrix interactions, like elastin microfibril interfacer 1, have also been identified in patients with valve abnormalities.[3]
In higher vertebrates, the heart is not only vital for sustaining life but also the first functional organ in the developing embryo, beginning to beat spontaneously by the fourth week of human development. The primitive bulbus cordis is crucial in forming several vital structures of the developing heart. Specifically, the proximal third of the bulbus cordis becomes the trabeculated part of the right ventricle (RV); the midportion, known as the conus cordis, develops into the outflow tracts of both ventricles; and the distal third, the truncus arteriosus, forms the proximal aorta and pulmonary trunk. The area where the primitive ventricle meets the bulbus cordis is the primary interventricular foramen. As ventricular septation progresses, the primitive ventricle evolves into the significant portion of the definitive LV. At the same time, the proximal third of the bulbus cordis develops into the major portion of the definitive RV (see Image. Bending of the Cardiac Tube).[2]
During the fifth to seventh weeks of development, neural crest cells migrate into the region of the outflow tract, giving rise to the conotruncal ridges, which develop along the inner walls of the conotruncus and spiral toward each other, ultimately fusing to form the aorticopulmonary septum. This septum divides the outflow tract into the aorta and pulmonary trunk. Concurrently, the endocardial cushions within the outflow tract contribute to forming the aortic and pulmonary semilunar valves, ensuring the unidirectional flow of blood from the ventricles to the great arteries. By the seventh to eighth weeks, the aortic and pulmonary roots align with their respective ventricles: the aorta with the left, forming the LVOT, and the pulmonary trunk with the right.
The LVOT is incorporated into the developing LV as the interventricular septum forms, consisting of muscular and membranous parts that close the ventricular septal defect of earlier stages. The aortic valve, aortic root, and ascending aorta undergo further refinement, with the aortic valve emerging from the endocardial cushions, which experience proliferation, programmed cell death, and remodeling to form its mature 3-leaflet structure. Abnormalities in these developmental processes can lead to congenital heart defects involving the LVOT, such as AS, aortic coarctation, or double outlet RV.
Congenital AS can be classified into 4 main types based on the location of the narrowing (see Image. Types of Left Ventricular Outflow Tract Obstruction).[4] They are:
- Valvular AS
- This is characterized by hypoplastic, dysplastic, or numerically abnormal valve cusps, with the bicuspid valve being the most common form. Occurring 4 times more frequently in males than females, it results from abnormal development of the endocardial cushions. This type often involves the fusion of the right coronary cusp with an adjacent cusp. In 20% of cases, bicuspid valves are associated with other congenital heart defects, such as coarctation of the aorta.
- Bicuspid aortic valve (BAV) is the most common form of congenital AS, and its prevalence ranges from 0.77% to 1.4%, which may be higher when asymptomatic individuals are considered. Although patients with BAV typically do not experience significant issues during infancy and childhood, they may develop various aortic valve abnormalities, such as stenosis and insufficiency, as well as aortic complications like root dilatation, rupture, and dissection later in life. Consequently, BAV is associated with the highest mortality rate among congenital heart diseases.
- BAV can be categorized into 3 main types using the Sievers classification (see Image. Sievers Classification System for Bicuspid Aortic Valve):
- Type-0: This type has 2 equal cusps without a raphe.
- Type-1: This is the most common type, accounting for 90% to 95% of cases, and is characterized by the fusion of 2 cusps. Type-1 is further divided into 3 subgroups:
- R-L
- This is the fusion of the right and left coronary cusps.
- The R-L subgroup is the most prevalent and linked to neural crest cell migration issues.[5]
- R-N
- This is the fusion of the right coronary and noncoronary cusps.
- L-N
- This is the fusion of the left coronary and noncoronary cusps.
- This is the rarest form.
- R-L
- Type-2: This is the rarest, involving the fusion of 3 leaflets.
- A new international consensus classification for BAV has been recently introduced, which categorizes BAV based on the type and phenotype of the valve, its function, the presence and characteristics of the raphe, cusp shape, cusp size, BAV symmetry, and the presence or absence of aortopathy or coarctation. The international consensus offers several advantages over the Sievers classification, including a) the ability to define all BAV phenotypes, such as fused, 2-sinus, and partial fusion (forme fruste) phenotypes; b) the recognition of fused BAV without a raphe, distinguishing it from 2-sinus BAV; c) providing a symmetry assessment crucial for surgical repair planning of fused BAV; d) the inclusion of aortic phenotypes (root, ascending, and extended); and e) the use of more straightforward and descriptive language (see Image. International Consensus Classification for Bicuspid Aortic Valve).[6]
- Subvalvular AS
- This type features a thickened ring or collar of dense endocardial fibrous tissue below the valve cusps, narrowing the LVOT. Accounting for 10% of AS cases, it is twice as common in males and is the second most common form of congenital AS.
- Patients often present with mild aortic regurgitation due to the thickening and immobility of the aortic cusps, sometimes exacerbated by bacterial endocarditis. A prominent systolic murmur, occasionally accompanied by a vibratory "thrill," is common.
- This condition can lead to LV hypertrophy and an increased risk of sudden death during exertion. The muscular form of subvalvular AS, a hypertrophic cardiomyopathy variant, is hypertrophic obstructive cardiomyopathy (HOCM). Historically referred to as idiopathic hypertrophic subaortic stenosis, HOCM is a genetic disease caused by mutations in contractile proteins.
- Supravalvular AS
- This type is caused by the narrowing of the ascending aorta just above the aortic valve and is the least common variant of LVOT obstruction, occurring with an estimated frequency of 1 in 20,000 live births.
- This condition most frequently presents as a discrete, hourglass-like constriction of the ascending aorta near the sinotubular junction. In approximately one-quarter to one-third of cases, however, the narrowing is more diffuse, extending into the aortic arch. Modern understanding now recognizes this condition as a generalized arteriopathy marked by thickening the aortic media or intima layers. This leads to a progressive constriction of the ascending aorta and other systemic and pulmonary arteries.[7]
- This congenital anomaly arises from the incomplete expression of the ELN gene located on chromosome 7q11.23. The ELN gene encodes tropoelastin, a precursor of elastin expressed in smooth muscle cells during early development. Elastin, in harmony with other extracellular proteins, forms elastic fibers within the arterial media, imparting necessary distensibility to the vasculature. Disruption of the ELN gene results in reduced and disorganized elastin fibers, increased hypertrophied smooth muscle cells, and excessive collagen deposition within the arterial media. The condition follows an autosomal dominant inheritance pattern with incomplete penetrance and variable expressivity.
- A microdeletion of the ELN gene is responsible for Williams-Beuren syndrome, characterized by an elastin arteriopathy alongside other manifestations, such as intellectual disability, hypercalcemia, and distinctive facial features. In this syndrome, the prevalence of supravalvular AS is estimated to be approximately 70%. Point mutations in the ELN gene give rise to familial forms of supravalvular AS, in which the patient exhibits a similar arteriopathy as those with Williams-Beuren syndrome but generally retains normal cognitive function and lacks dysmorphic features.[8][9]
- Unicommissural and acommissural valves
- These types cause symptoms early in life. Congenital AS presenting in infancy and childhood often results from a severely deformed valve associated with a hypoplastic aortic annulus. This anomaly may be part of hypoplastic left heart syndrome.
Epidemiology
Congenital heart defects are among the most prevalent congenital malformations, affecting about 9 out of every 1000 live births. There are notable geographical variations in the prevalence of congenital heart defects. The highest prevalence is reported in Asia, with 9.3 cases per 1000 live births. At the same time, Europe has the second highest prevalence, at 8.2 cases per 1000 live births, and Africa has the lowest prevalence at 1.9 per 1000 live births.[10]
The distribution of various cardiac malformations is as follows: ventricular septal defect constitutes 42%, atrial septal defect accounts for 10%, pulmonary stenosis represents 8%, and patent ductus arteriosus makes up 7%. Tetralogy of Fallot and coarctation of the aorta occur in 5% of cases. Atrioventricular septal defect and AS are each found in 4% of cases, while transposition of the great arteries also appears in 4%. Persistent truncus arteriosus, total anomalous pulmonary venous connection, and tricuspid atresia represent 1% of cardiac malformations.[11]
Pathophysiology
Despite being a short channel, the LVOT involves intricate anatomy, mechanical function, and performance, all of which are perfectly integrated (see Image. Complex Anatomy of the Left Ventricular Outflow Tract). The crown-shaped aortic annulus supports and anchors the aortic valve within the aortic root. As an anchor for the synchronized movement of both the inlet mitral valve and the outlet semilunar valve, the aortic root also supports the intricate motions of the aortic valve cusps.[12] Over the past 2 decades, most morphological studies of stenotic, purely regurgitant, and infected congenitally malformed aortic valves have focused on calcium analysis in operatively excised specimens. Study results on these excised valves have shown that unicuspid valves contain the most calcium, making them the heaviest and most stenotic. Bicuspid valves exhibit the next highest level of calcification, while stenotic tricuspid valves have the least calcific deposits, rendering them the lightest and least stenotic.[13]
Subvalvular AS includes various conditions and is known for its progressive nature. Subvalvular AS may present as a solitary subvalvular membrane within the LVOT or as a small fibrous muscular ridge on the subvalvular ventricular septum, called discrete subvalvular AS. In more severe forms, subvalvular AS manifests as a narrow fibromuscular tunnel-like obstruction in the LVOT. This stenosis leads to turbulent blood flow, which can damage the aortic valve, potentially causing aortic regurgitation due to valve prolapse. Membranous subaortic tissue can also extend onto the aortic valve leaflets, restricting their mobility and alignment. Aortic regurgitation often progresses after subaortic resection, with reoperation rates for valve repair or replacement following initial resection reaching up to 20%. However, long-term outcomes regarding reoperation and aortic valve intervention in pediatric patients with subvalvular AS remain poorly defined.[14]
History and Physical
Clinical manifestations of valvular AS vary depending on the patient's presentation age, the stenosis's severity, and any associated cardiac conditions. Valvular AS can be detected in fetuses through fetal echocardiography, where a thickened or domed aortic valve with increased Doppler flow velocity greater than 1 m/s indicates the condition. Valvular AS is a progressive disorder, and in some cases, it may advance to HLHS. This progression can be identified by reversed flow in the transverse aortic arch and foramen ovale, monophasic mitral inflow, and LV dysfunction during the second trimester. A slowed growth rate of left heart structures can also predict hypoplastic left heart syndrome development.
Neonatal and routine well-baby check-ups often fail to effectively screen for severe valvular AS. Infants with severe valvular AS typically present with signs of congestive heart failure by around 2 months of age. These infants may exhibit symptoms such as pallor, mottled skin, hypotension, and dyspnea. Clinical examination may reveal a normal first heart sound, an ejection click, and a gallop in approximately 50% of affected infants. An ejection systolic murmur of varying intensity is often detected along the mid-left and right upper sternal borders, with radiation to the carotid arteries. The presence of hypoxia (PaO2 30–40 mm Hg) and metabolic acidosis signals an urgent need for medical intervention.
Older children and adolescents with valvular AS are generally asymptomatic, but about 10% may experience symptoms such as dyspnea, angina, or syncope, particularly during exercise. The appearance of these symptoms warrants immediate and thorough evaluation due to the risk of sudden death, which has been observed in 1% to 10% of individuals aged 5 to 15 years with moderate to severe valvular AS. In children, characteristic physical findings associated with valvular AS, such as a small amplitude and slow-rising pulse (pulsus parvus et tardus), carotid shudder, and a prominent jugular venous "a" wave, are not as consistently present as they are in adults. However, a precordial systolic thrill over the base of the heart is noted in over 65% of patients with more than mild valvular AS. Severe valvular AS may also be indicated by a narrowly split, single, or paradoxically split second and fourth heart sounds.
An ejection click, present in cases with a pliable valve and absent in severe valvular AS, is not influenced by respiration. The typical low-pitched crescendo-decrescendo systolic murmur of valvular AS, which begins just after the ejection click, is best heard at the base of the heart and radiates to the carotid arteries. This murmur becomes more pronounced with maneuvers that increase stroke volume, such as isotonic exercise and premature ventricular contractions.[15] Older children and adolescents with valvular AS are often asymptomatic, but approximately 10% may exhibit symptoms of congestive heart failure, such as dyspnea, angina, or syncope, particularly during exercise. To assess the severity of stenosis, a combination of maximum aortic velocity, mean pressure gradient, and aortic valve area is used, as outlined in the latest guidelines from the American College of Cardiology and the American Heart Association (AHA).[16][17]
The inclusion criteria, aligned with the AHA operative guidelines, are as follows for critical AS:
- Patients with a small aortic valve annulus, with or without subvalvular narrowing, and either a peak instantaneous gradient of ≥60 mm Hg, a mean pressure gradient of ≥40 mm Hg, or an aortic valve area of ≤0.5 cm²/m²
- Patients with a peak gradient of ≥50 mm Hg or an aortic valve area of 0.5 to 0.8 cm²/m² accompanied by symptoms such as angina, syncope, or electrocardiographic changes, or those planning for pregnancy
- Patients with a peak gradient of ≥35 mm Hg or an aortic valve area of 0.8 to 1.5 cm²/m² with moderate or greater aortic regurgitation due to valve deformity or previous ballooning [18]
Evaluation
Imaging Recommendations
Aortic dilation in patients with BAV can involve the aortic root, ascending aorta, or both, with the ascending aorta most commonly affected. In some cases, the dilation may extend into the aortic arch. The reported prevalence of aortic dilation in BAV varies widely, ranging from 20% to 84%, depending on the population studied and the specific definition of aortic dilation used. Patients with both BAV and aortic dilation are at an increased risk for aortic dissection. To evaluate aortic valve function, aortic dilation, and potential aortic coarctation, imaging of the aortic root, ascending aorta, arch, and proximal descending aorta should be performed using transthoracic echocardiography (TTE). Additionally, in patients undergoing TTE, detecting unexplained aortic root or ascending aortic dilation should raise suspicion for underlying BAV. If TTE results for the aortic valve are inconclusive, further imaging with cardiac magnetic resonance imaging (MRI), cardiac computed tomography angiography (CTA), or transesophageal echocardiography (TEE) can be used for a more detailed assessment.[19] Cardiac-gated CT or MRI is particularly effective for visualizing the aortic root and ascending aorta when TTE does not provide sufficient detail. The choice between CT and MRI depends on various factors, including patient characteristics, institutional expertise, renal function, cost considerations, and concerns about radiation exposure.[20]
Z-score Calculation
A method for describing clinical and echocardiographic variables is a Z-score, which quantifies how many standard deviations a particular measurement is from the mean of a size- or age-specific population. The Z-score is calculated using the formula:
Z= (x −μ) / σ
where x represents the observed measurement, μ is the expected measurement (population mean), and σ is the population's standard deviation. A positive Z-score indicates a value above the population mean, while a negative Z-score indicates a value below it, reflecting the extent of deviation from the mean. For instance, if the mean size of the aortic valve is 20 mm with a standard deviation of 3 mm, a valve with an annulus measuring 14 mm would have a Z-score of:
Z= (14 − 20) / 3= −2
To accurately calculate a Z-score, one must establish the population mean and standard deviation for each body size.[21]
Hemodynamic Parameters
In pediatrics, the severity of valve stenosis, which helps determine the need and timing for intervention, is often quantified by measuring the aortic valve pressure gradient. This transvalvular pressure gradient (P) is typically calculated using peak instantaneous Doppler echocardiographic velocity (V) measurements from various angles. The simplified Bernoulli equation is applied to estimate the pressure gradient:
P= 4V2
This equation is a common method for assessing the severity of valvular AS in this patient population.[15]
Familial Screening Recommendations
Certain hereditary thoracic aortic diseases show an increased prevalence of BAV. For instance, about 10% of patients with Loeys-Dietz syndrome, associated with pathogenic variants in genes such as TGFBR1, TGFBR2, SMAD3, TGFB2, and TGFB3, have BAV. Hereditary thoracic aortic diseases are linked to pathogenic variants in NOTCH1, ACTA2, MAT2A, SMAD6, and LOX, which also show a higher prevalence of BAV.[22] Notably, despite the familial nature of BAV and thoracic aortic aneurysms, most patients with these conditions will not have an identifiable pathogenic genetic variant even after genetic testing. However, when the condition is familial, patients with BAV and aortopathy should be evaluated, counseled, and genetically tested by a medical geneticist or a specialist in genetic aortopathy.[23]
The prevalence of BAV among first-degree relatives of individuals with this condition is approximately 10% to 15%. Due to the hemodynamic consequences and the genetic predisposition to aortic dilation associated with BAV, early detection, and regular monitoring are crucial. Consequently, American and European guidelines from the past decade recommend screening primary family members using TEE. The evidence supporting these recommendations varies, generally falling within class IIa/b.[24]
Treatment / Management
Congenital AS is a progressive condition, and guidelines have been established to outline the indications for intervention. These interventions include balloon valvuloplasty in the cardiac catheterization lab and transcatheter or surgical aortic valve replacement. Results from a 30-year follow-up study revealed that diagnosing mild AS before 6 months of age significantly increases the risk of requiring aortic valvotomy and balloon valvuloplasty as the patient ages. The probability that the stenosis remains mild is less than 20%, highlighting the necessity for long-term follow-up of mild AS into adulthood. Recently, it was shown that the likelihood of requiring balloon valvuloplasty is 20% for patients with catheter-measured peak pressure gradients less than 25 mm Hg and increases to 40% and 70% for those with gradients of 25 to 49 mm Hg and over 50 mm Hg, respectively.[6]
LVOT obstruction (LVOTO) is an instantaneous peak pressure gradient of 30 mm Hg or greater within the LVOT, as measured by Doppler echocardiography. LVOTO is dynamic and sensitive to ventricular preload, afterload, and myocardial contractility variations. For patients with hypertrophic cardiomyopathy (HCM) who do not exhibit LVOTO at rest, a provocation test is necessary to assess latent LVOTO. This can be done using methods such as the Valsalva maneuver, transitioning from a supine to standing or squatting to a standing position or performing an upright treadmill or semisupine bicycle test. Current guidelines advise against using dobutamine due to its lack of specificity.[25][26](B3)
Study results have shown that LVOTO significantly increases the risk of adverse events, including sudden cardiac death, progression to moderate to severe heart failure, stroke, and death in patients with HCM. A resting or provoked LVOT pressure gradient of 50 mm Hg or greater is typically the threshold for considering septal reduction therapy in patients who have drug-refractory symptoms.[27][28] Short- and mid-term outcomes for LVOT abnormalities have improved due to the implementation of increasingly complex surgical procedures, ie, the Ross procedure, the Ross–Konno procedure, and the Konno–Rastan procedure.[29]
The Ross procedure is a well-established surgical technique to replace a severely dysfunctional aortic valve with the patient's pulmonary valve, known as an autograft, with the native pulmonary valve replaced with a homograft or a conduit.[30] When the native annulus is too small, the Konno incision can enlarge the LVOT by incision from the annulus into the ventricular septum.[31] This combined Ross-Konno procedure offers distinct advantages, particularly for infants and neonates with competent pulmonic valves. The autograft in the new aortic position can grow with the patient and does not require anticoagulation.[32] Alternative treatments, such as serial balloon dilations, aortic valve repair, and mechanical valve replacements, have significant drawbacks. However, there are concerns about the potential for neoaortic root dilatation due to the pulmonary valve being exposed to unfamiliar systemic pressures, the need for repeated replacements of the RV to pulmonary artery (RV-PA) conduit or valve, and cardiac performance in the immediate postoperative period.[33](B2)
Neonates and young infants considered for the Ross-Konno procedure are often critically ill; they may present with multiple left-sided anomalies, including complex LVOTO, mitral stenosis, and aortic arch abnormalities. These patients might have previously undergone partially successful procedures, such as aortic valve balloon dilatation. Study results from single and multiple institutions have reported early survival rates ranging from 78% to 96% for this age group.[34][35] The traditional Konno–Rastan procedure involves enlarging the aortic root through anterior aortoventriculoplasty and replacing the aortic valve with a mechanical or biological prosthetic valve. Reoperation after the Konno–Rastan procedure presents significant challenges and carries a risk of high mortality and morbidity, with potential complications such as heart block, need for anticoagulation, possible prosthesis patient mismatch later in life, and excessive bleeding.[18]
The peak LVOT gradient determines the indication for surgical intervention. Surgical treatment is recommended for patients with a peak LVOT gradient exceeding 50 mm Hg. For those with a gradient between 30 and 50 mm Hg, the decision to proceed with surgery depends on the presence of symptoms and the progression of aortic regurgitation. While a subvalvular membrane may be identified during preoperative echocardiography, the final surgical approach is determined intraoperatively after inspecting the LVOT.
The specific approach depends on the anatomy of the stenosis, including the presence of a membrane and the degree of septal hypertrophy. The extent of myectomy is tailored to the individual's anatomy and LVOT size, with careful attention to the conduction system. At the center, the standard approach for pediatric patients involves transaortic septal myectomy, with no transmitral or apical septal myectomy performed during the study period. During surgery, access to the LVOT is achieved through an oblique aortotomy. The aortic valve is retracted, enabling resection of the membrane and septal myectomy in cases of myocardial hypertrophy. Any redundant subvalvular membrane tissue that extends onto the aortic valve, mitral valve, or accessory papillary muscle attachments is also resected. Multiple levels, including subvalvular stenosis and aortic valve dysfunction, may benefit from anterior aortoventriculoplasty, ie, the Ross-Konno or Konno-Rastan procedures.[14]
An example of the Ross-Kono procedure is as follows:
- Standard surgical techniques for the Ross-Konno procedure involve initiating cardiopulmonary bypass via a midline sternotomy, bicaval cannulation, and moderate hypothermia. Deep hypothermic circulatory arrest is used for patients requiring concurrent aortic arch repair. Myocardial protection is achieved using antegrade or retrograde delivery of cold blood cardioplegia or modified St Thomas cardioplegia solution into the coronary sinuses.
- The surgery begins by transecting the aorta above the sinotubular junction, securing commissural stay sutures, and inspecting the aortic valve. After confirming the Ross-Konno procedure is suitable and ensuring the pulmonary valve is a trileaflet and viable as an autograft, the aortic valve leaflets are excised, and the coronary buttons are mobilized, along with the proximal epicardial coronary course. The pulmonary autograft is harvested from the right ventricular outflow tract and interventricular septum while preserving septal perforators. Antegrade cardioplegia identifies and oversees any bleeding in the autograft bed.
- A Konno incision is made to address the size mismatch between the pulmonary and aortic valves, extending 5 to 30 mm into the interventricular septum to enlarge the LVOT and accommodate the larger pulmonary autograft (see Image. Ross-Konno Procedure). The RV free wall patch beneath the left anterior pulmonary sinus on the autograft is integrated into the LVOT. In contrast, the remaining 2 pulmonary sinuses are sewn horizontally around the inflow, supporting the left anterior sinus with the free-grafted RV muscle. A separate ventricular septal defect patch is rarely necessary. The autograft commissures are marked externally, and a 5-0 or 6-0 polypropylene continuous suture technique is employed, using a new suture for each sinus. The autograft is positioned, suture tension is confirmed with a nerve hook, and the sutures are secured. The autograft is implanted at the previous annulus level, taking care to avoid the conduction tissue in the membranous septum. An additional 6-0 polypropylene continuous suture reinforces the proximal suture line.
- The coronary buttons are implanted with continuous sutures into punch holes, and trapdoor flaps are rarely used. In this age group, specific annulus reinforcement techniques are not applied. The reconstructed neocortical root is anastomosed end-to-end to the ascending aorta. A cryopreserved pulmonary homograft, sized 12 to 14 mm, is preferred for the RV-PA conduit. An aortic homograft or bovine jugular vein valved conduit is used if unavailable. The distal conduit anastomosis typically precedes the distal aortic suture line, using a continuous polypropylene suture. Before completing the proximal homograft suture line and with the PA vented, the atrial septal defect is closed primarily, and the heart is deaired with aortic root suction. The right atrial and proximal conduit anastomoses are then completed. In some cases, a bovine pericardial skirt is sewn to cover the rightward part of the orifice around the neoaorta due to a size mismatch between the proximal conduit and the autograft harvest site. The conduit and the ventriculotomy edge are then attached to this.
- A postoperative TEE confirms the autograft's competence, ventricular performance, and mitral valve function. A prolonged hemostasis period is often required, and delayed sternal closure is routinely performed in neonates and those with extended aortic cross-clamp time or marginal postoperative status. Extracorporeal membrane oxygenation is employed postoperatively in patients with significant inotrope requirements, volume-dependent circulation, or extended cross-clamp time or who could not be successfully separated from bypass to avoid the need for emergent cannulation.
Differential Diagnosis
The differential diagnoses for congenital AS include:
- Aortic valve masses
- Masses on the aortic valve are extremely rare in children, with potential diagnoses including vegetation, thrombus, and primary cardiac tumors. This condition may manifest as valvular AS, potentially causing sudden hemodynamic compromise due to aortic vegetation extending into the ascending aorta and eroding through its posterior wall.[36]
- The rising survival rates of children with congenital heart conditions and extended hospital stays in newborns have contributed to an increase in infective endocarditis cases. In these instances, the mass on the aortic valve or in the ascending aorta could be a vegetation, tumor, or thrombus.
- Aortic thrombosis
- In the neonatal population, symptomatic aortic thrombosis occurs at 0.1 to 1.1 per 100,000 newborns. The clinical presentation can vary widely, from nonspecific irritability to embolic severe complications such as stroke, acute limb-threatening ischemia, mesenteric ischemia, and renal failure.
- Lambl excrescences, an anatomical variant, are typically limited to older adults.
- Papillary fibroelastoma
- This is a primary cardiac tumor with a preference for the valvular endocardium, primarily affecting older adults, with only a few cases reported in children.
Prognosis
The prognosis following surgical approaches to congenital AS varies depending on the type and severity of the stenosis, the patient's age, and the specific surgical procedure performed. In general, surgical interventions such as balloon valvuloplasty, aortic valve repair, or replacement, and more complex procedures like the Ross-Konno procedure have significantly improved survival rates and long-term outcomes in patients with congenital AS. However, patients require lifelong follow-up, as the condition can recur, and there are risks of reintervention. The prognosis for patients undergoing the Ross-Konno procedure is generally favorable, especially in pediatric patients, due to using the patient's pulmonary valve as an autograft. This technique provides good long-term hemodynamic performance and reduces the need for anticoagulation. However, patients may face complications such as autograft dilation or dysfunction, which may necessitate reoperation. Despite these risks, the Ross-Konno procedure has shown excellent durability, with survival rates around 90% at 10 years and a low risk of sudden cardiac death.
Results from a recent study on patients who underwent a Ross or Ross-Konno procedure reported no early deaths, with all patients surviving until discharge. However, there was 1 late death caused by cardiac arrest due to a pulmonary hypertensive crisis. Out of the total, 27 patients (77.1%) left the operating room with an open sternum, typically closed after a median of 5 days. Delayed chest closure was almost universally utilized in neonates, with 12 out of 13 (92.3%) undergoing this approach. Additionally, 6 patients (17.1%) required mechanical circulatory support. Since undergoing their Ross or Ross-Konno procedure, 12 patients (34.3%) required at least 1 reoperation, with a median age of 3.4 years (interquartile range, 0.6-8.9 years). These reoperations included 10 reinterventions for RV-PA conduit issues, 2 mitral valve repairs, and 1 mechanical mitral valve replacement.
Additionally, 1 patient needed a reoperation on the LVOT. Another patient required right PA stenting and angioplasty at 16 months of age due to progressive proximal right PA stenosis. Ten patients required RV-PA conduit replacement at a median of 10.6 years following their Ross or Ross-Konno procedure. All 34 surviving patients exhibited normal LV systolic function at the most recent follow-up. Of these, 9 patients (26.5%) had mild neoaortic regurgitation, while the remaining 25 (73.5%) had none or only minimal regurgitation. None of the patients developed neoaortic stenosis.[35] Overall, the prognosis of surgical approaches to congenital AS has improved with advances in surgical techniques, perioperative care, and long-term management. However, ongoing surveillance is essential due to the risk of reoperation and complications such as valve degeneration, aortic root dilation, and arrhythmias.
Complications
In a recent study of operative complications following the Ross-Konno procedure, the results showed that low cardiac output syndrome was observed in 21 patients (60%), with 4 experiencing postoperative cardiac arrests due to various causes: tamponade, hypokalemia, transient complete heart block, and an unidentified cause. One infant required immediate reoperation for bleeding. Clinically significant arrhythmias were noted in 11 patients (31.4%), with 1 needing a permanent pacemaker 2 weeks postsurgery due to a complete atrioventricular heart block (in a patient who had undergone a Konno incision). Mediastinitis developed in 4 patients (11.4%), and 1 of these cases progressed to fungal endocarditis following a prolonged extracorporeal membrane oxygenation (ECMO) period with an open chest, leading to RV-PA conduit replacement at 10 weeks after a Ross/Konno procedure. Five patients (14%) experienced cerebrovascular events, with 3 occurring during ECMO. Two patients (5.7%) had seizures. Additionally, 13 patients (37.1%) required peritoneal dialysis, 18 (51.4%) experienced significant bleeding, and 12 (34.3%) needed factor VII administration.[35]
Following the Ross-Konno or Konno-Rastan procedure, replacing the RV with the PA conduit is the most common reoperation. Most patients with pulmonary regurgitation do not exhibit symptoms. However, those who do may initially experience exertional dyspnea due to decreased cardiac output caused by volume overload in the right heart. Over time, these patients might notice a gradual reduction in their ability to exercise. In more severe cases, pulmonary regurgitation can lead to symptoms indicative of right heart failure, such as swelling in the legs (pedal edema), an enlarged congested liver (congestive hepatomegaly), and occasionally, distended neck veins (jugular vein distention). Similarly, most individuals with mild pulmonary stenosis typically do not exhibit symptoms. Those who do experience symptoms usually have moderate to severe pulmonary stenosis and often present with exertional dyspnea or fatigue, which varies according to the degree of obstruction and the heart's ability to compensate. In severe pulmonary stenosis, where RV dysfunction develops, systemic venous congestion may occur.
The results of a recent study showed that freedom from reoperation was 66%. Twelve patients (34.3%) required at least 1 reoperation since their initial Ross or Ross-Konno procedure, with the median age at reoperation being 3.4 years. These reoperations included 10 RV-PA conduit interventions, 2 mitral valve repairs, and 1 mechanical mitral valve replacement. Additionally, 1 patient required reintervention on the LVOT, another needed right PA stenting and angioplasty at 16 months for progressive proximal right PA stenosis, and 1 patient had a permanent pacemaker implanted 2 weeks after a Ross-Konno due to complete atrioventricular block.[35]
Aortic valve disease can increase the risk of ventricular arrhythmias. The pressure or volume overload associated with the disease often leads to ventricular hypertrophy, which elevates myocardial oxygen demand while reducing coronary flow reserve. This combination creates a higher susceptibility to ischemia and scar tissue formation, both of which can trigger ventricular arrhythmias. Although aortic valve surgery, such as the Ross procedure, aims to address these issues, it may not eliminate the risk of arrhythmias. Furthermore, the surgical procedures, including the Ross procedure and additional interventions, may introduce proarrhythmic factors. For example, excision of the pulmonary root, enlargement of the aortic annulus (via Ross-Konno), and muscle resection in the subaortic area can all lead to scar formation, providing a substrate for reentrant arrhythmias. Additionally, the first septal perforator of the left anterior descending coronary artery may be damaged during the pulmonary root excision, resulting in ischemia and subsequent ventricular arrhythmias. The process of explanting and reimplanting the coronary arteries during surgery also carries a risk of creating ischemic areas, further contributing to the potential for ventricular arrhythmias.[37]
Deterrence and Patient Education
Deterrence and patient education regarding surgical approaches to congenital AS are essential for improving outcomes, minimizing complications, and ensuring long-term management. Although congenital AS is typically a structural defect that cannot be entirely prevented, early prenatal and newborn screening detection is critical. Regular monitoring of valve function LVOTO and associated cardiovascular anomalies can help clinicians intervene at the optimal time, potentially delaying the need for surgical intervention. In cases where congenital AS progresses to severe stenosis, timely referral to a cardiac specialist is essential to avoid complications such as heart failure or sudden cardiac death.
Preventive strategies also include managing modifiable risk factors that can exacerbate the condition, such as hypertension or infections like bacterial endocarditis. Patients with a history of congenital AS or BAV should receive prophylactic antibiotics before certain dental or surgical procedures to prevent infective endocarditis. Patient education focuses on helping patients and families understand the nature of congenital AS, its potential progression, and the importance of regular follow-ups. Clear communication about the various surgical options—such as balloon valvuloplasty, aortic valve repair, or the Ross-Konno procedure—along with the risks, benefits, and recovery expectations—is essential.
Patients should also be informed about postoperative care, including the importance of medication adherence, particularly for those on anticoagulants after mechanical valve replacement. Education on recognizing signs of complications, such as shortness of breath, chest pain, or palpitations, ensures prompt medical attention when needed. Long-term lifestyle guidance is equally important. Patients should understand the need for lifelong monitoring, exercise limitations, and potential future reoperations. For young patients, this education extends to school participation, sports activities, and the psychological impact of living with a congenital heart defect. Educating families empowers them to support the patient’s health and improve their quality of life while mitigating future risks.
Pearls and Other Issues
An urgent Ross-Konno procedure can be done safely in neonates and infants, where survival and LV recovery are excellent, but reintervention for pulmonary valve replacement is high.
Enhancing Healthcare Team Outcomes
Effective surgical management of congenital aortic stenosis demands a coordinated, interprofessional team approach to enhance patient-centered care and outcomes. The team, including congenital cardiac surgeons, pediatric cardiologists, anesthesiologists, and intensivists, must work closely together. Surgeons use detailed preoperative cardiologist assessments, including echocardiographic and imaging studies, to define left ventricular outflow tract anatomy. Advanced clinicians and nurses ensure precise perioperative monitoring, managing patient stability, fluids, and vital signs. Nurses also provide essential emotional support to patients and families. At the same time, pharmacists oversee anticoagulation therapy in cases involving mechanical valve replacements to mitigate risks such as thrombosis or hemorrhage.
Interprofessional communication and collaboration improve patient safety and outcomes through structured decision-making. Daily rounds, preoperative discussions, and postoperative follow-ups foster real-time adjustments to care plans and ensure that the entire team, from surgeons to recovery nurses, is aligned in addressing complications like infection or arrhythmias. The heart team approach, observed in coronary artery disease management, illustrates the value of diverse perspectives in clinical decision-making, as noted in United States and European guidelines.[29] This coordinated care continues into outpatient follow-up, with cardiologists and primary care clinicians monitoring long-term patient progress. Patient outcomes are enhanced through teamwork, communication, and shared decision-making, and surgical risks are minimized.
Media
(Click Image to Enlarge)
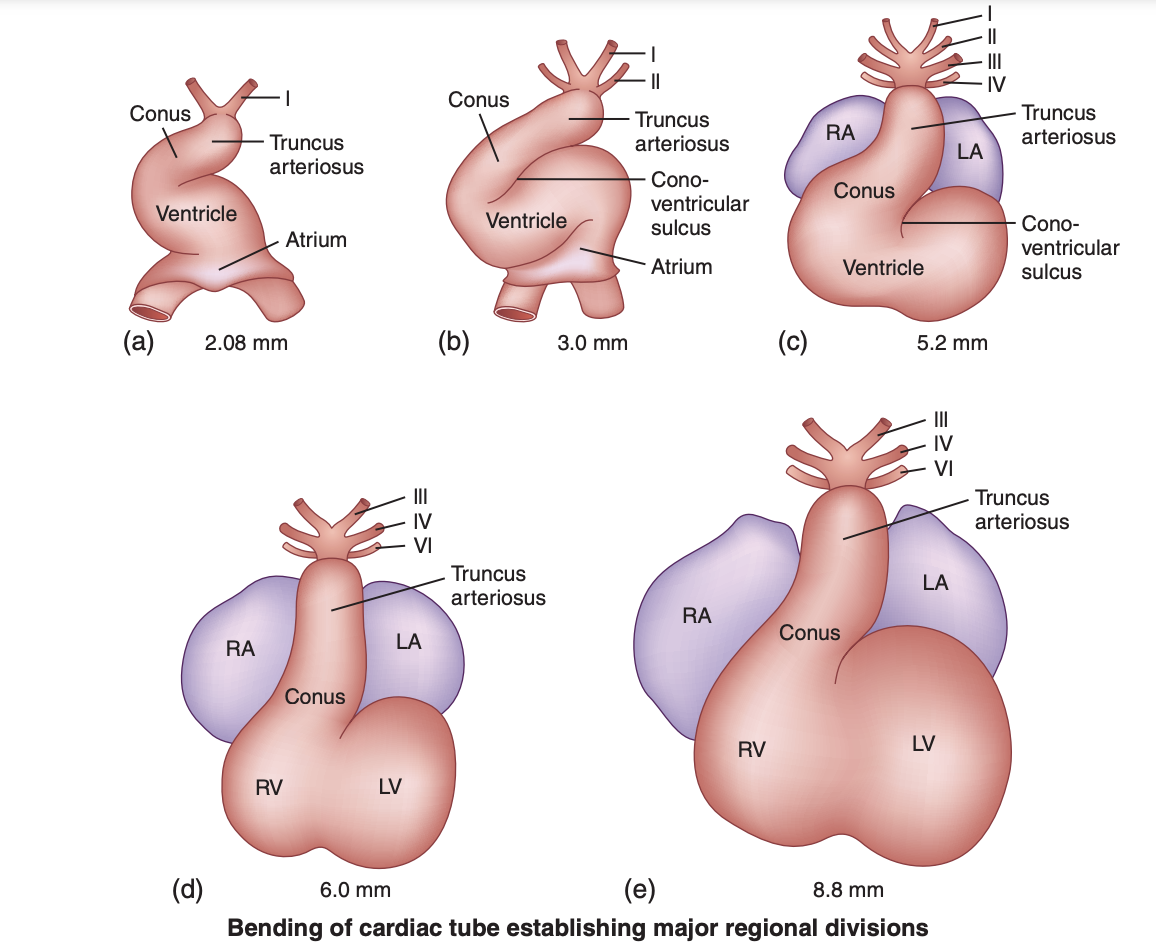
Bending of the Cardiac Tube. This image illustrates the process during embryonic development where the initially straight heart tube curves and twists into a "C" shape, known as "cardiac looping.”
Contributed by L Maximilian Buja, MD
(Click Image to Enlarge)
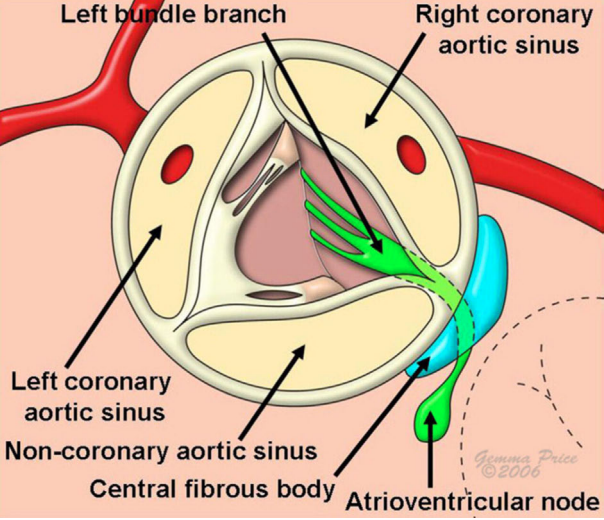
Complex Anatomy of the Left Ventricular Outflow Tract. This illustration shows the anatomy of the left ventricular outflow tract.
Contributed by V Tsang, MS, MSc, FRCS
(Click Image to Enlarge)
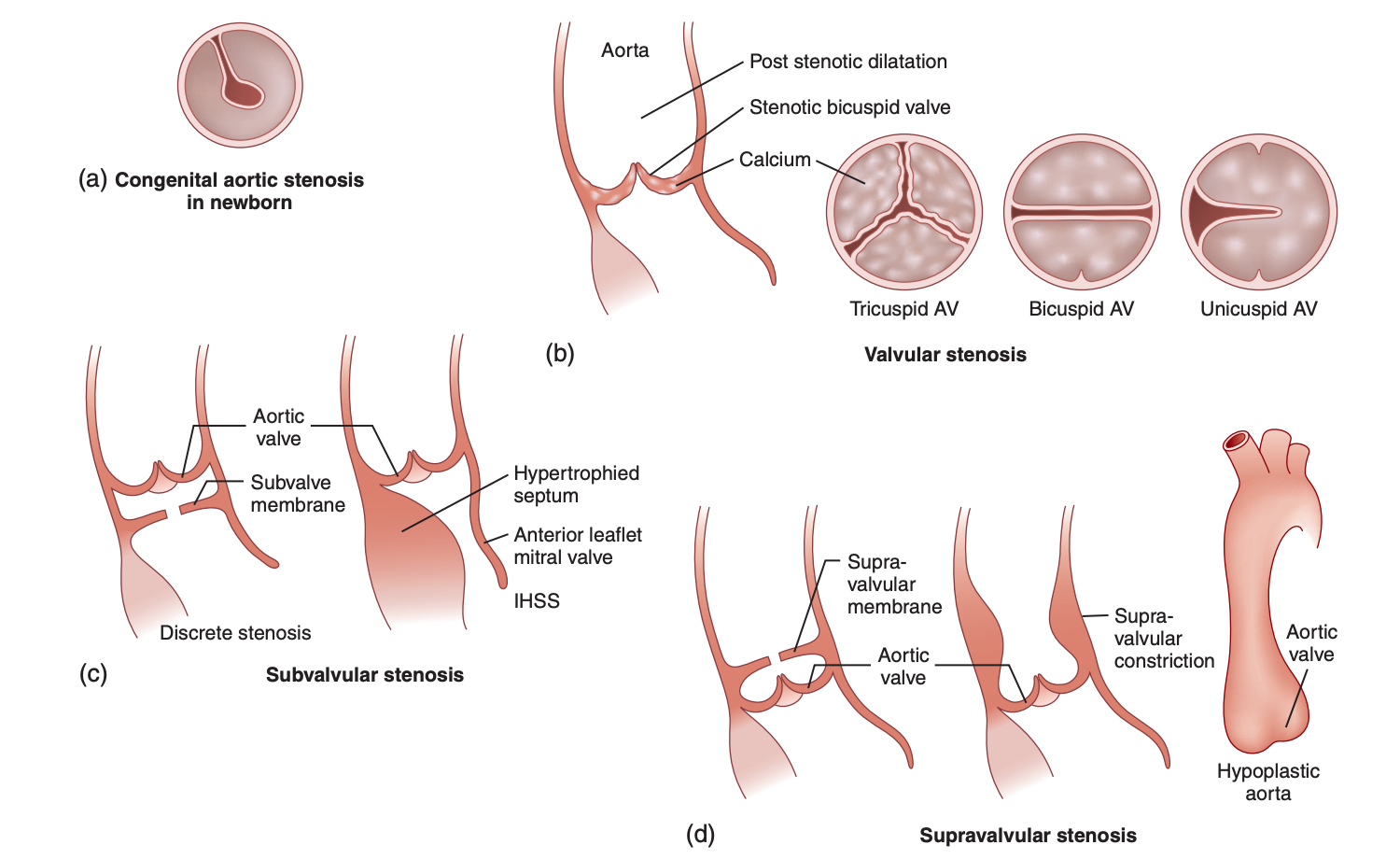
Types of Left Ventricular Outflow Tract Obstruction. Congenital aortic stenosis can be classified into 4 main types based on the location of the narrowing.
Contributed by L Maximilian Buja, MD
(Click Image to Enlarge)
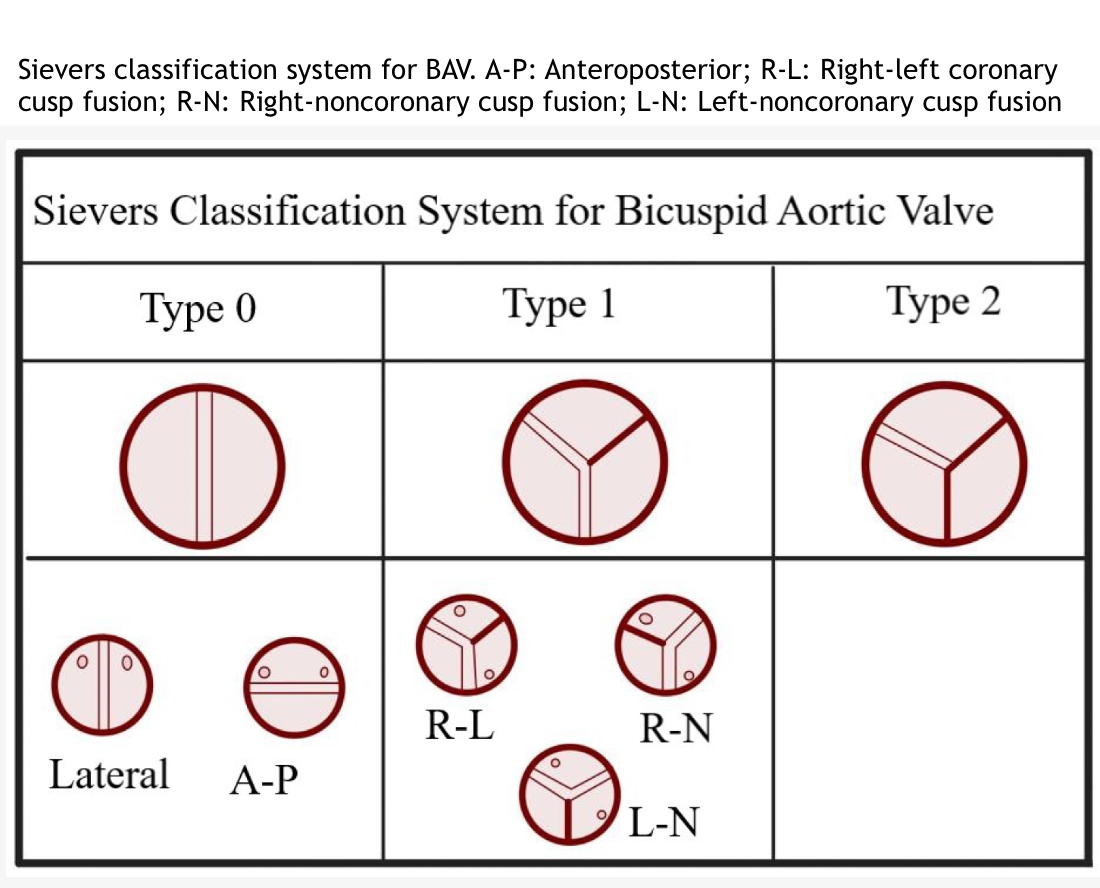
Sievers Classification System for Bicuspid Aortic Valve. This classification categorizes the bicuspid aortic valve into 3 main types.
Contributed by H Ibrahim Bulut, MD
(Click Image to Enlarge)
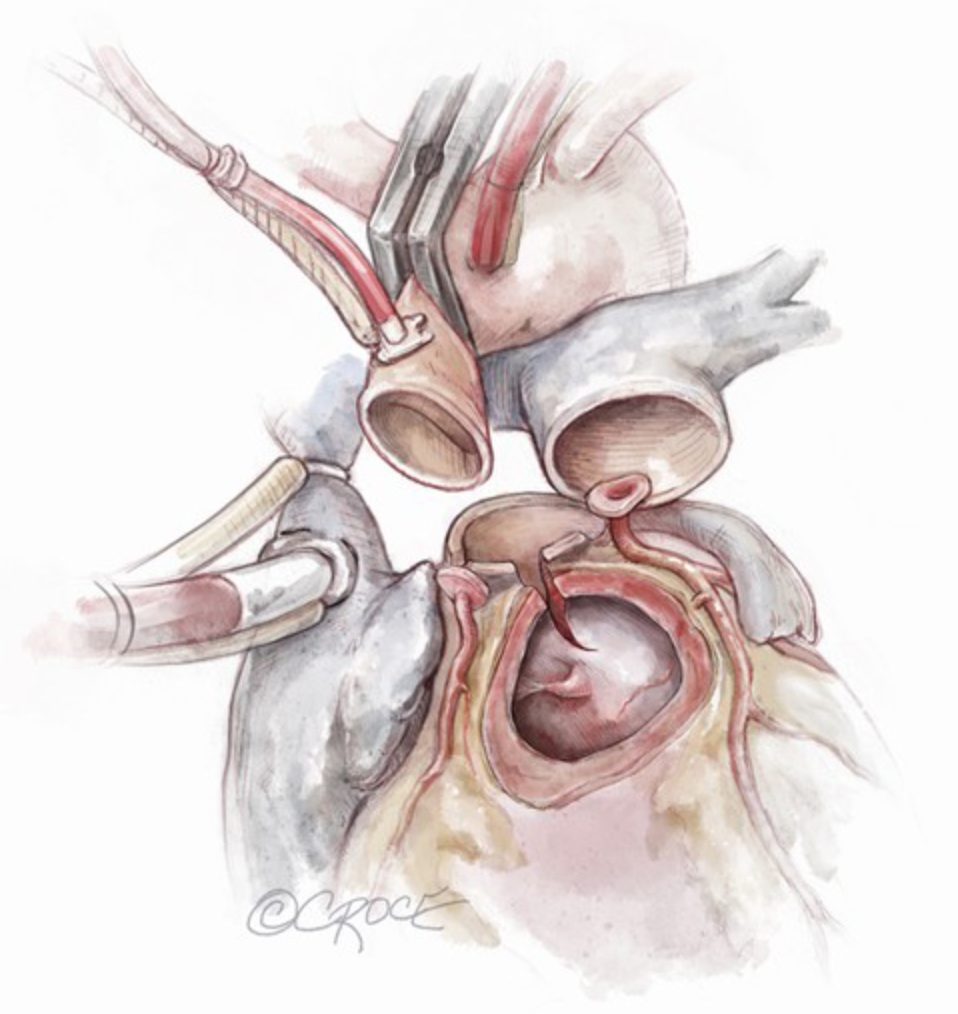
Ross-Konno Procedure. This illustration depicts a Konno incision in the interventricular septum.
Contributed by S Said, MD
(Click Image to Enlarge)
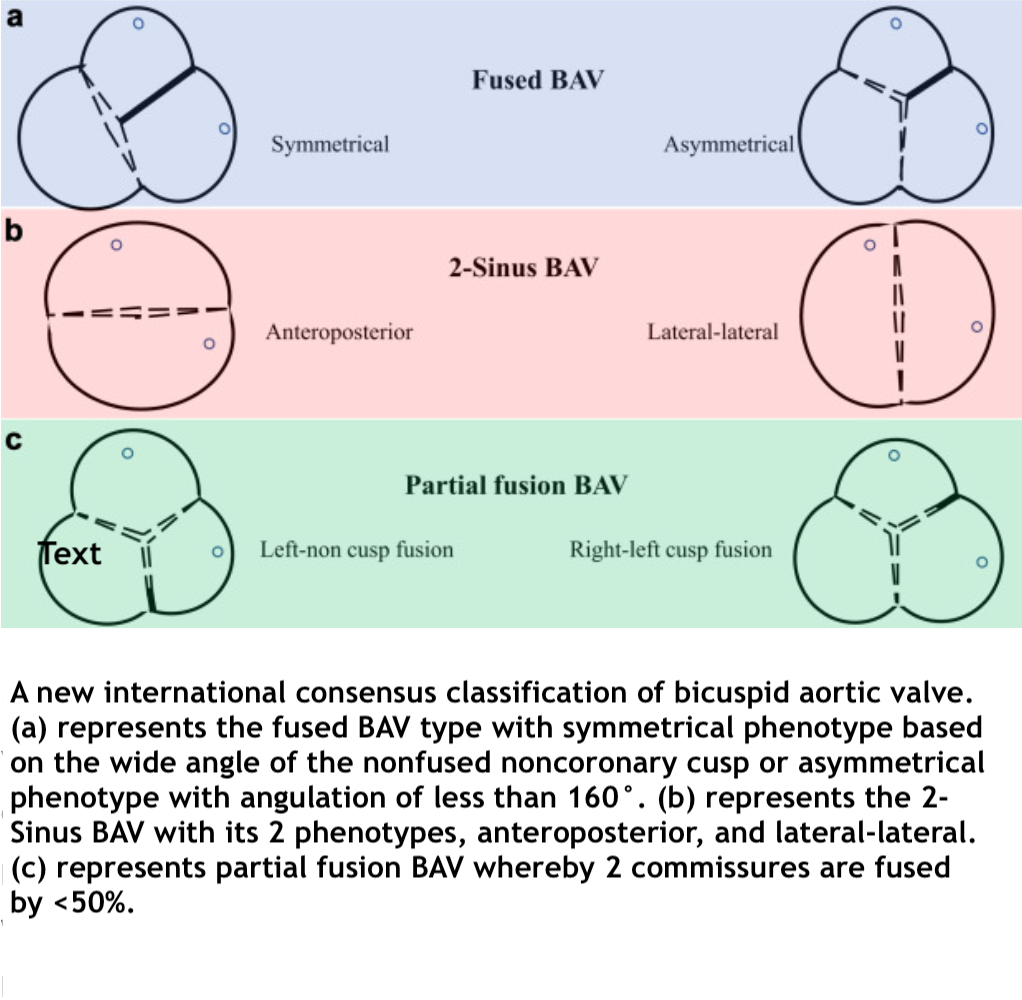
International Consensus Classification for Bicuspid Aortic Valve. This table shows the 3 types of bicuspid aortic valve classifications: fused, 2-sinus, and partial fusion.
Contributed by R Puri, MBBS, PhD
References
Amirghofran AA, Salimi M, Kamran H, Bazrafshan H, Navaei MR, Shokrollahi A, Nirooei E, Edraki M, Amoozgar H, Ajami G, Arabi H. Long-term outcomes of the classic Konno-Rastan procedure in paediatric and adult patients: impact of aortic annulus size on patient outcomes. Interdisciplinary cardiovascular and thoracic surgery. 2023 Oct 4:37(4):. doi: 10.1093/icvts/ivad151. Epub [PubMed PMID: 37665750]
Rana MS, Christoffels VM, Moorman AF. A molecular and genetic outline of cardiac morphogenesis. Acta physiologica (Oxford, England). 2013 Apr:207(4):588-615. doi: 10.1111/apha.12061. Epub 2013 Feb 1 [PubMed PMID: 23297764]
Ahmed SH, Deng AT, Huntley RP, Campbell NH, Lovering RC. Capturing heart valve development with Gene Ontology. Frontiers in genetics. 2023:14():1251902. doi: 10.3389/fgene.2023.1251902. Epub 2023 Oct 17 [PubMed PMID: 37915827]
Hochstrasser L, Ruchat P, Sekarski N, Hurni M, von Segesser LK. Long-term outcome of congenital aortic valve stenosis: predictors of reintervention. Cardiology in the young. 2015 Jun:25(5):893-902. doi: 10.1017/S1047951114001085. Epub 2014 Jul 1 [PubMed PMID: 24983130]
Bulut HI, Arjomandi Rad A, Syrengela AA, Ttofi I, Djordjevic J, Kaur R, Keiralla A, Krasopoulos G. A Comprehensive Review of Management Strategies for Bicuspid Aortic Valve (BAV): Exploring Epidemiology, Aetiology, Aortopathy, and Interventions in Light of Recent Guidelines. Journal of cardiovascular development and disease. 2023 Sep 18:10(9):. doi: 10.3390/jcdd10090398. Epub 2023 Sep 18 [PubMed PMID: 37754827]
Kalra A, Das R, Alkhalil M, Dykun I, Candreva A, Jarral O, Rehman SM, Majmundar M, Patel KN, Rodes-Cabau J, Reardon MJ, Puri R. Bicuspid Aortic Valve Disease: Classifications, Treatments, and Emerging Transcatheter Paradigms. Structural heart : the journal of the Heart Team. 2024 Jan:8(1):100227. doi: 10.1016/j.shj.2023.100227. Epub 2023 Oct 25 [PubMed PMID: 38283572]
Merla G, Brunetti-Pierri N, Piccolo P, Micale L, Loviglio MN. Supravalvular aortic stenosis: elastin arteriopathy. Circulation. Cardiovascular genetics. 2012 Dec:5(6):692-6. doi: 10.1161/CIRCGENETICS.112.962860. Epub [PubMed PMID: 23250899]
Pober BR, Johnson M, Urban Z. Mechanisms and treatment of cardiovascular disease in Williams-Beuren syndrome. The Journal of clinical investigation. 2008 May:118(5):1606-15. doi: 10.1172/JCI35309. Epub [PubMed PMID: 18452001]
Stamm C, Friehs I, Ho SY, Moran AM, Jonas RA, del Nido PJ. Congenital supravalvar aortic stenosis: a simple lesion? European journal of cardio-thoracic surgery : official journal of the European Association for Cardio-thoracic Surgery. 2001 Feb:19(2):195-202 [PubMed PMID: 11167112]
van der Linde D, Konings EE, Slager MA, Witsenburg M, Helbing WA, Takkenberg JJ, Roos-Hesselink JW. Birth prevalence of congenital heart disease worldwide: a systematic review and meta-analysis. Journal of the American College of Cardiology. 2011 Nov 15:58(21):2241-7. doi: 10.1016/j.jacc.2011.08.025. Epub [PubMed PMID: 22078432]
Level 1 (high-level) evidenceHoffman JI, Kaplan S. The incidence of congenital heart disease. Journal of the American College of Cardiology. 2002 Jun 19:39(12):1890-900 [PubMed PMID: 12084585]
Tran PK, Tsang V. When and How to Enlarge the Small Aortic Root. Seminars in thoracic and cardiovascular surgery. Pediatric cardiac surgery annual. 2016:19(1):55-8. doi: 10.1053/j.pcsu.2015.11.007. Epub [PubMed PMID: 27060044]
Roberts WC, Ko JM. Relation of weights of operatively excised stenotic aortic valves to preoperative transvalvular peak systolic pressure gradients and to calculated aortic valve areas. Journal of the American College of Cardiology. 2004 Nov 2:44(9):1847-55 [PubMed PMID: 15519018]
Schlein J, Wollmann F, Kaider A, Wiedemann D, Gabriel H, Hornykewycz S, Base E, Michel-Behnke I, Laufer G, Zimpfer D. Long-term outcomes after surgical repair of subvalvular aortic stenosis in pediatric patients. Frontiers in cardiovascular medicine. 2022:9():1033312. doi: 10.3389/fcvm.2022.1033312. Epub 2022 Dec 2 [PubMed PMID: 36531724]
Singh GK. Congenital Aortic Valve Stenosis. Children (Basel, Switzerland). 2019 May 13:6(5):. doi: 10.3390/children6050069. Epub 2019 May 13 [PubMed PMID: 31086112]
Otto CM, Nishimura RA, Bonow RO, Carabello BA, Erwin JP 3rd, Gentile F, Jneid H, Krieger EV, Mack M, McLeod C, O'Gara PT, Rigolin VH, Sundt TM 3rd, Thompson A, Toly C. 2020 ACC/AHA Guideline for the Management of Patients With Valvular Heart Disease: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation. 2021 Feb 2:143(5):e72-e227. doi: 10.1161/CIR.0000000000000923. Epub 2020 Dec 17 [PubMed PMID: 33332150]
Yasuhara J, Schultz K, Bigelow AM, Garg V. Congenital aortic valve stenosis: from pathophysiology to molecular genetics and the need for novel therapeutics. Frontiers in cardiovascular medicine. 2023:10():1142707. doi: 10.3389/fcvm.2023.1142707. Epub 2023 Apr 28 [PubMed PMID: 37187784]
Alsoufi B. The Konno-Rastan procedure in children and young adults: efficiency versus effectiveness. Interdisciplinary cardiovascular and thoracic surgery. 2023 Oct 4:37(4):. doi: 10.1093/icvts/ivad171. Epub [PubMed PMID: 37847656]
Masri A, Kalahasti V, Svensson LG, Alashi A, Schoenhagen P, Roselli EE, Johnston DR, Rodriguez LL, Griffin BP, Desai MY. Aortic Cross-Sectional Area/Height Ratio and Outcomes in Patients With Bicuspid Aortic Valve and a Dilated Ascending Aorta. Circulation. Cardiovascular imaging. 2017 Jun:10(6):e006249. doi: 10.1161/CIRCIMAGING.116.006249. Epub [PubMed PMID: 28592593]
Level 2 (mid-level) evidenceWriting Committee Members, Otto CM, Nishimura RA, Bonow RO, Carabello BA, Erwin JP 3rd, Gentile F, Jneid H, Krieger EV, Mack M, McLeod C, O'Gara PT, Rigolin VH, Sundt TM 3rd, Thompson A, Toly C. 2020 ACC/AHA Guideline for the Management of Patients With Valvular Heart Disease: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Journal of the American College of Cardiology. 2021 Feb 2:77(4):e25-e197. doi: 10.1016/j.jacc.2020.11.018. Epub 2020 Dec 17 [PubMed PMID: 33342586]
Chubb H, Simpson JM. The use of Z-scores in paediatric cardiology. Annals of pediatric cardiology. 2012 Jul:5(2):179-84. doi: 10.4103/0974-2069.99622. Epub [PubMed PMID: 23129909]
Gillis E, Kumar AA, Luyckx I, Preuss C, Cannaerts E, van de Beek G, Wieschendorf B, Alaerts M, Bolar N, Vandeweyer G, Meester J, Wünnemann F, Gould RA, Zhurayev R, Zerbino D, Mohamed SA, Mital S, Mertens L, Björck HM, Franco-Cereceda A, McCallion AS, Van Laer L, Verhagen JMA, van de Laar IMBH, Wessels MW, Messas E, Goudot G, Nemcikova M, Krebsova A, Kempers M, Salemink S, Duijnhouwer T, Jeunemaitre X, Albuisson J, Eriksson P, Andelfinger G, Dietz HC, Verstraeten A, Loeys BL, Mibava Leducq Consortium. Candidate Gene Resequencing in a Large Bicuspid Aortic Valve-Associated Thoracic Aortic Aneurysm Cohort: SMAD6 as an Important Contributor. Frontiers in physiology. 2017:8():400. doi: 10.3389/fphys.2017.00400. Epub 2017 Jun 13 [PubMed PMID: 28659821]
Musunuru K, Hershberger RE, Day SM, Klinedinst NJ, Landstrom AP, Parikh VN, Prakash S, Semsarian C, Sturm AC, American Heart Association Council on Genomic and Precision Medicine; Council on Arteriosclerosis, Thrombosis and Vascular Biology; Council on Cardiovascular and Stroke Nursing; and Council on Clinical Cardiology. Genetic Testing for Inherited Cardiovascular Diseases: A Scientific Statement From the American Heart Association. Circulation. Genomic and precision medicine. 2020 Aug:13(4):e000067. doi: 10.1161/HCG.0000000000000067. Epub 2020 Jul 23 [PubMed PMID: 32698598]
Isselbacher EM, Preventza O, Hamilton Black J 3rd, Augoustides JG, Beck AW, Bolen MA, Braverman AC, Bray BE, Brown-Zimmerman MM, Chen EP, Collins TJ, DeAnda A Jr, Fanola CL, Girardi LN, Hicks CW, Hui DS, Schuyler Jones W, Kalahasti V, Kim KM, Milewicz DM, Oderich GS, Ogbechie L, Promes SB, Gyang Ross E, Schermerhorn ML, Singleton Times S, Tseng EE, Wang GJ, Woo YJ, Peer Review Committee Members. 2022 ACC/AHA Guideline for the Diagnosis and Management of Aortic Disease: A Report of the American Heart Association/American College of Cardiology Joint Committee on Clinical Practice Guidelines. Circulation. 2022 Dec 13:146(24):e334-e482. doi: 10.1161/CIR.0000000000001106. Epub 2022 Nov 2 [PubMed PMID: 36322642]
Level 1 (high-level) evidenceAyoub C, Geske JB, Larsen CM, Scott CG, Klarich KW, Pellikka PA. Comparison of Valsalva Maneuver, Amyl Nitrite, and Exercise Echocardiography to Demonstrate Latent Left Ventricular Outflow Obstruction in Hypertrophic Cardiomyopathy. The American journal of cardiology. 2017 Dec 15:120(12):2265-2271. doi: 10.1016/j.amjcard.2017.08.047. Epub 2017 Sep 19 [PubMed PMID: 29054275]
Reant P, Dufour M, Peyrou J, Reynaud A, Rooryck C, Dijos M, Vincent C, Cornolle C, Roudaut R, Lafitte S. Upright treadmill vs. semi-supine bicycle exercise echocardiography to provoke obstruction in symptomatic hypertrophic cardiomyopathy: a pilot study. European heart journal. Cardiovascular Imaging. 2018 Jan 1:19(1):31-38. doi: 10.1093/ehjci/jew313. Epub [PubMed PMID: 28329285]
Level 3 (low-level) evidenceElliott PM, Gimeno JR, Tomé MT, Shah J, Ward D, Thaman R, Mogensen J, McKenna WJ. Left ventricular outflow tract obstruction and sudden death risk in patients with hypertrophic cardiomyopathy. European heart journal. 2006 Aug:27(16):1933-41 [PubMed PMID: 16754630]
Avegliano G, Politi MT, Costabel JP, Kuschnir P, Trivi M, Ronderos R. Differences in the extent of fibrosis in obstructive and nonobstructive hypertrophic cardiomyopathy. Journal of cardiovascular medicine (Hagerstown, Md.). 2019 Jun:20(6):389-396. doi: 10.2459/JCM.0000000000000800. Epub [PubMed PMID: 30994509]
Lee G, Chikwe J, Milojevic M, Wijeysundera HC, Biondi-Zoccai G, Flather M, Gaudino MFL, Fremes SE, Tam DY. ESC/EACTS vs. ACC/AHA guidelines for the management of severe aortic stenosis. European heart journal. 2023 Mar 7:44(10):796-812. doi: 10.1093/eurheartj/ehac803. Epub [PubMed PMID: 36632841]
Ross DN. Replacement of aortic and mitral valves with a pulmonary autograft. Lancet (London, England). 1967 Nov 4:2(7523):956-8 [PubMed PMID: 4167516]
Elkins RC, Knott-Craig CJ, Ward KE, McCue C, Lane MM. Pulmonary autograft in children: realized growth potential. The Annals of thoracic surgery. 1994 Jun:57(6):1387-93; discussion 1393-4 [PubMed PMID: 8010778]
Hörer J, Hanke T, Stierle U, Takkenberg JJ, Bogers AJ, Hemmer W, Rein JG, Hetzer R, Hübler M, Robinson DR, Sievers HH, Lange R. Neoaortic root diameters and aortic regurgitation in children after the Ross operation. The Annals of thoracic surgery. 2009 Aug:88(2):594-600; discussion 600. doi: 10.1016/j.athoracsur.2009.04.077. Epub [PubMed PMID: 19632419]
Pradegan N, Castaldi B, Azzolina D, Stellin G, Vida VL. Long-Term Fate of the Neoaortic Root After Neonatal Ross Operation: A Case Series. World journal for pediatric & congenital heart surgery. 2019 May:10(3):364-366. doi: 10.1177/2150135119840023. Epub [PubMed PMID: 31084313]
Level 2 (mid-level) evidenceSames-Dolzer E, Wickenhauser E, Kreuzer M, Benedikt P, Gitter R, Prandstetter C, Gierlinger G, Tulzer G, Mair R. The Ross-Konno procedure in neonates and infants less than 3 months of age. European journal of cardio-thoracic surgery : official journal of the European Association for Cardio-thoracic Surgery. 2018 Jul 1:54(1):71-77. doi: 10.1093/ejcts/ezy030. Epub [PubMed PMID: 29444227]
Luxford JC, Ayer JG, Betts K, Salve GG, Orr Y, Chard RB, Roberts P, Sholler GF, Winlaw DS. The Ross/Ross-Konno procedure in infancy is a safe and durable solution for aortic stenosis. The Journal of thoracic and cardiovascular surgery. 2022 Feb:163(2):365-375. doi: 10.1016/j.jtcvs.2021.06.066. Epub 2021 Sep 8 [PubMed PMID: 34600763]
Kadiyani L, Ramakrishnan S, Verma M, Kumar S, Hote M. Unusual large mass on aortic valve in an infant. Annals of pediatric cardiology. 2022 Sep-Dec:15(5-6):529-532. doi: 10.4103/apc.apc_240_21. Epub 2023 Mar 1 [PubMed PMID: 37152505]
Pasquali SK, Marino BS, Kaltman JR, Schissler AJ, Wernovsky G, Cohen MS, Spray TL, Tanel RE. Rhythm and conduction disturbances at midterm follow-up after the ross procedure in infants, children, and young adults. The Annals of thoracic surgery. 2008 Jun:85(6):2072-8. doi: 10.1016/j.athoracsur.2008.02.051. Epub [PubMed PMID: 18498822]