 Batten Disease (Juvenile Neuronal Ceroid Lipofuscinosis)
Batten Disease (Juvenile Neuronal Ceroid Lipofuscinosis)
Introduction
Neuronal ceroid lipofuscinoses (NCLs) are a group of devastating and lethal neurodegenerative lysosomal storage diseases that usually affect children.[1] The disease is known to have 14 forms, each resulting from mutations in a distinct gene. While the NCLs are individually rare conditions, together, they form the most common neurodegenerative disorder in children.[1] These disorders affect the cellular removal of a waste product called ceroid lipofuscin.[2] Cellular accumulation of ceroid lipofuscin indicates lysosomal dysfunction.
Although Batten disease was historically regarded as the juvenile form of NCL, some physicians used, and still use, the term "Batten disease" to describe all forms of NCL.[3] The disease is named after Frederick Eustace Batten, an English pediatrician and neurologist who is also called the father of pediatric neurology.[4]
Each form of the disease is identified as ceroid lipofuscinosis neuronal (CLN) and given a different number based on the subtype (ie, CLN1 and CLN2).[1] Each form of Batten disease, along with the associated gene variant, has different severity levels, average ages of onset, and progresses at different rates.
Problems with the lysosomes lead to the death of neurons over time. Patients with NCLs usually grow and develop normally in the initial phases of life, reaching developmental milestones at appropriate times. However, at the onset of the disease, the progress stops, and there is a decline in acquired skills. Depending on the onset of such symptoms, the disease may be congenital (onset at birth), infantile-onset (usual onset within the first year of life or within 6 months to 18 months), late-infantile onset (usually onset at 2 to 4 years of age), juvenile-onset (onset at 4 to 10 years of age, usually at 4 to 7 years of age), late juvenile onset (usual onset at age 8 to 16), and adult-onset (onset during late teenage years to mid-adulthood).[5]
Diagnostic methods include genetic analysis, enzyme assay, histopathology, electron microscopy, electrophysiological tests, and neuroimaging. Management is primarily symptomatic, though approved disease-specific therapy also exists.[6]
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
Batten disease is primarily caused by genetic mutations; the possibility of an autoimmune etiology has also been described.[7]
Genetic: More than a dozen genes containing over 430 mutations underlying human NCLs have been identified (see Table 1. Genes Known to Cause NCL).[8] These genes encode lysosomal enzymes (CLN1, CLN2, CLN10, CLN13), a soluble lysosomal protein (CLN5), a protein in the secretory pathway (CLN11), 2 cytoplasmic proteins that also peripherally associate with membranes (CLN4, CLN14), and many transmembrane proteins with different subcellular locations (CLN3, CLN6, CLN7, CLN8, CLN12).
For the majority of NCLs, the function of the causative gene has not been fully defined. Most of the mutations in these genes are associated with a typical disease phenotype, but some result in variable disease onset, severity, and progression, including distinct clinical phenotypes.[9] CLN3 disease is caused by bi-allelic mutations in CLN3, encoding CLN3, a presumed lysosomal protein.[10] Without proper CLN3 functioning, ceroid lipopigments accumulate in all body cells, predominantly affecting the retina and the brain.[11]
Table 1. Genes Known to Cause NCL
| DISEASE | CHROMOSOME LOCATION | GENE AFFECTED | PROTEIN |
| Congenital NCL (CNCL) | 11p15.5 | CTSD | Cathepsin D [8] |
| Santavuori-Haltia or Infantile-Onset NCL (INCL) | 1p32 | CLN1 | Palmitoyl protein thioesterase I [12] |
| Jansky-Bielschowsky or Late Infantile-Onset NCL (LINCL) | 11p15 | CLN2 | Tripeptidylpeptidase protein I [13] |
| Batten, Spielmeyer-Sjogren or Juvenile-Onset NCL (JNCL) | 16p12 | CLN3 [14] | Unknown |
| Kufs or Adult-Onset NCL (ANCL) | Unknown | CLN4 (DNAJC5)[15] | Unknown |
| Variant Finnish LINCL (vfinLINCL) | 13q31-32 | CLN5 [16] | Unknown |
| Costa Rican or Variant LINCL (VLINCL) | 15q21-23 | CLN6 [17] | Unknown |
| Variant Turkish LINCL (vturkLINCL) | 4q28.1-q28.2 |
CLN7 (MFSD8) [18] |
Unknown |
| Northern Epilepsy/Epilepsy with Progressive Mental Retardation | 8p23 | CLN8 [19] | Unknown |
Autoimmunity and Inflammation: In studying a mouse model for Batten disease, Chattopadhyay et al reported the presence of an autoantibody to glutamic acid decarboxylase (GAD65) in CLN3-knockout mice serum that associates with brain tissue but is not present in sera or brain of unaffected mice.[20] This autoantibody to GAD65 was seen to inhibit the activity of glutamic acid decarboxylase, resulting in brain samples from CLN3-knockout mice having elevated levels of glutamate compared to normal levels. Excessive excitatory neurotransmitters, including glutamate, may cause toxicity to the neural tissue and features such as seizures.[21] An autoantibody to GAD65 was found in individuals with Batten disease.[22] Hence, the authors suggested that an autoimmune response to GAD65 may contribute to a preferential loss of GABAergic neurons associated with Batten disease.[22] However, the association between Batten disease and anti-GAD antibodies is still being explored.
Autoimmunity may also target other neuronal antigens.[23] Changes in antigen-presenting cells and microglia (including increased expression of sialoadhesin) are noted in those with Batten disease, which might predispose to neural tissue inflammation.[24][25] Also, an anti-alpha fetoprotein antibody has been noted in the sera of some patients with Batten disease.[26]
Epidemiology
Batten disease is the most common neurodegenerative disorder in childhood.[3] This disease can result from mutations in various genes, and the worldwide prevalence of Batten disease is approximately 1 in 100,000 live births.[27] The maximum prevalence of NCL disease is noted in Scandinavian countries, including Finland.[28]
JNCL (CLN3) is the most common form of NCL worldwide.[29] Other common types of NCL include adult NCL (CLN4, Kufs, or Parry disease) and late infantile NCL (CLN2, Jansky-Bielschowsky disease). CLN1 (Santavuori-Haltia disease) is common in Finland, with a 1 in 20,000 incidence and a carrier frequency of 1 in 70.[29] About 20% of NCL cases in the United States are due to CLN1.[29]
The JNCL Batten disease is caused by a mutation in the CLN3 gene inherited in an autosomal recessive pattern, mostly in White populations.[30] The incidences of the CLN3 mutation have been reported to range from 0.02 to 4.8 per 100,000 worldwide.[31][32] The carrier frequencies for the most common mutation have been estimated with advancements in genetic analyses and databases. In Finnish and non-Finnish Europeans, the estimated frequencies are 1/558 and 1/380 individuals, whereas in Latinos and Americans, it is estimated to be 1/1169 and 1/506 individuals.[33]
Pathophysiology
Each form of NCL has distinct defects in genes encoding proteins in the endo-lysosomal system.[30] These can be classified into soluble lysosomal enzyme defects, cytosolic protein deficiencies, and defects in insoluble transmembrane proteins.[2] These transmembrane proteins may either be present in the lysosomal membrane or the endoplasmic reticulum, which is involved in the endo-lysosomal system. However, the exact mechanisms by which these mutations lead to the common downstream phenotypes remain largely unresolved. These include the pathognomic accumulation of autofluorescent storage material, dysregulated autophagy, significant and progressive glial activation, and neuronal death within the nervous system.[34]
Since NCLs result from distinct monogenic mutations, each form may have unique pathomechanisms affecting the endo-lysosomal system, leading to different phenotypes.[35] Many of the pathological phenotypes seen in the NCLs are common to other lysosomal storage disorders as well as other neurodegenerative disorders.[36]
Recently, research into glial cells, including astrocytes, microglia, and oligodendrocytes, has revealed many of their critical functions in brain homeostasis and potential contributions to neurodegenerative diseases.[37] A consideration of glial dysfunction and its contribution to the pathogenesis of CLN1, CLN2, and CLN3 diseases (the 3 most common forms of NCL) is being explored.[38]
Histopathology
Histological anomalies are diverse and occasionally incoherent. Early degeneration of dendritic spines in apical dendrites may elicit a microglial response and cause neuro-inflammation. Together with microglial activation, macrophages phagocytose cellular debris and lipopigments. Loss of neurons and their axons causes the breakdown of myelin and, thus, demyelination, although mainly of a secondary nature. Eventually, cellular astrocytosis and later fibrillar astrocytosis ensue, transforming areas of preceding primary nerve cell loss into scarred regions. At autopsy, the cerebrum and cerebellum cortices may variably deplete neurons.[39] The limited data on cardio-pathological findings highlights the frequently heavy production of NCL lipopigments, with vacuolation, particularly in the conduction system, and also reveals degenerative myocardial fibrosis.[40]
The retina is involved in the pathological process of NCL in all childhood forms. Retinal degeneration probably starts at the outer segments of the photoreceptors and proceeds inward during the disease. Loss of photoreceptors is more prominent in central regions. Details of autopsied retinas in adult NCL patients are less often reported, as visual failure or blindness is less prevalent in adult NCL.[41] A postmortem study on a 33-year-old woman who had late adolescence onset of molecularly proven CLN5 revealed preservation of all retinal layers, including those of the photoreceptors, but lipopigment formation was encountered in each neuronal cell type, suggesting that retinal degeneration and lipopigment formation may not be closely associated.[42] Retinal atrophy is most pronounced in CLN1 and CLN3, less so in CLN2. In CLN5, less severe pathology has briefly been described.[35]
History and Physical
NCL has varied presentations based on the 14 types of NCL that have been described in the literature. The distinguishing features are as follows:
CLN1 (classic infantile-onset form): The affected child may have microcephaly, seizures, motor weakness, and speech deterioration. Infantile NCL typically spares most of the anterior portion of the eye with no gross changes in eyelids, conjunctiva, cornea, iris, and even vitreous. The intraocular pressure is also usually normal. The fundus may show atrophy of the retinal pigment epithelium (RPE), attenuation of retinal vessels, and optic disc pallor secondary to optic atrophy. Aggregation of pigment in the macula has been noted with characteristic “patchy ring-like defects.” Peripheral retinal pigmentation is not classically seen in patients with CLN1 disease.[43]
CLN2 (classic late infantile-onset form): In most patients, the onset of ocular symptoms occurs late in the disease at an average age of 4 to 5.[44] The child starts having a language delay and epilepsy at around 2 to 3 years of age, which can be accompanied by ataxia and global developmental delay by 5 years of age. No anterior segment manifestation has been reported in the literature. The fundus may exhibit fulminant bull’s eye maculopathy.[45][44] Patients should be evaluated for motor, seizure, language, and visual symptoms.
CLN3 (classic juvenile-onset form): Visual symptoms precede or coincide with neuropsychiatric manifestations. The mean age of presentation to an ophthalmologist is 5.5 to 8.5 years.[46] The child may exhibit rotatory nystagmus, along with eccentric viewing/overlooking. Normal to severe pigmentary retinopathy with optic disc pallor, arteriolar attenuation, and peripheral pigmentation may be seen on fundus examination.[47] Peripheral retinal pigment accumulation occurs later. The progression of retinal disease and visual symptoms is usually faster than other retinal degenerations.[48] Rarely, exudative retinal vasculopathy like Coats disease may be seen.[49][50] Other motor symptoms include myoclonus, parkinsonism, and severe dysarthria, resulting in mutism during their twenties. Most patients develop behavioral problems with angry outbursts, physical violence, and features of depression.[51]
CLN4 (Kufs disease): Unlike other variants of NCL, which are autosomal recessive, CLN4 is autosomal dominantly inherited. It has 2 clinical phenotypes as follows:
- Type A presents with the onset of generalized tonic-clonic seizures followed by progressive myoclonic epilepsy, ataxia, and dysarthria.
- Neuropsychiatric symptoms, including dementia, behavioral symptoms, and cerebellar or extrapyramidal signs, dominate type B. Vision is usually preserved, and there is no retinal involvement.[52] However, the retina may show ceroid lipofuscin on histology in some cases.[53]
CLN5 (Finnish variant late infantile NCL [LINCL]): An affected child usually has ataxia at 2 to 6 years of age. This is followed by psychomotor regression with the inability to walk by age 10. Most of the patients do not survive until their third decade. Vision loss is progressive, and retinal degeneration occurs between 6 and 10 years of age. The fundus shows macular dystrophy and optic nerve atrophy.[47]
CLN6 (Costa Rican variant LINCL): The age of disease onset is highly variable from 18 months to 8 years. Affected individuals exhibit progressive cerebellar, pyramidal, and extrapyramidal signs, with seizures typically leading to death by their mid-twenties.[47]
CLN7 (earlier known as the Turkish variant LINCL): Affected children have tremors and lower limb weakness. Hypermetria, along with bradykinesia, occurs during the later stages. The visual impairment is diagnosed between 4 and 5.5 years of age.[47]
CLN8 (previously known as northern epilepsy syndrome, or progressive epilepsy with mental retardation [EPMR]): The EPMR variant is characterized by the onset of generalized seizures, often refractory to treatment between 5 and 10 years of age, and cognitive decline but no vision loss. The juvenile-onset variant, which is called northern epilepsy, presents around 3 to 7 years of age with a motor decline, severe myoclonic epilepsy, and severe vision loss, along with ataxia and dysarthria.[54]
CLN9: This form is extremely rare. Like most of the other types, this form also presents with progressive ataxia and seizures. The underlying genetic causes of the disease are yet to be delineated. Therefore, CLN-9 has not been confirmed as a distinct clinical entity.[47]
CLN10 (previously known as congenital neuronal ceroid lipofuscinosis): This form is a severe neurodegenerative variant. The disease manifests around birth as respiratory insufficiency and status epilepticus, followed by death within hours to weeks. The patients are microcephalic with severe brain atrophy and exhibit an absence of neonatal reflexes.[55] The fundus may show pigmentary retinopathy.[56]
CLN11: This form presents between 13 and 25 years of age. Affected individuals experience cerebellar ataxia, seizures, and cognitive disorders along with retinitis pigmentosa and retinal dystrophy.[57]
CLN12: The child may have learning difficulties and show extrapyramidal signs with progressive loss of muscle strength, like that of Parkinson disease. The eyes may show slow vertical movements; however, the retina remains uninvolved.[58]
CLN13: The neurological presentation develops between 12 and 16 years of age and is characterized by a lack of coordination, muscular weakness, and premature death.[59]
CLN14: This form is characterized by rapidly progressing neurodegeneration and intractable myoclonic seizures before 2 years of age. Vision loss is also rapidly progressive, with signs of optic atrophy on fundoscopy.[60]
Evaluation
At least 14 types of Batten disease (NCL1 through NCL14) have been identified; however, the most commonly observed forms are NCL1, NCL2, and NCL3.[61] Key diagnostic criteria, symptomatology, and available testing methods are essential for accurately identifying the various forms of this complex neurodegenerative disorder.
Clinical symptoms:
- Common clinical hallmarks of Batten disease include visual impairment, inability to achieve typical developmental milestones and/or developmental regression, behavioral problems, progressive cerebral atrophy, seizures, cognitive decline, and dementia.[62]
- However, as discussed in the previous section, the order and frequency of these symptoms vary between the different subtypes and variants for each genetic mutation.
Genetic testing:
- NCLs are now recognized for their significant genotype-phenotype heterogeneity, where different mutations in the same gene or across genes can produce identical phenotypes. Similarly, the same mutation in a single gene may result in variable phenotypes among individuals within a family.[43]
- The gene defect causing the infantile form (CLN1) has been assigned to 1p32 (1p34.2, according to OMIM) in Finnish families.[63] In contrast, the disease locus of the juvenile (CLN3) form has been localized to 16p12 in European and Canadian families.[63] The gene defect causing the late infantile form (CLN2) has been mapped to chromosome 11p15.[13]
Biochemical testing:
- For CLN1, palmitoyl protein thioesterase (PPT) levels can be measured in leukocytes, cultured fibroblasts, dried blood spots, and saliva. Lymphoblast PPT is less than 0.2 pmoles/min/mg (normal levels are 1 to 3).[64]
- For CLN2, tripeptidyl peptidase 1 (TTP1) levels can be measured in leukocytes, cultured fibroblasts, dried blood spots, and saliva. Fibroblast TTP1 activity is approximately 17,000 micromoles of amino acids produced per hour per milligram of protein. The TTP1 activity in CLN2 NCL is less than 4% of normal levels.[65]
Neuroimaging:
- Magnetic resonance imaging (MRI) & MR Spectroscopy- NCL shows predominant cerebellar-over-cerebral atrophy in CLN2, CLN5, and CLN7 diseases and minimal abnormality until early adolescence in CLN3 disease.[66] Marked cerebellar atrophy is visible at an early stage of CLN2 disease.[67]
- Positron emission tomography (PET) Scan: PET with 2-deoxy-2 [18F]-fluoro-D-glucose measures functional brain activity, particularly in the dendritic field.[68] In CLN3, hypometabolism slowly spreads from calcarine to anterior areas, sparing subcortical structures and the brainstem. In CLN2, degeneration is rapid with generalized cortical and subcortical hypometabolism. This is associated with rapidly progressive cerebral atrophy on anatomical neuroimaging.[69]
Other diagnostic modalities:
- Electron microscopy of muscle shows curvilinear bodies within the muscle fibers, skin, and conjunctival biopsies. Histopathological changes include loss of nerve cells at the cortices of the cerebrum and cerebellum. Lipopigments are present.[35] The ultrastructural patterns of lipopigments include fingerprint, rectilinear or curvilinear, and granular profiles.[35] The contents of these lipopigments include varying proportions of saposin A, saposin D, subunit C of ATP synthase, and beta-amyloid proteins.[70] Histological retinal atrophy is noted in the childhood forms.
- Ultrastructural examination of peripheral blood lymphocytes in the NCLs reveals different specific cytoplasmic inclusions, with curvilinear profiles typically occurring in classic CLN2 disease.[71]
- Detection of specific antibodies like those against TPP1: This protein is absent or markedly reduced in lymphocytes, lymphoblasts, and fibroblasts.[72]
- Blood film microscopy may show vacuolated lymphocytes, and electron microscopy may reveal the typical lysosomal inclusions in classic patterns, including fingerprint profiles.[50]
Ophthalmic assessment:
- Fundoscopy may reveal optic disc pallor, arteriolar attenuation, peripheral granular appearance of the fundus, macular mottling, or typical Bull's eye maculopathy (see Image. Fundus Photos of Patient with Batten Disease).
- Optical coherence tomography (OCT) of the macula usually shows foveal thinning, loss of outer retinal layers, and thinning of the nerve fiber layer and ganglion cell layer.[50]
Electrophysiology:
- Electroencephalogram (EEG) features common to various NCL subtypes include progressive slowing of background activity, the disappearance of sleep spindles during sleep, and bursts of diffuse or focal slow waves prevalent over temporal and occipital regions.[73]
- Visually Evoked Potentials (VEP)- In CLN1 disease, VEP is unrecordable at age 4; CLN2 disease shows abnormally enhanced but diminished potentials in the final stages, while the CLN3 variant shows early abnormalities.[73]
- Electroretinogram (ERG)- The ERG may show a reduction of amplitudes in the scotopic condition. Specifically, the b-to-a ratio may be reduced, suggesting that the inner retina may primarily be involved, which correlates with the localization of CLN3 product in the inner retina.[74][48] Pattern ERG is usually undetectable.
Treatment / Management
Several therapies are developing to treat NCL, although not for all subtypes. Notably, CLN4, CLN9, CLN12, CLN13, and CLN14 diseases need further therapeutic studies. Most of these therapies in development are potentially disease-modifying in that they may slow or even halt the progression of the disease. However, few therapies are hypothesized to partially reverse the disease, though a complete reversal is improbable. Therefore, early diagnosis and treatment are more important than ever as these damage-limiting interventions become available.
The primary barrier to effectively treating Batten disease is the need for central nervous system (CNS) access.[75] These treatments primarily target defects in soluble lysosomal proteins and act via enzyme replacement, gene therapy, neural stem cell therapy, or small-molecule pharmaceuticals.
The treatment of Batten disease encompasses several categories, including symptom management, targeted therapies, and supportive care. Each aims to improve patients' quality of life and slow disease progression.
Symptomatic: Most of the management of NCL focuses on treating the symptoms.
- Anticonvulsants- Carbamazepine, oxcarbazepine, phenytoin, valproic acid, gabapentin, lamotrigine, topiramate and levetiracetam can be used.[76]
- Antiepileptics- Isradipine is a potentially viable neuroprotective agent that can help patients with neurological disorders, such as Batten disease.[77]
- Antispasmodics- Baclofen treats generalized spasticity, and due to the intensity of spasticity, a high dose is usually necessary for patients with CLN2.[78] (A1)
Targeted:
- Enzyme Replacement Therapy (ERT) with intraventricular recombinant pro-enzyme of TPP-1, ie, cerliponase alfa, has been approved by the US Food and Drug Administration (FDA) as an enzyme replacement therapy in CLN2 disease. A multicenter, open-label study evaluated the effect of intraventricular infusion of cerliponase alfa every 2 weeks in children with CLN2 disease who were between the ages of 3 and 16. Treatment was initiated at 30 mg, 100 mg, or 300 mg; all the patients then received the 300 mg dose for at least 96 weeks. It was evident that this therapy resulted in less decline in motor and language function than that in historical controls. Serious adverse events included failure of the intraventricular device and device-related infections.[79] Visual symptoms may not improve or slow down despite this therapy.
- Gene Therapy- Chen et al reported an adeno-associated virus (AAV) gene therapy that showed marked benefit in a mouse model of CLN7 Batten disease. Unfortunately, exactly what these results may suggest for individuals living with CLN7 Batten disease is unclear.[80]
Supportive:
- Balanced diet- Adequate caloric intake, divided into smaller volumes, is recommended to prevent aspiration.
- Sleep management- Melatonin helps regulate the sleep and wake cycle, especially in patients with impaired vision.[77]
- Physical rehabilitation- Maintaining physical strength and managing pain is essential.
- Psychological rehabilitation- These patients may need emotional support and cognitive behavioral therapy.[81] (B3)
Differential Diagnosis
The differential diagnosis for Batten disease includes the following:
- Inherited metabolic disorders: Niemann Pick type C may present with eye movement defects in conjunction with developmental regression and splenomegaly in the early infantile forms.[82] Mitochondrial disorders can present with retinitis pigmentosa (RP) with visual failure as a primary feature, for example, in neuropathy and ataxia with retinitis pigmentosa (NARP).[83]
- Retinitis pigmentosa (RP): The classic clinical triad of this disease is arteriolar attenuation, retinal pigmentary changes (could be either hypopigmentation or hyperpigmentation in the form of bone-spicule and pigment clumpings), and waxy disc pallor.[84] The characteristic pigmentary changes occur in the mid-peripheral fundus, predominantly populated by rods. The fundus findings often have a high degree of symmetry between the 2 eyes.[84]
- Stargardt disease: This is characterized by juvenile-onset foveal atrophy, taking on a “beaten-bronze” appearance, with surrounding distinct yellow flecks that are round or pisciform at the level of the RPE.[85] These flecks have been shown to follow a pattern of radial expansion from the fovea to the peripheral fundus. Mutations in the ABCA4 gene most commonly cause Stargardt disease and are typically inherited in an autosomal recessive fashion.[85]
- Cone rod dystrophies (CRDs): These are inherited retinal dystrophies belonging to the pigmentary retinopathies group. CRDs are characterized by retinal pigment deposits visible on fundus examination, predominantly localized to the macular region.[86]
Pertinent Studies and Ongoing Trials
The therapeutic horizon for NCLs is expanding, although there is controversy regarding the best methods of modifying these diseases. Given that NCLs represent multiple pathobiological processes, any single therapeutic approach will likely not be definitive for all forms. Although the primary treatment is primarily symptomatic, some promising preclinical studies and planned and ongoing clinical trials exist.[87]
Cerliponase alfa (recombinant tripeptidyl-peptidase 1; TPP1) was approved for use in CLN2 disease based on efficacy data from a phase I/II clinical trial that showed a decreased rate of decline, but not improvement, on the motor/language score of the Hamburg Rating Scale.[88]
Trehalose is a disaccharide composed of 2 glucose molecules. Studies have shown that trehalose, an activator of transcription factor EB (TFEB), reduces the buildup of lipofuscin in both cell and mouse models.[89] The enzyme trehalase lyses trehalose in the small intestine, so a barrier to using trehalose therapeutically is that it cannot be administered enterally. Preclinical studies are being conducted to evaluate the intravenous delivery of trehalose and the oral delivery of miglustat, which inhibits trehalase.[90]
Peroxisome proliferator-activated receptor–alpha (PPARα) agonists have been shown to induce autophagy, which is thought to be impaired in many forms of NCL. Preclinical CLN2 mouse model studies of gemfibrozil (an FDA-approved PPARα agonist used to regulate cholesterol) found decreased cellular accumulation of lipofuscin, increased motor coordination, and increased longevity.[91]
Staging
Various stagings systems for NCL are described as follows:
The unified Batten disease rating scale (UBDRS) is a global disease severity scale used to measure disease progression in all variants of Batten disease.[92]
- The UBDRS consists of 4 categories: physical assessment (score 0–112), seizure assessment (score 0–54), behavior assessment (score 0–55), and capability assessment (score 0–14).
- Higher scores correspond to increased severity.
- The UBDRS also contains a Batten disease history section to record the approximate age at the onset of clinical features from a caregiver interview.
- The UBDRS is a valid and reliable tool for quantifying disease severity and progression in Batten (CLN3) disease.
CLN3 disease staging system (CLN3SS):
- The CLN3SS is a disease-specific staging system that can classify individuals into strata based on age and disease severity.
- It consists of 4 stages (0–3) and is based on genetic confirmation, vision loss, presence of seizures, and loss of independent walking.[93]
- The CLN3SS has potential applications in clinical trials for cohort stratification.
Hamburg scale for quantifying loss of function in CLN3 disease:
- Using the findings from the ocular examinations and ancillary testing, the Hamburg CLN3 ophthalmic rating scale was established.[94]
- The total score is calculated by adding visual acuity, fundus, macular striae, macular pigmentation, peripheral bony spicules, vessel rarefaction, and OCT (optical coherence tomography) of the macula score (see Table 2. Hamburg Scale).
- Grading ranges from 0 to 3 based on the total score obtained. For example, a total score of 14 means Grade 0 (unaffected), 10-13 is assigned Grade 1 (affected), 5-9 implies Grade 3 (severely affected), and a total score of 3 is assigned Grade 3 (end-stage).
- A CLN3 ophthalmic grade of 0 represents a normal fundus, regular macular profile and thickness on OCT scans, and a BCVA ≥20/25. In contrast, a grade of 3 denotes nearly complete retinal atrophy in both the posterior pole and the peripheral retina.
Table 2. Hamburg Scale
| BCVA (Best corrected visual acuity) | Score | |
| Visual Acuity |
≥20/25 ≥20/63 ≥20/160 ≥20/400 ≥20/1000 ≥Counting fingers ≥Hand motion Light perception/No light perception |
7 6 5 4 3 2 1 0 |
| Fundus Score (optic pallor) |
None Severe |
1 0 |
| Macular Striae |
None Presence |
1 0 |
| Macular Orange Pigment |
None Presence |
1 0 |
| Peripheral Bony Spicules |
None Presence |
1 0 |
| Vessel Rarefaction |
None Presence |
1 0 |
|
Optical Coherence Tomography (OCT) of the Macula (Ellipsoid Zone) |
Intact
Disruption in a diameter < 2000 um
Complete loss
|
2
1
0
|
Prognosis
The only treatment currently approved by the U FDA specifically to treat Batten disease is cerliponase alfa, an enzyme replacement therapy designed to slow the loss of walking ability in children with a late-infantile NCL, CLN2.[79] While no cure exists, other treatments, such as antiseizure medications, can help control disease symptoms. Physiotherapy and occupational therapy may aid in maintaining patients’ ability to carry out day-to-day activities.[95] These treatments cannot cure Batten disease but help children retain their abilities for as long as possible.
Most forms of Batten disease cause vision loss, seizures, delayed developmental milestones, behavioral and learning problems, and loss of language and motor skills. Some children with infantile Batten disease also have microcephaly.[95] Vision loss is often the first symptom and can rapidly progress. Parents also usually notice clumsiness and stumbling in older children due to a loss of motor coordination. Eventually, children with Batten disease become blind, unable to walk, talk, or swallow, and are confined to a wheelchair or bed.[96] Research into ways to possibly treat Batten disease is ongoing, including gene therapy, enzyme replacement therapy, small molecule carriers, stem cell therapy, and RNA and lysosomal modifiers.[97]
The prognosis highly depends on the age of onset. The earlier the onset, the more severe the disease and the higher the risk of disability and early death. Most forms of NCL are fatal diseases with reduced life expectancy despite treatment.[6][98] Most individuals with Batten disease die between the ages of 15 and 30.[3] Most become bilaterally blind by age 10 to 14. However, the adult variants (Kufs disease) may have milder symptoms and normal life expectancy.[99]
Complications
Living with Batten disease is challenging for both patients and their caregivers. Treatment, diet, lifestyle changes, and support groups can help maintain a higher quality of life.
The time when vision loss occurs and how fast it develops can vary from 1 patient to another. For example, partial or complete loss of vision often develops in individuals with childhood forms of the disease, while it is usually preserved in those with adult-onset Batten disease. There currently is no effective treatment to prevent vision loss in patients with Batten disease. However, early diagnosis of vision impairment is essential so that interventions can be implemented to allow children to achieve their full developmental potential.[100]
Due to a loss of balance and poor coordination, children with Batten disease may have changes in how they walk or their posture. The ability to perform fine motor skills, which are used in activities of daily living such as writing, dressing, bathing, and feeding, may also be affected.[101]
Mood changes such as anxiety and depression may occur and increase as the patient becomes more worried and frustrated as the disease develops.[101] People with Batten disease may experience a progressive deterioration in speech ability. Due to vision impairment, seizures, learning, and communication problems, children rely more and more on their sense of hearing, so teaching resources and methods should be adapted accordingly.[102]
Deterrence and Patient Education
Deterrence of Batten disease primarily focuses on genetic counseling and early detection, as there are currently no preventive measures to stop the onset of this inherited disorder. Educating patients and families is crucial for managing expectations, understanding the progressive nature of the disease, and making informed decisions about care. Comprehensive patient education should cover the importance of regular monitoring, potential treatment options, and the availability of supportive resources. Additionally, addressing the emotional and psychological aspects through continuous education can empower families to cope with the challenges posed by the disease and improve overall quality of life.
Support groups and disease associations can greatly assist. The Beyond Batten Disease Foundation was established to eradicate juvenile Batten disease by raising awareness and funds to accelerate research for a treatment or cure. The Batten Disease Support and Research Association (BDSRA) is an international support and research organization for families of children and young adults with the disorder. In the United Kingdom, the Batten Disease Family Association (BDFA) and Metabolic Support UK (formerly known as Climb) are national organizations providing disease information to families, professionals, and others.
Enhancing Healthcare Team Outcomes
Managing Batten disease necessitates a cohesive and interprofessional healthcare team to provide patient-centered care. Physicians, advanced practitioners, nurses, pharmacists, and other health professionals must possess specialized knowledge and skills in diagnosing and managing Batten disease. Strategically, this involves integrating the latest evidence-based practices and individualized care plans to optimize clinical outcomes. Professionals should be adept at recognizing the signs and symptoms of Batten disease and implementing appropriate interventions to slow disease progression and manage symptoms.
Since Batten disease has numerous genetic origins, clinical genetics testing should be carried out to determine the underlying mutation and the particular subtype. This will assist in forecasting the disease's trajectory and could be useful in treatment selection. In this cooperative approach, patient-centered treatment and experts in neurology, ophthalmology, physiotherapy, psychotherapy, and speech therapy are essential.
Whether a child can continue attending a typical school or needs to transfer to a specialized school is primarily determined by the age at which the condition first manifests. Children with Batten disease typically need an individualized education plan (IEP) because of their vision impairment, seizures, learning difficulties, and communication issues.
Given the progressive and debilitating nature of Batten disease, ethical considerations are paramount. Healthcare providers must ensure that patients and families are fully informed about the disease, treatment options, and potential outcomes, respecting their autonomy in decision-making. Responsibilities include providing compassionate care, maintaining patient confidentiality, and advocating for the patient's best interests while balancing the complex needs of the disease.
Placing the patient and family at the center of care is crucial in managing those affected by Batten disease. This includes addressing not only the physical symptoms but also the emotional, psychological, and social needs of the patient and their caregivers. By prioritizing patient-centered care, the healthcare team can improve quality of life, provide meaningful support, and empower families to participate actively in the care process.
Media
(Click Image to Enlarge)
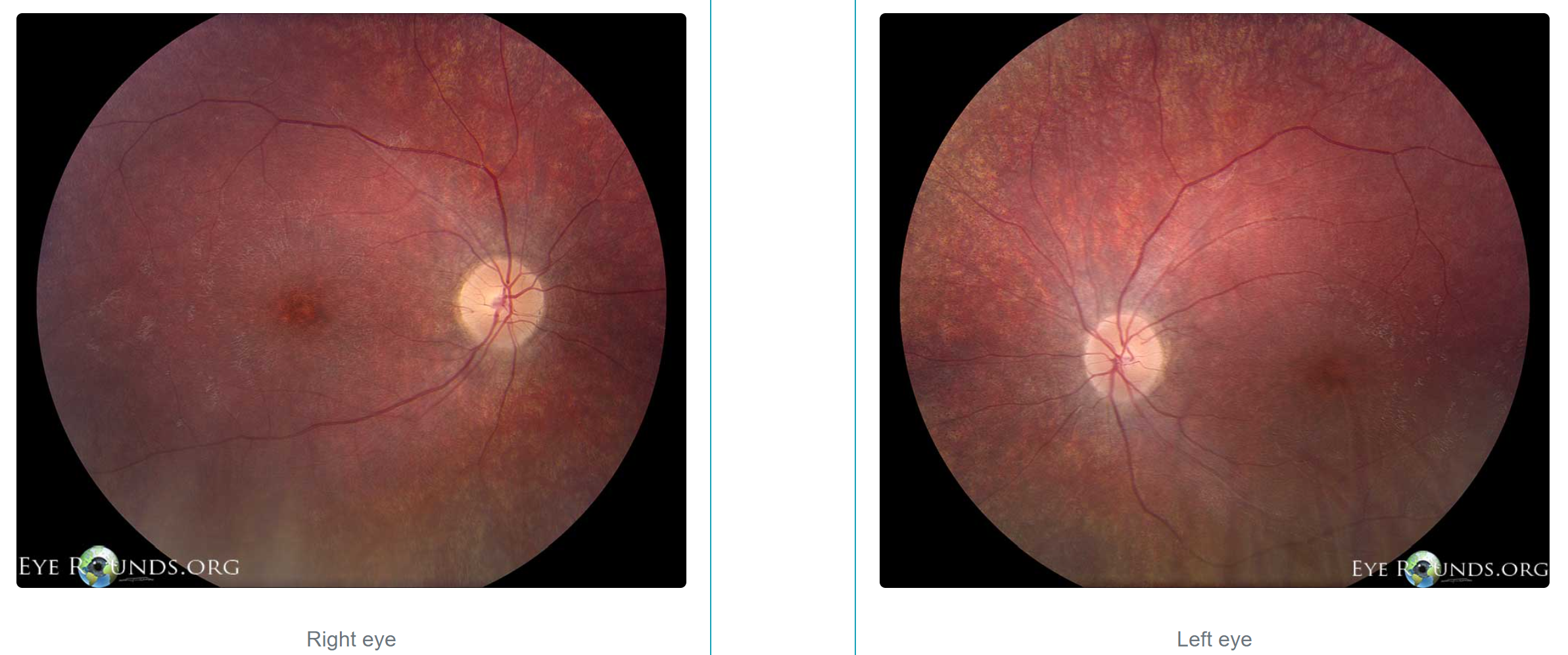
Fundus Photos of Patient with Batten Disease. See mild optic nerve pallor, central macular mottling, attenuated arterioles, and peripheral retinal granularity.
University of Iowa, Eyerounds.org
References
Rahim AA, Russell C, Mole SE. Special edition: The NCLs/Batten disease. Biochimica et biophysica acta. Molecular basis of disease. 2020 Sep 1:1866(9):165824. doi: 10.1016/j.bbadis.2020.165824. Epub 2020 May 5 [PubMed PMID: 32387426]
Mukherjee AB, Appu AP, Sadhukhan T, Casey S, Mondal A, Zhang Z, Bagh MB. Emerging new roles of the lysosome and neuronal ceroid lipofuscinoses. Molecular neurodegeneration. 2019 Jan 16:14(1):4. doi: 10.1186/s13024-018-0300-6. Epub 2019 Jan 16 [PubMed PMID: 30651094]
Level 3 (low-level) evidenceOstergaard JR. Juvenile neuronal ceroid lipofuscinosis (Batten disease): current insights. Degenerative neurological and neuromuscular disease. 2016:6():73-83. doi: 10.2147/DNND.S111967. Epub 2016 Aug 1 [PubMed PMID: 30050370]
Chaves-Carballo E. Eponym: Frederick E. Batten: father of pediatric neurology. Southern medical journal. 1978 Nov:71(11):1428-9 [PubMed PMID: 362542]
Simonati A, Williams RE. Neuronal Ceroid Lipofuscinosis: The Multifaceted Approach to the Clinical Issues, an Overview. Frontiers in neurology. 2022:13():811686. doi: 10.3389/fneur.2022.811686. Epub 2022 Mar 11 [PubMed PMID: 35359645]
Level 3 (low-level) evidenceRosenberg JB, Chen A, Kaminsky SM, Crystal RG, Sondhi D. Advances in the Treatment of Neuronal Ceroid Lipofuscinosis. Expert opinion on orphan drugs. 2019:7(11):473-500. doi: 10.1080/21678707.2019.1684258. Epub 2019 Nov 27 [PubMed PMID: 33365208]
Level 3 (low-level) evidenceSeehafer SS, Ramirez-Montealegre D, Wong AM, Chan CH, Castaneda J, Horak M, Ahmadi SM, Lim MJ, Cooper JD, Pearce DA. Immunosuppression alters disease severity in juvenile Batten disease mice. Journal of neuroimmunology. 2011 Jan:230(1-2):169-72. doi: 10.1016/j.jneuroim.2010.08.024. Epub [PubMed PMID: 20937531]
Gardner E, Mole SE. The Genetic Basis of Phenotypic Heterogeneity in the Neuronal Ceroid Lipofuscinoses. Frontiers in neurology. 2021:12():754045. doi: 10.3389/fneur.2021.754045. Epub 2021 Oct 18 [PubMed PMID: 34733232]
Aungaroon G, Hallinan B, Jain P, Horn PS, Spaeth C, Arya R. Correlation Among Genotype, Phenotype, and Histology in Neuronal Ceroid Lipofuscinoses: An Individual Patient Data Meta-Analysis. Pediatric neurology. 2016 Jul:60():42-48.e4. doi: 10.1016/j.pediatrneurol.2016.03.018. Epub 2016 Apr 8 [PubMed PMID: 27238410]
Level 1 (high-level) evidenceMole SE, Anderson G, Band HA, Berkovic SF, Cooper JD, Kleine Holthaus SM, McKay TR, Medina DL, Rahim AA, Schulz A, Smith AJ. Clinical challenges and future therapeutic approaches for neuronal ceroid lipofuscinosis. The Lancet. Neurology. 2019 Jan:18(1):107-116. doi: 10.1016/S1474-4422(18)30368-5. Epub 2018 Nov 21 [PubMed PMID: 30470609]
Anderson GW, Goebel HH, Simonati A. Human pathology in NCL. Biochimica et biophysica acta. 2013 Nov:1832(11):1807-26. doi: 10.1016/j.bbadis.2012.11.014. Epub 2012 Nov 29 [PubMed PMID: 23200925]
Das AK, Becerra CH, Yi W, Lu JY, Siakotos AN, Wisniewski KE, Hofmann SL. Molecular genetics of palmitoyl-protein thioesterase deficiency in the U.S. The Journal of clinical investigation. 1998 Jul 15:102(2):361-70 [PubMed PMID: 9664077]
Steigerwald C, Borsuk J, Pappas J, Galey M, Scott A, Devaney JM, Miller DE, Abreu NJ. CLN2 disease resulting from a novel homozygous deep intronic splice variant in TPP1 discovered using long-read sequencing. Molecular genetics and metabolism. 2023 Dec:140(4):107713. doi: 10.1016/j.ymgme.2023.107713. Epub 2023 Oct 30 [PubMed PMID: 37922835]
. Isolation of a novel gene underlying Batten disease, CLN3. The International Batten Disease Consortium. Cell. 1995 Sep 22:82(6):949-57 [PubMed PMID: 7553855]
Jedličková I, Cadieux-Dion M, Přistoupilová A, Stránecký V, Hartmannová H, Hodaňová K, Barešová V, Hůlková H, Sikora J, Nosková L, Mušálková D, Vyleťal P, Sovová J, Cossette P, Andermann E, Andermann F, Kmoch S, Adult NCL Gene Discovery Consortium. Autosomal-dominant adult neuronal ceroid lipofuscinosis caused by duplication in DNAJC5 initially missed by Sanger and whole-exome sequencing. European journal of human genetics : EJHG. 2020 Jun:28(6):783-789. doi: 10.1038/s41431-019-0567-2. Epub 2020 Jan 9 [PubMed PMID: 31919451]
Xin W, Mullen TE, Kiely R, Min J, Feng X, Cao Y, O'Malley L, Shen Y, Chu-Shore C, Mole SE, Goebel HH, Sims K. CLN5 mutations are frequent in juvenile and late-onset non-Finnish patients with NCL. Neurology. 2010 Feb 16:74(7):565-71. doi: 10.1212/WNL.0b013e3181cff70d. Epub [PubMed PMID: 20157158]
Gao H, Boustany RM, Espinola JA, Cotman SL, Srinidhi L, Antonellis KA, Gillis T, Qin X, Liu S, Donahue LR, Bronson RT, Faust JR, Stout D, Haines JL, Lerner TJ, MacDonald ME. Mutations in a novel CLN6-encoded transmembrane protein cause variant neuronal ceroid lipofuscinosis in man and mouse. American journal of human genetics. 2002 Feb:70(2):324-35 [PubMed PMID: 11791207]
Level 2 (mid-level) evidenceKousi M, Siintola E, Dvorakova L, Vlaskova H, Turnbull J, Topcu M, Yuksel D, Gokben S, Minassian BA, Elleder M, Mole SE, Lehesjoki AE. Mutations in CLN7/MFSD8 are a common cause of variant late-infantile neuronal ceroid lipofuscinosis. Brain : a journal of neurology. 2009 Mar:132(Pt 3):810-9. doi: 10.1093/brain/awn366. Epub 2009 Feb 5 [PubMed PMID: 19201763]
Ranta S, Zhang Y, Ross B, Lonka L, Takkunen E, Messer A, Sharp J, Wheeler R, Kusumi K, Mole S, Liu W, Soares MB, Bonaldo MF, Hirvasniemi A, de la Chapelle A, Gilliam TC, Lehesjoki AE. The neuronal ceroid lipofuscinoses in human EPMR and mnd mutant mice are associated with mutations in CLN8. Nature genetics. 1999 Oct:23(2):233-6 [PubMed PMID: 10508524]
Eliason SL, Stein CS, Mao Q, Tecedor L, Ding SL, Gaines DM, Davidson BL. A knock-in reporter model of Batten disease. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2007 Sep 12:27(37):9826-34 [PubMed PMID: 17855597]
Pearce DA, Atkinson M, Tagle DA. Glutamic acid decarboxylase autoimmunity in Batten disease and other disorders. Neurology. 2004 Dec 14:63(11):2001-5 [PubMed PMID: 15596740]
Chattopadhyay S, Ito M, Cooper JD, Brooks AI, Curran TM, Powers JM, Pearce DA. An autoantibody inhibitory to glutamic acid decarboxylase in the neurodegenerative disorder Batten disease. Human molecular genetics. 2002 Jun 1:11(12):1421-31 [PubMed PMID: 12023984]
Lim MJ, Beake J, Bible E, Curran TM, Ramirez-Montealegre D, Pearce DA, Cooper JD. Distinct patterns of serum immunoreactivity as evidence for multiple brain-directed autoantibodies in juvenile neuronal ceroid lipofuscinosis. Neuropathology and applied neurobiology. 2006 Oct:32(5):469-82 [PubMed PMID: 16972881]
Hersrud SL, Kovács AD, Pearce DA. Antigen presenting cell abnormalities in the Cln3(-/-) mouse model of juvenile neuronal ceroid lipofuscinosis. Biochimica et biophysica acta. 2016 Jul:1862(7):1324-36. doi: 10.1016/j.bbadis.2016.04.011. Epub 2016 Apr 19 [PubMed PMID: 27101989]
Groh J, Ribechini E, Stadler D, Schilling T, Lutz MB, Martini R. Sialoadhesin promotes neuroinflammation-related disease progression in two mouse models of CLN disease. Glia. 2016 May:64(5):792-809. doi: 10.1002/glia.22962. Epub 2016 Jan 17 [PubMed PMID: 26775238]
Castaneda JA, Pearce DA. Identification of alpha-fetoprotein as an autoantigen in juvenile Batten disease. Neurobiology of disease. 2008 Jan:29(1):92-102 [PubMed PMID: 17931875]
Rider JA, Rider DL. Batten disease: past, present, and future. American journal of medical genetics. Supplement. 1988:5():21-6 [PubMed PMID: 3146319]
Uvebrant P, Hagberg B. Neuronal ceroid lipofuscinoses in Scandinavia. Epidemiology and clinical pictures. Neuropediatrics. 1997 Feb:28(1):6-8 [PubMed PMID: 9151309]
Haltia M. The neuronal ceroid-lipofuscinoses. Journal of neuropathology and experimental neurology. 2003 Jan:62(1):1-13 [PubMed PMID: 12528813]
Mole SE, Cotman SL. Genetics of the neuronal ceroid lipofuscinoses (Batten disease). Biochimica et biophysica acta. 2015 Oct:1852(10 Pt B):2237-41. doi: 10.1016/j.bbadis.2015.05.011. Epub 2015 May 27 [PubMed PMID: 26026925]
Mitchison HM, O'Rawe AM, Taschner PE, Sandkuijl LA, Santavuori P, de Vos N, Breuning MH, Mole SE, Gardiner RM, Järvelä IE. Batten disease gene, CLN3: linkage disequilibrium mapping in the Finnish population, and analysis of European haplotypes. American journal of human genetics. 1995 Mar:56(3):654-62 [PubMed PMID: 7887419]
Elleder M, Franc J, Kraus J, Nevsímalová S, Sixtová K, Zeman J. Neuronal ceroid lipofuscinosis in the Czech Republic: analysis of 57 cases. Report of the 'Prague NCL group'. European journal of paediatric neurology : EJPN : official journal of the European Paediatric Neurology Society. 1997:1(4):109-14 [PubMed PMID: 10728204]
Level 3 (low-level) evidenceSleat DE, Gedvilaite E, Zhang Y, Lobel P, Xing J. Analysis of large-scale whole exome sequencing data to determine the prevalence of genetically-distinct forms of neuronal ceroid lipofuscinosis. Gene. 2016 Nov 30:593(2):284-91. doi: 10.1016/j.gene.2016.08.031. Epub 2016 Aug 20 [PubMed PMID: 27553520]
Hawkins-Salsbury JA, Cooper JD, Sands MS. Pathogenesis and therapies for infantile neuronal ceroid lipofuscinosis (infantile CLN1 disease). Biochimica et biophysica acta. 2013 Nov:1832(11):1906-9. doi: 10.1016/j.bbadis.2013.05.026. Epub 2013 Jun 6 [PubMed PMID: 23747979]
Radke J, Stenzel W, Goebel HH. Human NCL Neuropathology. Biochimica et biophysica acta. 2015 Oct:1852(10 Pt B):2262-6. doi: 10.1016/j.bbadis.2015.05.007. Epub 2015 May 16 [PubMed PMID: 25989315]
Palmer DN, Barry LA, Tyynelä J, Cooper JD. NCL disease mechanisms. Biochimica et biophysica acta. 2013 Nov:1832(11):1882-93. doi: 10.1016/j.bbadis.2013.05.014. Epub 2013 May 23 [PubMed PMID: 23707513]
Sheeler C, Rosa JG, Ferro A, McAdams B, Borgenheimer E, Cvetanovic M. Glia in Neurodegeneration: The Housekeeper, the Defender and the Perpetrator. International journal of molecular sciences. 2020 Dec 2:21(23):. doi: 10.3390/ijms21239188. Epub 2020 Dec 2 [PubMed PMID: 33276471]
Takahashi K, Nelvagal HR, Lange J, Cooper JD. Glial Dysfunction and Its Contribution to the Pathogenesis of the Neuronal Ceroid Lipofuscinoses. Frontiers in neurology. 2022:13():886567. doi: 10.3389/fneur.2022.886567. Epub 2022 Apr 4 [PubMed PMID: 35444603]
Hofman IL, van der Wal AC, Dingemans KP, Becker AE. Cardiac pathology in neuronal ceroid lipofuscinoses--a clinicopathologic correlation in three patients. European journal of paediatric neurology : EJPN : official journal of the European Paediatric Neurology Society. 2001:5 Suppl A():213-7 [PubMed PMID: 11589001]
Tomiyasu H, Takahashi W, Ohta T, Yoshii F, Shibuya M, Shinohara Y. [An autopsy case of juvenile neuronal ceroid-lipofuscinosis with dilated cardiomyopathy]. Rinsho shinkeigaku = Clinical neurology. 2000 Apr:40(4):350-7 [PubMed PMID: 10967652]
Level 3 (low-level) evidenceMink JW, Augustine EF, Adams HR, Marshall FJ, Kwon JM. Classification and natural history of the neuronal ceroid lipofuscinoses. Journal of child neurology. 2013 Sep:28(9):1101-5. doi: 10.1177/0883073813494268. Epub 2013 Jul 9 [PubMed PMID: 23838030]
Goebel HH, Schochet SS, Jaynes M, Gutmann L. Ultrastructure of the retina in adult neuronal ceroid lipofuscinosis. Acta anatomica. 1998:162(2-3):127-32 [PubMed PMID: 9831759]
Kamate M, Reddy N, Detroja M, Hattiholi V. Neuronal Ceroid Lipofuscinoses in Children. Annals of Indian Academy of Neurology. 2021 Mar-Apr:24(2):192-197. doi: 10.4103/aian.AIAN_61_20. Epub 2021 Apr 28 [PubMed PMID: 34220062]
Orlin A, Sondhi D, Witmer MT, Wessel MM, Mezey JG, Kaminsky SM, Hackett NR, Yohay K, Kosofsky B, Souweidane MM, Kaplitt MG, D'Amico DJ, Crystal RG, Kiss S. Spectrum of ocular manifestations in CLN2-associated batten (Jansky-Bielschowsky) disease correlate with advancing age and deteriorating neurological function. PloS one. 2013:8(8):e73128. doi: 10.1371/journal.pone.0073128. Epub 2013 Aug 28 [PubMed PMID: 24015292]
Johnson AM, Mandelstam S, Andrews I, Boysen K, Yaplito-Lee J, Fietz M, Nagarajan L, Rodriguez-Casero V, Ryan MM, Smith N, Scheffer IE, Ellaway C. Neuronal ceroid lipofuscinosis type 2: an Australian case series. Journal of paediatrics and child health. 2020 Aug:56(8):1210-1218. doi: 10.1111/jpc.14890. Epub 2020 Apr 24 [PubMed PMID: 32329550]
Level 2 (mid-level) evidenceCollins J, Holder GE, Herbert H, Adams GG. Batten disease: features to facilitate early diagnosis. The British journal of ophthalmology. 2006 Sep:90(9):1119-24 [PubMed PMID: 16754648]
Singh RB, Gupta P, Kartik A, Farooqui N, Singhal S, Shergill S, Singh KP, Agarwal A. Ocular Manifestations of Neuronal Ceroid Lipofuscinoses. Seminars in ophthalmology. 2021 Oct 3:36(7):582-595. doi: 10.1080/08820538.2021.1936571. Epub 2021 Jun 9 [PubMed PMID: 34106804]
Bozorg S, Ramirez-Montealegre D, Chung M, Pearce DA. Juvenile neuronal ceroid lipofuscinosis (JNCL) and the eye. Survey of ophthalmology. 2009 Jul-Aug:54(4):463-71. doi: 10.1016/j.survophthal.2009.04.007. Epub [PubMed PMID: 19539834]
Level 3 (low-level) evidenceGupta A, Paulbuddhe VS, Shukla UV, Tripathy K. Exudative Retinitis (Coats Disease). StatPearls. 2024 Jan:(): [PubMed PMID: 32809517]
Wright GA, Georgiou M, Robson AG, Ali N, Kalhoro A, Holthaus SK, Pontikos N, Oluonye N, de Carvalho ER, Neveu MM, Weleber RG, Michaelides M. Juvenile Batten Disease (CLN3): Detailed Ocular Phenotype, Novel Observations, Delayed Diagnosis, Masquerades, and Prospects for Therapy. Ophthalmology. Retina. 2020 Apr:4(4):433-445. doi: 10.1016/j.oret.2019.11.005. Epub 2019 Nov 13 [PubMed PMID: 31926949]
Boustany RM. Neurology of the neuronal ceroid-lipofuscinoses: late infantile and juvenile types. American journal of medical genetics. 1992 Feb 15:42(4):533-5 [PubMed PMID: 1609833]
Nardocci N, Verga ML, Binelli S, Zorzi G, Angelini L, Bugiani O. Neuronal ceroid-lipofuscinosis: a clinical and morphological study of 19 patients. American journal of medical genetics. 1995 Jun 5:57(2):137-41 [PubMed PMID: 7668317]
Dom R, Brucher JM, Ceuterick C, Carton H, Martin JJ. Adult ceroid-lipofuscinosis (Kufs' disease) in two brothers. Retinal and visceral storage in one; diagnostic muscle biopsy in the other. Acta neuropathologica. 1979 Jan 12:45(1):67-72 [PubMed PMID: 760366]
Zelnik N, Mahajna M, Iancu TC, Sharony R, Zeigler M. A novel mutation of the CLN8 gene: is there a Mediterranean phenotype? Pediatric neurology. 2007 Jun:36(6):411-3 [PubMed PMID: 17560505]
Siintola E, Partanen S, Strömme P, Haapanen A, Haltia M, Maehlen J, Lehesjoki AE, Tyynelä J. Cathepsin D deficiency underlies congenital human neuronal ceroid-lipofuscinosis. Brain : a journal of neurology. 2006 Jun:129(Pt 6):1438-45 [PubMed PMID: 16670177]
Steinfeld R, Reinhardt K, Schreiber K, Hillebrand M, Kraetzner R, Bruck W, Saftig P, Gartner J. Cathepsin D deficiency is associated with a human neurodegenerative disorder. American journal of human genetics. 2006 Jun:78(6):988-98 [PubMed PMID: 16685649]
Smith KR, Damiano J, Franceschetti S, Carpenter S, Canafoglia L, Morbin M, Rossi G, Pareyson D, Mole SE, Staropoli JF, Sims KB, Lewis J, Lin WL, Dickson DW, Dahl HH, Bahlo M, Berkovic SF. Strikingly different clinicopathological phenotypes determined by progranulin-mutation dosage. American journal of human genetics. 2012 Jun 8:90(6):1102-7. doi: 10.1016/j.ajhg.2012.04.021. Epub 2012 May 17 [PubMed PMID: 22608501]
Marcos AL, Corradi GR, Mazzitelli LR, Casali CI, Fernández Tome MDC, Adamo HP, de Tezanos Pinto F. The Parkinson-associated human P5B-ATPase ATP13A2 modifies lipid homeostasis. Biochimica et biophysica acta. Biomembranes. 2019 Oct 1:1861(10):182993. doi: 10.1016/j.bbamem.2019.05.015. Epub 2019 May 24 [PubMed PMID: 31132336]
Di Fabio R, Moro F, Pestillo L, Meschini MC, Pezzini F, Doccini S, Casali C, Pierelli F, Simonati A, Santorelli FM. Pseudo-dominant inheritance of a novel CTSF mutation associated with type B Kufs disease. Neurology. 2014 Nov 4:83(19):1769-70. doi: 10.1212/WNL.0000000000000953. Epub 2014 Oct 1 [PubMed PMID: 25274848]
Saleh MM, Hamhom AM, Al-Otaibi A, AlGhamdi M, Housawi Y, Aljadhai YI, Alameer S, Almannai M, Jad LA, Alwadei AH, Tabassum S, Alsaman A, AlAsmari A, Al Mutairi F, Althiyab H, Bashiri FA, AlHumaidi S, Alfadhel M, Mink JW, AlHashim A, Faqeih EA, Saudi NCL Study Consortium. Clinical and Molecular Characteristics of Neuronal Ceroid Lipofuscinosis in Saudi Arabia. Pediatric neurology. 2024 Jun:155():149-155. doi: 10.1016/j.pediatrneurol.2024.03.004. Epub 2024 Mar 7 [PubMed PMID: 38653183]
Bennett MJ, Rakheja D. The neuronal ceroid-lipofuscinoses. Developmental disabilities research reviews. 2013:17(3):254-9. doi: 10.1002/ddrr.1118. Epub [PubMed PMID: 23798013]
Goebel HH, Wisniewski KE. Current state of clinical and morphological features in human NCL. Brain pathology (Zurich, Switzerland). 2004 Jan:14(1):61-9 [PubMed PMID: 14997938]
Hellsten E, Vesa J, Speer MC, Mäkelä TP, Järvelä I, Alitalo K, Ott J, Peltonen L. Refined assignment of the infantile neuronal ceroid lipofuscinosis (INCL, CLN1) locus at 1p32: incorporation of linkage disequilibrium in multipoint analysis. Genomics. 1993 Jun:16(3):720-5 [PubMed PMID: 8325646]
Kohan R, Noher de Halac I, Tapia Anzolini V, Cismondi A, Oller Ramírez AM, Paschini Capra A, de Kremer RD. Palmitoyl Protein Thioesterase1 (PPT1) and Tripeptidyl Peptidase-I (TPP-I) are expressed in the human saliva. A reliable and non-invasive source for the diagnosis of infantile (CLN1) and late infantile (CLN2) neuronal ceroid lipofuscinoses. Clinical biochemistry. 2005 May:38(5):492-4 [PubMed PMID: 15820783]
Chang X, Huang Y, Meng H, Jiang Y, Wu Y, Xiong H, Wang S, Qin J. Clinical study in Chinese patients with late-infantile form neuronal ceroid lipofuscinoses. Brain & development. 2012 Oct:34(9):739-45. doi: 10.1016/j.braindev.2011.12.005. Epub 2012 Jan 14 [PubMed PMID: 22245569]
Level 2 (mid-level) evidenceBiswas A, Krishnan P, Amirabadi A, Blaser S, Mercimek-Andrews S, Shroff M. Expanding the Neuroimaging Phenotype of Neuronal Ceroid Lipofuscinoses. AJNR. American journal of neuroradiology. 2020 Oct:41(10):1930-1936. doi: 10.3174/ajnr.A6726. Epub 2020 Aug 27 [PubMed PMID: 32855186]
Santavuori P, Vanhanen SL, Autti T. Clinical and neuroradiological diagnostic aspects of neuronal ceroid lipofuscinoses disorders. European journal of paediatric neurology : EJPN : official journal of the European Paediatric Neurology Society. 2001:5 Suppl A():157-61 [PubMed PMID: 11588989]
Pauwels EK, Coumou AW, Kostkiewicz M, Kairemo K. [¹⁸F]fluoro-2-deoxy-d-glucose positron emission tomography/computed tomography imaging in oncology: initial staging and evaluation of cancer therapy. Medical principles and practice : international journal of the Kuwait University, Health Science Centre. 2013:22(5):427-37. doi: 10.1159/000346303. Epub 2013 Jan 26 [PubMed PMID: 23363934]
Philippart M, da Silva E, Chugani HT. The value of positron emission tomography in the diagnosis and monitoring of late infantile and juvenile lipopigment storage disorders (so-called Batten or neuronal ceroid lipofuscinoses). Neuropediatrics. 1997 Feb:28(1):74-6 [PubMed PMID: 9151330]
Elleder M, Sokolová J, Hrebícek M. Follow-up study of subunit c of mitochondrial ATP synthase (SCMAS) in Batten disease and in unrelated lysosomal disorders. Acta neuropathologica. 1997 Apr:93(4):379-90 [PubMed PMID: 9113203]
Anderson GW, Smith VV, Brooke I, Malone M, Sebire NJ. Diagnosis of neuronal ceroid lipofuscinosis (Batten disease) by electron microscopy in peripheral blood specimens. Ultrastructural pathology. 2006 Sep-Oct:30(5):373-8 [PubMed PMID: 17090516]
Kurachi Y, Oka A, Mizuguchi M, Ohkoshi Y, Sasaki M, Itoh M, Hayashi M, Goto Y, Takashima S. Rapid immunologic diagnosis of classic late infantile neuronal ceroid lipofuscinosis. Neurology. 2000 Apr 25:54(8):1676-80 [PubMed PMID: 10762513]
Trivisano M, Ferretti A, Calabrese C, Pietrafusa N, Piscitello L, Carfi' Pavia G, Vigevano F, Specchio N. Neurophysiological Findings in Neuronal Ceroid Lipofuscinoses. Frontiers in neurology. 2022:13():845877. doi: 10.3389/fneur.2022.845877. Epub 2022 Feb 25 [PubMed PMID: 35280270]
Katz ML, Gao CL, Prabhakaram M, Shibuya H, Liu PC, Johnson GS. Immunochemical localization of the Batten disease (CLN3) protein in retina. Investigative ophthalmology & visual science. 1997 Oct:38(11):2375-86 [PubMed PMID: 9344361]
Specchio N, Ferretti A, Trivisano M, Pietrafusa N, Pepi C, Calabrese C, Livadiotti S, Simonetti A, Rossi P, Curatolo P, Vigevano F. Neuronal Ceroid Lipofuscinosis: Potential for Targeted Therapy. Drugs. 2021 Jan:81(1):101-123. doi: 10.1007/s40265-020-01440-7. Epub [PubMed PMID: 33242182]
Reddy S, Brahmbhatt H. Application of Anticonvulsants, Antiepileptic Drugs, and Vitamin C in the Treatment and Analysis of Batten Disease. Cureus. 2022 Jan:14(1):e21745. doi: 10.7759/cureus.21745. Epub 2022 Jan 30 [PubMed PMID: 35145828]
Ilijic E, Guzman JN, Surmeier DJ. The L-type channel antagonist isradipine is neuroprotective in a mouse model of Parkinson's disease. Neurobiology of disease. 2011 Aug:43(2):364-71. doi: 10.1016/j.nbd.2011.04.007. Epub 2011 Apr 16 [PubMed PMID: 21515375]
Goyal V, Laisram N, Wadhwa RK, Kothari SY. Prospective Randomized Study of Oral Diazepam and Baclofen on Spasticity in Cerebral Palsy. Journal of clinical and diagnostic research : JCDR. 2016 Jun:10(6):RC01-5. doi: 10.7860/JCDR/2016/17067.7975. Epub 2016 Jun 1 [PubMed PMID: 27504360]
Level 1 (high-level) evidenceMarkham A. Cerliponase Alfa: First Global Approval. Drugs. 2017 Jul:77(11):1247-1249. doi: 10.1007/s40265-017-0771-8. Epub [PubMed PMID: 28589525]
Brudvig JJ, Weimer JM. CLN7 gene therapy: hope for an ultra-rare condition. The Journal of clinical investigation. 2022 Mar 1:132(5):. doi: 10.1172/JCI157820. Epub [PubMed PMID: 35229731]
Honingh AK, Kruithof YL, Kuper WFE, van Hasselt PM, Sterkenburg PS. Towards Understanding Behaviour and Emotions of Children with CLN3 Disease (Batten Disease): Patterns, Problems and Support for Child and Family. International journal of environmental research and public health. 2022 May 12:19(10):. doi: 10.3390/ijerph19105895. Epub 2022 May 12 [PubMed PMID: 35627432]
Level 3 (low-level) evidenceDavison JE. Eye involvement in inherited metabolic disorders. Therapeutic advances in ophthalmology. 2020 Jan-Dec:12():2515841420979109. doi: 10.1177/2515841420979109. Epub 2020 Dec 29 [PubMed PMID: 33447730]
Level 3 (low-level) evidenceZeviani M, Carelli V. Mitochondrial Retinopathies. International journal of molecular sciences. 2021 Dec 25:23(1):. doi: 10.3390/ijms23010210. Epub 2021 Dec 25 [PubMed PMID: 35008635]
O'Neal TB, Luther EE. Retinitis Pigmentosa. StatPearls. 2024 Jan:(): [PubMed PMID: 30137803]
Kohli P, Tripathy K, Kaur K. Stargardt Disease. StatPearls. 2024 Jan:(): [PubMed PMID: 36508525]
Kohl S. [Genetic causes of hereditary cone and cone-rod dystrophies]. Der Ophthalmologe : Zeitschrift der Deutschen Ophthalmologischen Gesellschaft. 2009 Feb:106(2):109-15. doi: 10.1007/s00347-008-1864-2. Epub [PubMed PMID: 19184602]
Masten MC, Mink JW, Augustine EF. Batten disease: an expert update on agents in preclinical and clinical trials. Expert opinion on investigational drugs. 2020 Dec:29(12):1317-1322. doi: 10.1080/13543784.2020.1837110. Epub 2020 Nov 1 [PubMed PMID: 33135495]
Level 3 (low-level) evidenceSchulz A, Ajayi T, Specchio N, de Los Reyes E, Gissen P, Ballon D, Dyke JP, Cahan H, Slasor P, Jacoby D, Kohlschütter A, CLN2 Study Group. Study of Intraventricular Cerliponase Alfa for CLN2 Disease. The New England journal of medicine. 2018 May 17:378(20):1898-1907. doi: 10.1056/NEJMoa1712649. Epub 2018 Apr 24 [PubMed PMID: 29688815]
Palmieri M, Pal R, Nelvagal HR, Lotfi P, Stinnett GR, Seymour ML, Chaudhury A, Bajaj L, Bondar VV, Bremner L, Saleem U, Tse DY, Sanagasetti D, Wu SM, Neilson JR, Pereira FA, Pautler RG, Rodney GG, Cooper JD, Sardiello M. mTORC1-independent TFEB activation via Akt inhibition promotes cellular clearance in neurodegenerative storage diseases. Nature communications. 2017 Feb 6:8():14338. doi: 10.1038/ncomms14338. Epub 2017 Feb 6 [PubMed PMID: 28165011]
Cendret V, Legigan T, Mingot A, Thibaudeau S, Adachi I, Forcella M, Parenti P, Bertrand J, Becq F, Norez C, Désiré J, Kato A, Blériot Y. Synthetic deoxynojirimycin derivatives bearing a thiolated, fluorinated or unsaturated N-alkyl chain: identification of potent α-glucosidase and trehalase inhibitors as well as F508del-CFTR correctors. Organic & biomolecular chemistry. 2015 Nov 21:13(43):10734-44. doi: 10.1039/c5ob01526j. Epub [PubMed PMID: 26356422]
Ghosh A, Rangasamy SB, Modi KK, Pahan K. Gemfibrozil, food and drug administration-approved lipid-lowering drug, increases longevity in mouse model of late infantile neuronal ceroid lipofuscinosis. Journal of neurochemistry. 2017 May:141(3):423-435. doi: 10.1111/jnc.13987. Epub 2017 Apr 3 [PubMed PMID: 28199020]
Wibbeler E, Nickel M, Schwering C, Schulz A, Mink JW. The Unified Batten Disease Rating Scale (UBDRS): Validation and reliability in an independent CLN3 disease sample. European journal of paediatric neurology : EJPN : official journal of the European Paediatric Neurology Society. 2022 May:38():62-65. doi: 10.1016/j.ejpn.2022.03.005. Epub 2022 Apr 4 [PubMed PMID: 35427884]
Level 1 (high-level) evidenceMasten MC, Williams JD, Vermilion J, Adams HR, Vierhile A, Collins A, Marshall FJ, Augustine EF, Mink JW. The CLN3 Disease Staging System: A new tool for clinical research in Batten disease. Neurology. 2020 Jun 9:94(23):e2436-e2440. doi: 10.1212/WNL.0000000000009454. Epub 2020 Apr 16 [PubMed PMID: 32300063]
Dulz S, Atiskova Y, Wibbeler E, Wildner J, Wagenfeld L, Schwering C, Nickel M, Bartsch U, Spitzer MS, Schulz A. An Ophthalmic Rating Scale to Assess Ocular Involvement in Juvenile CLN3 Disease. American journal of ophthalmology. 2020 Dec:220():64-71. doi: 10.1016/j.ajo.2020.07.015. Epub 2020 Jul 21 [PubMed PMID: 32707205]
Bican R, Goddard V, Abreu N, Peifer D, Basinger A, Sveda M, Tanner K, de Los Reyes EC. Developmental Skills and Neurorehabilitation for Children With Batten Disease: A Retrospective Chart Review of a Comprehensive Batten Clinic. Pediatric neurology. 2024 Mar:152():107-114. doi: 10.1016/j.pediatrneurol.2023.12.001. Epub 2023 Dec 5 [PubMed PMID: 38242022]
Level 2 (mid-level) evidenceSchulz A, Patel N, Brudvig JJ, Stehr F, Weimer JM, Augustine EF. The parent and family impact of CLN3 disease: an observational survey-based study. Orphanet journal of rare diseases. 2024 Mar 18:19(1):125. doi: 10.1186/s13023-024-03119-8. Epub 2024 Mar 18 [PubMed PMID: 38500130]
Level 3 (low-level) evidenceKohlschütter A, Schulz A, Bartsch U, Storch S. Current and Emerging Treatment Strategies for Neuronal Ceroid Lipofuscinoses. CNS drugs. 2019 Apr:33(4):315-325. doi: 10.1007/s40263-019-00620-8. Epub [PubMed PMID: 30877620]
Schulz A, Kohlschütter A, Mink J, Simonati A, Williams R. NCL diseases - clinical perspectives. Biochimica et biophysica acta. 2013 Nov:1832(11):1801-6. doi: 10.1016/j.bbadis.2013.04.008. Epub 2013 Apr 17 [PubMed PMID: 23602993]
Level 3 (low-level) evidenceVelinov M, Dolzhanskaya N, Gonzalez M, Powell E, Konidari I, Hulme W, Staropoli JF, Xin W, Wen GY, Barone R, Coppel SH, Sims K, Brown WT, Züchner S. Mutations in the gene DNAJC5 cause autosomal dominant Kufs disease in a proportion of cases: study of the Parry family and 8 other families. PloS one. 2012:7(1):e29729. doi: 10.1371/journal.pone.0029729. Epub 2012 Jan 3 [PubMed PMID: 22235333]
Level 3 (low-level) evidenceOuseph MM, Kleinman ME, Wang QJ. Vision loss in juvenile neuronal ceroid lipofuscinosis (CLN3 disease). Annals of the New York Academy of Sciences. 2016 May:1371(1):55-67. doi: 10.1111/nyas.12990. Epub 2016 Jan 8 [PubMed PMID: 26748992]
Adams HR, Mink JW, University of Rochester Batten Center Study Group. Neurobehavioral features and natural history of juvenile neuronal ceroid lipofuscinosis (Batten disease). Journal of child neurology. 2013 Sep:28(9):1128-36. doi: 10.1177/0883073813494813. Epub [PubMed PMID: 24014508]
von Tetzchner S, Fosse P, Elmerskog B. Juvenile neuronal ceroid lipofuscinosis and education. Biochimica et biophysica acta. 2013 Nov:1832(11):1894-905. doi: 10.1016/j.bbadis.2013.02.017. Epub 2013 Mar 5 [PubMed PMID: 23470553]