
Introduction
Epidermolysis bullosa (EB) defines a prototypic group of rare, inherited dermatoses, characteristically featuring skin fragility secondary to structural defects in the dermo-epidermal junction. This skin fragility creates an impaired tolerance to mechanical stress. Trivial mechanical trauma and shear stress can provoke skin blistering, erosions, and ulceration.[1] This places patients with certain forms of epidermolysis bullosa at a greater risk of infection, disabling deformities secondary to heavy scarring, and aggressive cutaneous malignancy, leading to early fatality in some cases. An extensive phenotypic range is described, ranging from fragility and blistering localized to areas of weight-bearing or pressure to widespread generalized involvement, including extracutaneous disease. Thus, certain subtypes of EB confer high morbidity, with a risk of increased mortality due to multi-system pathology.[2][3]
Sixteen genes have been implicated in underpinning at least 30 observed epidermolysis bullosa subtypes. Each subtype features varying phenotypic severity and impact on morbidity and mortality. These subtypes have been organized into 4 major groups based on the ultrastructural plane within the dermo-epidermal junction that the defect impacts:
Epidermolysis Bullosa Simplex (EBS) comprises around 70% of all epidermolysis bullosa cases and features a fragility defect in the epidermis, mostly inherited in an autosomal dominant pattern.
Junctional Epidermolysis Bullosa (JEB) is an autosomal recessive fragility defect seen specifically within the lamina lucida and makes up around 5% of all epidermolysis bullosa cases.
Dystrophic Epidermolysis Bullosa (DEB) represents around 25% of all epidermolysis bullosa cases and may be autosomal dominant or recessive. Dystrophic epidermolysis bullosa features a fragility defect below the lamina densa of the basement membrane zone.
Kindler Epidermolysis Bullosa (KEB) is the rarest of the 4 major epidermolysis bullosa types inherited in an autosomal recessive pattern. The kindlin-1 protein is affected in Kindler epidermolysis bullosa, resulting in fragility in any plane of the dermo-epidermal junction. Around 400 cases have been reported worldwide.[4]
Inherited epidermolysis bullosa is distinct from epidermolysis bullosa aquisita, a separate, non-inherited, immunobullous disorder characterized by antibodies against type VII collagen.[1]
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
As epidermolysis bullosa results from defects within the dermo-epidermal junction, it is imperative to appreciate the components of this structure before understanding pathophysiology.[5]
The epidermis is the uppermost layer of the dermo-epidermal junction and overlies the dermis. The basement membrane zone is found between the lower (basal) layer of the epidermis and the upper (papillary) layer of the dermis. The basement membrane zone is multifunctional: it anchors the epidermis to the dermis, provides scaffolding for cellular migration and adhesion, forms a selectively permeable membrane for cells and molecules, and serves as a model for the differentiation, development, repair, and replacement of cells. The basement membrane zone consists of 2 layers: the lamina lucida, which lies below the basal epidermis, and the lamina densa, which lies below the lamina lucida.
The layers of the dermo-epidermal junction are dependent on one another, and function as a collective, as opposed to contributing individually. Protein structures are vital in maintaining the position and proximity of the dermo-epidermal junction layers. In epidermolysis bullosa, defective and deficient proteins arising from inherited or de novo genetic mutations result in dermo-epidermal junction malfunction. The phenotypic severity is often proportional to the degree of protein deficiency; therefore, relatively small quantities of functional protein reduce phenotypic severity as compared to an absolute loss of protein expression.[6]
The culmination of this dermo-epidermal junction malfunction is skin fragility. Erosions, blistering, and ulceration ensue in response to seemingly disproportionate mechanical trauma. The resultant wounds are frequently persistent, prone to chronic inflammation, susceptible to infection, and are associated with significant pruritis, pain, and diminished quality of life.[7]
Epidemiology
Approximately 70% of epidermolysis bullosa is epidermolysis bullosa simplex. Dystrophic epidermolysis bullosa accounts for around 25% of all cases, and junctional epidermolysis bullosa the remaining 5%. Kindler epidermolysis bullosa is notably the rarest of the 4 epidermolysis bullosa types, with only around 400 cases reported worldwide.[8][9]
Despite the challenges in formally diagnosing epidermolysis bullosa in resource-poor settings, robust epidemiological studies agree that epidermolysis bullosa prevalence is around 10 per one million population, with incidence at around 20 per one million live births.[10] While epidermolysis bullosa appears not to be sex-preferential, trials conducted in Scottish and Middle Eastern populations suggest a geographically biased distribution; this is thought to be attributable to fewer germ lines within small geographical confines, and sociocultural influences where consanguineous marriage is more common.[11][12]
Pathophysiology
Epidermolysis Bullosa Simplex
Epidermolysis bullosa simplex arises when there is a defect in genes encoding proteins governing epidermal integrity. While epidermolysis bullosa simplex can be inherited in an autosomal dominant or autosomal recessive pattern, the former is more common.
Several epidermolysis bullosa simplex subtypes exist: localized epidermolysis bullosa simplex, severe epidermolysis bullosa simplex, intermediate epidermolysis bullosa simplex, epidermolysis bullosa simplex with mottled pigmentation, migratory circinate epidermolysis bullosa simplex, intermediate epidermolysis bullosa simplex with cardiomyopathy, intermediate epidermolysis bullosa simplex with PLEC mutations, intermediate epidermolysis bullosa simplex with muscular dystrophy, severe epidermolysis bullosa simplex with pyloric atresia, autosomal recessive KRT5/KRT14 epidermolysis bullosa simplex, BP230 deficient epidermolysis bullosa simplex, exophilin 5 deficient epidermolysis bullosa simplex, and CD151 deficient epidermolysis bullosa simplex.[1]
Seven mutated genes (KRT5, KRT14, KLHL24, PLEC, DST, EXPH5, CD151) have been isolated as causes of epidermolysis bullosa simplex. 75% of epidermolysis bullosa simplex cases are due to mutations in genes KRT5 (keratin 5 protein) and KRT14 (keratin 14).[10] Mutated keratin proteins may lack mechanical stiffness (causing proteolysis, cellular stress, and cytolysis) and exhibit thermolability (causing increased blistering in warmer temperatures).[13] PLEC (plectin), DST (dystonin), and EXPH5 (exophilin 5) mutations affect the hemidesmosome and potentially disrupt the keratin cytoskeleton found within basal keratinocytes. Mutated KLHL24 and CD151 cause a more indirect disruption of keratin proteostasis.[14]
Epidermolysis bullosa simplex thus presents with blistering, pruritis, pain, nail abnormalities, dyspigmentation, keratoderma, erosions, and scarring.[14] Repeated chronic insult to basal keratinocytes is thought to predispose patients to a mildly elevated incidence of basal cell carcinoma.[15] Affected hemidesmosomal proteins may also be expressed in extracutaneous tissues, giving rise to varied manifestations, including cardiomyopathy, respiratory tract stenosis, pyloric atresia, muscular dystrophy, esophageal involvement, and nephropathy. Failure to thrive can also be seen in severely affected children.[1]
Junctional Epidermolysis Bullosa
Junctional epidermolysis bullosa is characterized by blistering within the plane of the lamina lucida. An autosomal recessive inheritance pattern is observed.
Eight junctional epidermolysis bullosa subtypes have been identified: severe junctional epidermolysis bullosa; intermediate junctional epidermolysis bullosa; junctional epidermolysis bullosa with pyloric atresia; localized junctional epidermolysis bullosa; junctional epidermolysis bullosa inversa; late-onset junctional epidermolysis bullosa; laryngo-onycho-cutaneous syndrome; and junctional epidermolysis bullosa with interstitial lung disease and nephrotic syndrome.[1]
Seven distinct mutated genes (LAMA3, LAMB3, LAMC2, COL17A, ITGA6, ITGB4, ITGA3) account for junctional epidermolysis bullosa. Mutations in either LAMA3, LAMB3, or LAMC2 result in a malfunctioning laminin 332 protein, hindering anchoring filament function and keratinocyte migration during wound healing.[16] Additionally, laminin 332 maintains epithelial function in the brain, eyes, lungs, kidneys, thymus, and gastrointestinal tract, explaining why dysfunction produces an array of extracutaneous complications. The absolute absence of laminin 332 in severe junctional epidermolysis bullosa creates extreme epithelial fragility, and severe granulation tissue within the airways can cause respiratory compromise, often limiting survival beyond 2 years of age. COL17A mutations create faulty collagen XVII proteins, which maintain hair follicle stem cells, melanocyte stem cells, cell mobility, and cell migration; consequently, skin atrophy, hair loss, dyspigmentation, and carcinogenesis ensue.[17] Malfunctioning integrin proteins form due to mutations in ITGA6, ITGB4, and ITGA3 genes. The resultant defective proteins alter cell-matrix adhesion, hemidesmosome, and keratinocyte function, ligand banding, and cell signaling, resulting in severe mucocutaneous fragility, overgranulation, and nail dystrophy. Their malfunction in the gastrointestinal mucosa results in pyloric atresia, while their impact on renal podocytes and alveolar epithelial cells propagates nephropathy and interstitial lung disease.[18][19]
Repeated injury in the lamina lucida increases squamous cell carcinoma risk, significantly contributing to early mortality in the junctional epidermolysis bullosa population.[20] Additionally, the proteins affected in junctional epidermolysis bullosa also play vital roles in the odontogenic epithelium; tooth enamel defects are therefore seen in all patients with junctional epidermolysis bullosa.[21]
Dystrophic Epidermolysis Bullosa
Dystrophic epidermolysis bullosa may be inherited in autosomal dominant or recessive patterns, with the latter giving rise to more severe phenotypes. Dystrophic epidermolysis bullosa features fragility below the lamina densa due to mutant COL7A1 (collagen VII protein).[22]
Four types of dominant dystrophic epidermolysis bullosa have been identified: intermediate dominant dystrophic epidermolysis bullosa, localized dominant dystrophic epidermolysis bullosa, dominant dystrophic epidermolysis bullosa pruriginosa, and self-improving dominant dystrophic epidermolysis bullosa. Six types of recessive dystrophic epidermolysis bullosa have been identified: intermediate recessive dystrophic epidermolysis bullosa, severe recessive dystrophic epidermolysis bullosa, recessive dystrophic epidermolysis bullosa inversa, localized recessive dystrophic epidermolysis bullosa, recessive dystrophic epidermolysis bullosa pruriginosa, and self-improving recessive dystrophic epidermolysis bullosa. One final subtype, severe dystrophic epidermolysis bullosa, features dominant and recessive compound heterozygosity.[1]
Deficiency in type VII collagen manifests with skin fragility, blistering, scarring, milia, and mucosal involvement. While all 4 types of epidermolysis bullosa cause repeated blistering, dystrophic epidermolysis bullosa features a defect much deeper in the dermo-epidermal junction, propagating profound mucocutaneous scarring and fibrosis.[23][24] Eventually, this significantly disables patients with contractures and pseudosyndactyly and encourages aggressive, metastatic squamous cell carcinoma development, the latter often proving fatal.[25][26]
Kindler Epidermolysis Bullosa
Kindler epidermolysis bullosa is a consequence of an autosomal recessive defect in the FERMT1 gene, which produces faulty kindlin-1 proteins. This alters keratinocyte cytoskeleton maintenance and integrin activation. In Kindler epidermolysis bullosa, blistering may occur in any plane of the dermo-epidermal junction.[9]
Due to defective kindlin-1, cutaneous mechanical trauma propagates cellular stress in keratinocytes. Consequently, an influx of growth factors and cytokines encourages dermal fibroblasts to over-synthesize the extracellular matrix, resulting in scarring and fibrosis. Uniquely amongst EB subtypes, Kindler epidermolysis bullosa features photosensitivity, with reduced kindlin-1 linked to UVB-mediated apoptosis.[27]
While childhood Kindler epidermolysis bullosa features blistering and photosensitivity, these symptoms eventually give way to poikiloderma, dyspigmentation, atrophy, erythema, mucocutaneous scarring and palmoplantar hyperkeratosis. Later in adulthood, epithelial cancers complicate Kindler epidermolysis bullosa, with squamous cell carcinoma typically presenting at acral and mucosal sites.[28] Beyond cutaneous expression, patients may suffer from gingivitis, esophageal stenosis, and colitis due to kindlin-1 protein utility in the oral mucosa and gastrointestinal tract.
Histopathology
Standard light microscopy is not relevant in diagnosing epidermolysis bullosa, as insufficient resolution and a failure to discriminate key ultrastructural diagnostic features can lead to misdiagnosis.
History and Physical
Epidermolysis bullosa initially presents at birth or early childhood. Common presenting complaints include blistering, skin fragility, and non-healing wounds disproportionate to mechanical trauma. A family history of similar complaints may be present. Additionally, pain and pruritis are common concerns. Blistering is distributed with bias for sites prone to pressure and friction, with the hands, feet, buttocks, and knees most susceptible. In neonates, infants, or children, the nappy region is also susceptible.[29] When epidermolysis bullosa is diagnosed later in life, it is milder or localized epidermolysis bullosa subtypes that are identified.[30]
As epidermolysis bullosa is a genetically heterozygous group of disorders, a broad phenotypic range is recognized.[31] The exact epidermolysis bullosa subtype dictates symptom distribution and severity. Subtle distinctions in the phenotype can be challenging to differentiate by clinical examination alone. Definitive diagnosis may be possible with specialized evaluation, including electron and immunofluorescence microscopy and genetic mutational analyses. Some classical subtypes are easily identifiable:
- Acrally distributed blistering with keratoderma is the hallmark of localized epidermolysis bullosa simplex.
- Confluent palmoplantar keratoderma features in severe epidermolysis bullosa simplex, with clustered arcuate herpetiform blisters.
- Neonates with a hoarse cry who develop overgranulation in the perioral, neck, and axillary areas and tooth enamel defects are likely to have junctional epidermolysis bullosa.
- Severe dystrophic epidermolysis bullosa features profound scarring causing microstomia, loss of linguinal papillae, digital contractures, and pseudosyndactyly.
- Kindler epidermolysis bullosa is distinct in displaying photosensitivity and features poikiloderma.[9][24][32]
The mode of inheritance (and therefore possible subtype) and consanguinity-related risk may be gleaned by establishing a thorough family history.
As mentioned previously, extracutaneous involvement is an important contributor to epidermolysis bullosa morbidity and mortality. The manifestations, as outlined in Figure 1, are varied, and their combination and severity are governed by specific genotypes.[2][3] Given the broad phenotypic range seen in extracutaneous disease, including a detailed history and physical examination is imperative. This will inform both the assessment and plan for treating clinicians.
Evaluation
Transmission Electron Microscopy and Immunofluorescence Antigen Mapping
Transmission electron microscopy and immunofluorescence antigen mapping are undertaken upon shave biopsy specimens from non-blistered skin subjected to gentle friction before the study. This is to induce progressive morphological changes of early blistering. Blistered skin is not used as reparative changes can lead to misdiagnosis.[33]
Following a biopsy, transmission electron microscopy, and immunofluorescence antigen mapping are vital in distinguishing the affected dermo-epidermal junction plane and morphological features specific to epidermolysis bullosa subtypes. Transmission electron microscopy details the planar location of the dermo-epidermal junction defect and morphological features (desmosomes, hemidesmosomes, anchoring filaments, fibrils, sub-basal dense plates, basal keratin filaments) aiding subtype diagnosis.[34] Conventional light microscopy reveals fewer fine details at the basement membrane zone and, therefore, has no diagnostic role in the setting of epidermolysis bullosa. Immunofluorescence antigen mapping and transmission electron microscopy help subtype epidermolysis bullosa by highlighting target antigens and quantifying their abundance (which shows a correlation with phenotypic severity).[35] Limitations of these processes are appreciated in resource-poor settings, given the cost, time, and expertise required.
Genetic Screening
Genetic screening can confirm the epidermolysis bullosa subtype. Next-generation sequencing has surpassed Sanger sequencing in epidermolysis bullosa diagnosis due to its lower cost, concurrent multiple gene screening efficiency, and operator-friendly data synthesis.[36][37] Mutational analyses also underpin genetic counseling for family members and may inform future family planning.
Treatment / Management
The absence of curative therapies has led to medical management oriented on supportive care, symptom control, and prevention of mild to severe complications.[29] Interdisciplinary management is necessary, guided by the subtype of EB that is diagnosed.(B3)
In epidermolysis bullosa, blisters are prevented by reducing exposure to mechanical trauma. Trauma provoked by cord clamps, clothing labels and seams, plastic patient identifying bands, and heel prick tests should be minimized in neonates. Hot, humid environments may also aggravate fragility.[38] Trauma-preventative dressings placed over body sites are more likely to meet with pressure or friction. Blisters may be pierced with sterile needles and drained, with the blister roof kept intact to minimize pain and prevent microbial colonization. Broken skin can be dressed with specialized non-adhesive foam and lipocolloid dressings.[39]
To reduce colonization, antiseptic washes, and ointments may be encouraged. Targeted antibiotic use should be reserved for proven infection, frank cellulitis, and sepsis management.[40] Wound overgranulation may be managed with short-term topical corticosteroid use.[41]
Squamous cell carcinoma is frequently a devastating complication in epidermolysis bullosa and notably a common cause of early mortality in junctional epidermolysis bullosa and dystrophic epidermolysis bullosa populations.[42] Wide local excision remains the recommended management; however, squamous cell carcinoma in epidermolysis bullosa is aggressive and highly metastatic regardless of conventional tumor histological grading or margin of surgical clearance. In severe recessive dystrophic epidermolysis bullosa, the risk of mortality due to squamous cell carcinoma rises to 78.8% at 55 years old from 38.7% at 35 years old, and the median survival is approximately 5 years following initial squamous cell carcinoma diagnosis.[15] Radiotherapy and chemotherapy can form palliative treatment options. Newer immunotherapy agents have been employed with varying degrees of success.(B2)
Pain is an important overarching symptom.[43] The source of pain may be the cutaneous disease, the extracutaneous manifestations, or secondary to wound dressing changes or bathing. Analgesia, involvement of a pain management team, and psychological support should aim to reduce pain, which frequently debilitates a patient's quality of life. Neuropathic pain medication can play a role in managing pruritic pain symptoms unresolved by emollient, topical corticosteroid, and antihistamine therapy.[44]
Very recently, beremagene geperpavec ('B-VEC'), a dosable topical gene therapy delivering COL7A1 using herpes simplex virus type 1 as a vector, has been approved by the FDA as a treatment for dystrophic epidermolysis bullosa.[45] Similarly, Oleogel-S10 (birch triterpene extract), a herbal gel remedy was recently approved by some regulatory authorities for wound management in junctional and dystrophic epidermolysis bullosa.
Differential Diagnosis
It is essential to rule out possible differential diagnoses, particularly in neonatal populations:
Pompholyx Eczema presents with acral pruritic blistering. A history of atopy may be present. Patch testing may be indicated to identify delayed hypersensitivity reactions to contact allergens.[46]
Porphyria presents with scarring, milia, and photosensitivity, with possible hypertrichosis. Skin biopsies and porphyrin testing may be indicated.[47]
Bullous Pemphigoid presents with large, tense bullae and itch. Clinical examination and skin biopsy can confirm bullous pemphigoid.[48]
Bullous Systemic Lupus Erythematosus presents with tense vesicles, bullae, and erosions, often in sun-exposed areas. Histopathology and immunofluorescence confirm this differential.[49]
Bullous Tinea Pedis presents with multilocular pedal blistering, with possible skin maceration between digits. Tinea pedis is diagnosed through mycological cultures from skin scrapings.[50]
Palmoplantar Keratoderma causes acral skin thickening, often with nail dystrophy. Genetic testing may be indicated, though acquired palmoplantar keratoderma may be associated with internal malignancy, thyroid abnormalities, inflammatory dermatoses such as lichen planus, and certain medications.[51]
Epidermolysis Bullosa Acquisita is an autoimmune disorder that may present similarly to dystrophic epidermolysis bullosa but later in life. Immunofluorescence studies on skin biopsies are indicated.[52]
Ehlers-Danlos Syndrome features joint hypermobility, hyperextensible skin, and easy bruising with skin fragility. Ehlers Danlos is diagnosed by skin biopsy and genetic testing.[53]
Incontinentia Pigmenti features Blaschkoid vesicles forming hyperpigmented verrucous whorls.[54]
Pertinent Studies and Ongoing Trials
Current clinical trials study the potential of a broad range of therapeutic strategies.[55]
Studies on gene replacement therapy explore how COL7A1 can be delivered cutaneously to patients with recessive dystrophic epidermolysis bullosa.[56] Recently, beremagene geperpavec ('B-VEC'), a topical gene therapy delivering COL7A1 using herpes simplex virus type 1 as a vector, has been approved by regulatory authorities.[45] Other gene replacement therapy trials explore the efficacy of COL7A1 transgenes and self-inactivating viral vectors expressing type VII collagen, which 'correct' cells ex-vivo before being reintroduced to patients cutaneously. Studies examining the effectiveness of grafting revertant skin to lesions in junctional epidermolysis bullosa suggest the potential of revertant mosaicism as a natural gene replacement therapy.[57] CRISPR/Cas9 technology has shown promise in gene-editing studies involving correcting COL7A1 mutations in fibroblasts and keratinocytes in mice.[58] Pre-clinical studies on exon-skipping in COL7A1 and COL17A1 genes show the effective formation of partly-functioning proteins, indicating another potential therapeutic strategy.[55] The use of induced pluripotent stem cells can encourage the expansion of genetically altered cells ex-vivo to produce sufficient skin grafts for patients.[59]
Repurposing existing therapies has also shown promise. When repurposed for junctional and dystrophic epidermolysis bullosa treatment, gentamycin, an aminoglycoside antibiotic, can 'read through' stop codon mutations in COL7A1 and LAMB3 when administered topically or intravenously, enabling the synthesis of full-length proteins.[60][61] Losartan, an angiotensin II receptor blocker, has shown improvement in inflammation, fibrosis, and tumor progression by altering TGF-beta signaling in dystrophic epidermolysis bullosa.[62] A trial investigating the repurposing of apremilast (a phosphodiesterase type-4 inhibitor currently used in psoriasis management) in epidermolysis bullosa simplex is underway.[63] Oleogel-S10 (birch triterpene extract), a herbal gel remedy recently approved by regulating authorities for junctional and dystrophic epidermolysis bullosa management, has demonstrated accelerated wound healing by altering pro-inflammatory mediator regulation, increasing migration of primary human keratinocytes and promoting keratinocyte differentiation.[64][65]
Ongoing protein therapy studies observe the efficacy of topical and intravenous recombinant type VII collagen in promoting wound healing, and HMGB1 proteins in suppressing inflammation by mobilizing cells from the bone marrow to damaged tissue.[66][67][66]
Literature on the management of squamous cell carcinoma in epidermolysis bullosa is limited. Case studies indicate that cetuximab, an epidermal growth factor receptor inhibitor, increases progression-free survival of head and neck squamous cell carcinoma.[68] Meanwhile, little research exists on the effectiveness of immune checkpoint inhibitors, such as nivolumab, pembrolizumab, and cemiplimab.[55] Rigosertib, a multikinase inhibitor, has so far shown successful apoptosis induction in recessive epidermolysis bullosa without impact on carcinoma-free keratinocytes.[69]
Advancement in epidermolysis bullosa trials requires a focus on epidermolysis bullosa severity scoring. While numerous severity scoring tools exist, the absence of a universally utilized tool creates discord when comparing epidermolysis bullosa severity and assessing symptomatic progression and remission.[70] A global initiative to develop a universal tool is in progress.
Prognosis
The prognosis in epidermolysis bullosa is variable and dictated by the specific causative subtype and the complications arising from cutaneous and extracutaneous manifestations.[71] In some subtypes, particularly those that are milder, epidermolysis bullosa has minimal impact on life expectancy. In other, more severe subtypes, infantile or childhood mortality is highly prevalent. For example, most patients with severe junctional epidermolysis bullosa will rarely survive beyond 2 years of age, and severe recessive dystrophic epidermolysis bullosa patients often face mortality in the third to fifth decades of life due to the development of aggressive squamous cell carcinoma.[72][73]
Complications
Interprofessional discussion should be prompted following diagnosis, guided by the specific extracutaneous complications present in the patient.
Oral and Gastrointestinal Disease
Repeated blistering, erosion, ulceration, and scarring in the oral mucosa secondary to dysfunctional laminin 332, kindlin-1, and types VII and XVII collagen can cause microstomia, tongue-tethering and vestibular sulcus loss. Junctional epidermolysis bullosa additionally features tooth enamel defect. Combined, these concerns increase the likelihood of dental caries formation due to difficulty maintaining oral hygiene.[74][75] Management may include dental scaling, root planning, and antibiotic therapy.
Nutrition is impacted by the aforementioned oral manifestations, as well as gastrointestinal involvement. Esophageal blistering contributes to poor nutrition by causing dysphagia. This is managed using analgesia, soft diet alteration, and occasionally inhaled steroids to reduce localized edema and erosion. Esophageal webbing and stricturing (including secondary to squamous cell carcinoma formation) hinder proper nutrition due to dysphagia and reflux symptoms. Non-malignant esophageal complications may warrant balloon dilation and proton pump inhibitor therapy.[76] Neonatal patients with pyloric atresia cannot develop nutrition at all without immediate surgical intervention.[77] Failure to thrive may be seen, together with vitamin and mineral deficiencies, anemia, hypoalbuminemia, delayed puberty, and poor wound healing. Long-term management warrants specialist dietician support and consideration of gastrostomy placement.[78]
Poor nutritional intake, alongside gastrointestinal and cutaneous losses, leads to microcytic or normocytic anemias, notably iron deficiency anemia or anemia of chronic disease.[79] Iron supplementation is an important consideration. Transfusion may be required in symptomatic anemia if iron is depleted. Intravenous iron infusion is preferred to oral supplementation, which can cause constipation. Other causes of constipation in epidermolysis bullosa include regular opioid use, poor fiber and fluid intake, and anal blistering. An osmotic laxative and dietary alteration can be prescribed prophylactically to manage constipation.[80]
Musculoskeletal Disease
Patients with epidermolysis bullosa have an increased risk of developing osteopenia and osteoporosis due to reduced mobility, unresolved inflammation, vitamin D deficiency, pubertal delay, and chronic opioid exposure.[81] Consequently, vitamin D supplements, physiotherapy, and induction of puberty may be considered when managing bone health. With regard to soft tissues, repeated blistering may propagate flexion contracture and pseudosyndactyly formation, significantly impacting manual dexterity and gait. Surgical release may be indicated to improve functionality if preventative measures, such as digit-separating dressings, physiotherapy, and podiatrist involvement, fail.[82][83] In subtypes with muscular dystrophy, only supportive therapy is recommended.[84]
Ophthalmic Involvement
While ophthalmological input is recommended for complications such as blepharitis, erosion, scarring, keratitis, symblepharon, and ectropion, lubricating preparations are recommended as prophylactic management. Around 6% of patients with recessive dystrophic epidermolysis bullosa develop blindness due to ophthalmic involvement.[85][86]
Genitourinary Involvement
Genito-urinary epidermolysis bullosa involvement includes stenosis of the urethra (causing hydroureter and hydronephrosis if unresolved), for which surgical stenting may be indicated.[87] Vaginal stenosis may also complicate epidermolysis bullosa.[88] Hemodialysis, peritoneal dialysis, and eventual transplantation may be warranted in epidermolysis bullosa-related IgA nephropathy, post-streptococcal glomerulonephritis, and renal amyloidosis.[89]
Cardiovascular and Respiratory Involvement
KLHL mutations causing dilated and hypertrophic cardiomyopathy require specialist cardiology input.[90] Interstitial lung disease confers a poor prognosis.[91] Laryngeal involvement requires emergent airway management.[92]
Psychosocial Impact
Epidermolysis bullosa is a frequently debilitating disease without a cure and has the potential to exert considerable psychological impact.[93] Chronic wounds, pain, itch, reduced mobility, and difficult social interaction can contribute to a markedly reduced quality of life.[26] The societal, familial, and financial consequences extend to caregivers, particularly those who are also relatives.[94] Psychological support may improve the emotional well-being of patients and caregivers.
Deterrence and Patient Education
As epidermolysis bullosa has genetic etiology, only genetic counseling, prenatal screening, and preimplantation screening can inform whether offspring could be affected.[95][96] While genetic counseling predicts the mode of inheritance and the likelihood of epidermolysis bullosa transmission to offspring, prenatal screening employs chorionic villus sampling or amniocentesis to guide expecting parents on inheritance risk.[97] Chorionic villus sampling is an effective method of screening though it carries a higher risk of miscarriage. Preimplantation screening and haplotyping identify mutations in early embryos and oocytes, respectively, allowing for genetically unaffected fertilization.[98]
Enhancing Healthcare Team Outcomes
Given that epidermolysis bullosa is presently incurable, the aims of current care are orientated around early and accurate diagnosis, information on prognosis, symptom relief, quality of life, prevention of complications, and genetic counseling.[99] Patients are generally managed in many settings. Though stable patients are usually managed in outpatient facilities, acute complications may require specialist assessment.
Based on severity, patients with epidermolysis bullosa should be scheduled for regular follow-up, at a minimum of annual, coordinated with their primary care clinician. A complete skin examination, with photography to monitor suspicious lesions, is recommended. The frequency of home visits by a specialist nurse is determined by phenotypic severity. Patients should be prescribed a supply of wound dressings as part of their prescription.
The epidermolysis bullosa team will almost always comprise a dermatologist and specialist nurse in coordination with primary care. There are several key members of the interdisciplinary team, depending on the phenotype, including general practitioners, pediatricians, podiatrists, oncologists, plastic surgeons, palliative care specialists, pain management specialists, psychologists, dentists, ophthalmologists, occupational therapists, physiotherapists, dietitians, gastroenterologists, urologists, speech and language specialists, otolaryngologists, cardiologists, endocrinologists, and orthotics. Multidisciplinary team meetings help to coordinate these interspecialty efforts.[98][100]
Media
(Click Image to Enlarge)
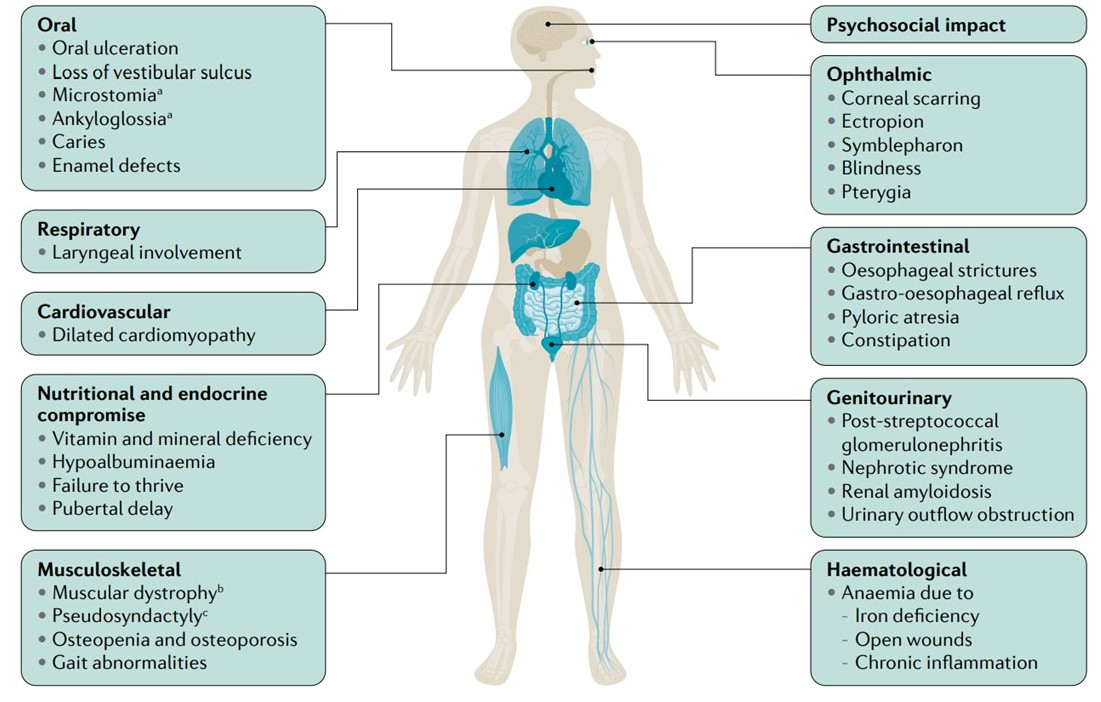
Extracutaneous manifestations of epidermolysis bullosa Contributed by Dr Ajoy Bardhan
(Click Image to Enlarge)
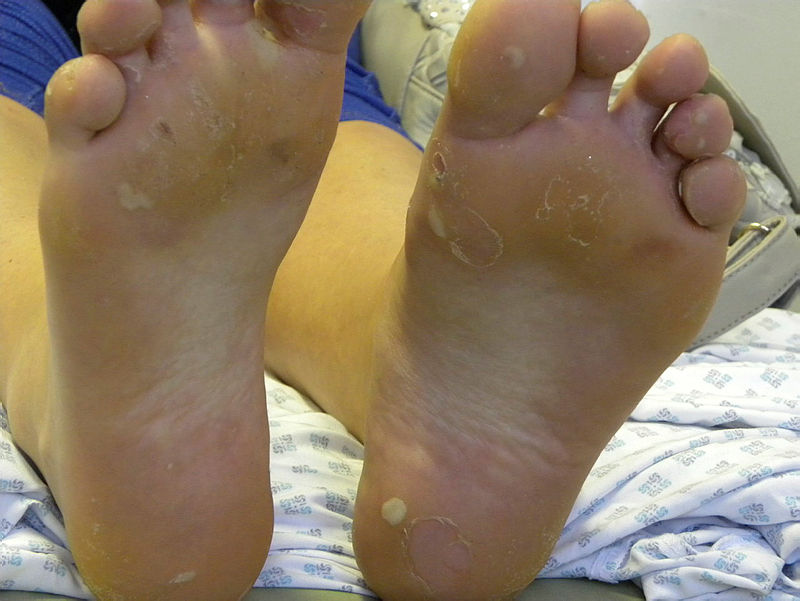
Plantar blistering and keratoderma in Epidermolysis Bullosa Simplex. Contributed by Dr Ajoy Bardhan BSc FRCP and Prof Adrian Heagerty BSc MD FRCP. Obtained with informed consent.
(Click Image to Enlarge)
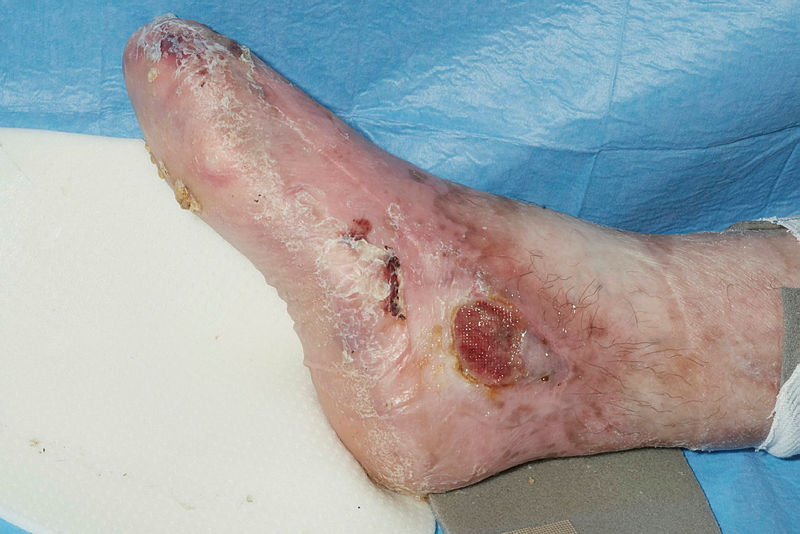
Pseudosyndactyly and ulceration of the lower limb in Recessive Dystrophic Epidermolysis Bullosa Contributed by Dr Ajoy Bardhan BSc FRCP and Prof Adrian Heagerty BSc MD FRCP. Obtained with informed consent.
(Click Image to Enlarge)
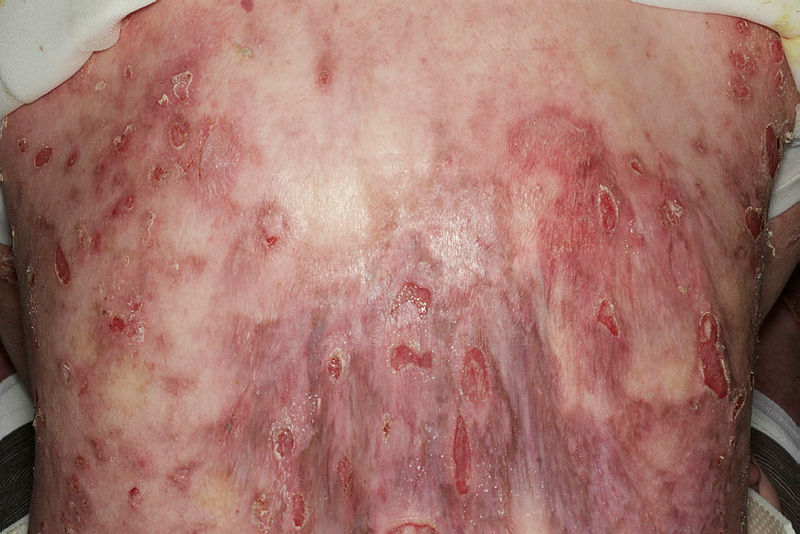
Truncal erosions and inflammation in Junctional Epidermolysis Bullosa. Contributed by Dr Ajoy Bardhan BSc FRCP and Prof Adrian Heagerty BSc MD FRCP. Obtained with informed consent.
References
Bardhan A, Bruckner-Tuderman L, Chapple ILC, Fine JD, Harper N, Has C, Magin TM, Marinkovich MP, Marshall JF, McGrath JA, Mellerio JE, Polson R, Heagerty AH. Epidermolysis bullosa. Nature reviews. Disease primers. 2020 Sep 24:6(1):78. doi: 10.1038/s41572-020-0210-0. Epub 2020 Sep 24 [PubMed PMID: 32973163]
Fine JD, Mellerio JE. Extracutaneous manifestations and complications of inherited epidermolysis bullosa: part I. Epithelial associated tissues. Journal of the American Academy of Dermatology. 2009 Sep:61(3):367-84; quiz 385-6. doi: 10.1016/j.jaad.2009.03.052. Epub [PubMed PMID: 19700010]
Fine JD, Mellerio JE. Extracutaneous manifestations and complications of inherited epidermolysis bullosa: part II. Other organs. Journal of the American Academy of Dermatology. 2009 Sep:61(3):387-402; quiz 403-4. doi: 10.1016/j.jaad.2009.03.053. Epub [PubMed PMID: 19700011]
Has C, Bauer JW, Bodemer C, Bolling MC, Bruckner-Tuderman L, Diem A, Fine JD, Heagerty A, Hovnanian A, Marinkovich MP, Martinez AE, McGrath JA, Moss C, Murrell DF, Palisson F, Schwieger-Briel A, Sprecher E, Tamai K, Uitto J, Woodley DT, Zambruno G, Mellerio JE. Consensus reclassification of inherited epidermolysis bullosa and other disorders with skin fragility. The British journal of dermatology. 2020 Oct:183(4):614-627. doi: 10.1111/bjd.18921. Epub 2020 Mar 11 [PubMed PMID: 32017015]
Level 3 (low-level) evidenceUitto J, Has C, Vahidnezhad H, Youssefian L, Bruckner-Tuderman L. Molecular pathology of the basement membrane zone in heritable blistering diseases:: The paradigm of epidermolysis bullosa. Matrix biology : journal of the International Society for Matrix Biology. 2017 Jan:57-58():76-85. doi: 10.1016/j.matbio.2016.07.009. Epub 2016 Aug 3 [PubMed PMID: 27496350]
Nakano A, Chao SC, Pulkkinen L, Murrell D, Bruckner-Tuderman L, Pfendner E, Uitto J. Laminin 5 mutations in junctional epidermolysis bullosa: molecular basis of Herlitz vs. non-Herlitz phenotypes. Human genetics. 2002 Jan:110(1):41-51 [PubMed PMID: 11810295]
Nyström A, Bruckner-Tuderman L. Injury- and inflammation-driven skin fibrosis: The paradigm of epidermolysis bullosa. Matrix biology : journal of the International Society for Matrix Biology. 2018 Aug:68-69():547-560. doi: 10.1016/j.matbio.2018.01.016. Epub 2018 Jan 31 [PubMed PMID: 29391280]
Petrof G, Papanikolaou M, Martinez AE, Mellerio JE, McGrath JA, Bardhan A, Harper N, Heagerty A, Ogboli M, Chiswell C, Moss C. The epidemiology of epidermolysis bullosa in England and Wales: data from the national epidermolysis bullosa database. The British journal of dermatology. 2022 May:186(5):843-848. doi: 10.1111/bjd.20958. Epub 2022 Mar 31 [PubMed PMID: 34927719]
Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, Youssefian L, Vahidnezhad H, Uitto J. Kindler Syndrome. GeneReviews(®). 1993:(): [PubMed PMID: 26937547]
Fine JD. Epidemiology of Inherited Epidermolysis Bullosa Based on Incidence and Prevalence Estimates From the National Epidermolysis Bullosa Registry. JAMA dermatology. 2016 Nov 1:152(11):1231-1238. doi: 10.1001/jamadermatol.2016.2473. Epub [PubMed PMID: 27463098]
Horn HM, Priestley GC, Eady RA, Tidman MJ. The prevalence of epidermolysis bullosa in Scotland. The British journal of dermatology. 1997 Apr:136(4):560-4 [PubMed PMID: 9155958]
Nakano A, Lestringant GG, Paperna T, Bergman R, Gershoni R, Frossard P, Kanaan M, Meneguzzi G, Richard G, Pfendner E, Uitto J, Pulkkinen L, Sprecher E. Junctional epidermolysis bullosa in the Middle East: clinical and genetic studies in a series of consanguineous families. Journal of the American Academy of Dermatology. 2002 Apr:46(4):510-6 [PubMed PMID: 11907499]
Morley SM, Dundas SR, James JL, Gupta T, Brown RA, Sexton CJ, Navsaria HA, Leigh IM, Lane EB. Temperature sensitivity of the keratin cytoskeleton and delayed spreading of keratinocyte lines derived from EBS patients. Journal of cell science. 1995 Nov:108 ( Pt 11)():3463-71 [PubMed PMID: 8586658]
Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, So JY, Teng J. Epidermolysis Bullosa Simplex. GeneReviews(®). 1993:(): [PubMed PMID: 20301543]
Fine JD, Johnson LB, Weiner M, Li KP, Suchindran C. Epidermolysis bullosa and the risk of life-threatening cancers: the National EB Registry experience, 1986-2006. Journal of the American Academy of Dermatology. 2009 Feb:60(2):203-11. doi: 10.1016/j.jaad.2008.09.035. Epub 2008 Nov 20 [PubMed PMID: 19026465]
Level 2 (mid-level) evidenceKiritsi D, Has C, Bruckner-Tuderman L. Laminin 332 in junctional epidermolysis bullosa. Cell adhesion & migration. 2013 Jan-Feb:7(1):135-41. doi: 10.4161/cam.22418. Epub 2012 Oct 17 [PubMed PMID: 23076207]
Level 3 (low-level) evidenceHas C, Kern JS. Collagen XVII. Dermatologic clinics. 2010 Jan:28(1):61-6. doi: 10.1016/j.det.2009.10.007. Epub [PubMed PMID: 19945617]
Mariath LM, Santin JT, Frantz JA, Doriqui MJR, Schuler-Faccini L, Kiszewski AE. Genotype-phenotype correlations on epidermolysis bullosa with congenital absence of skin: A comprehensive review. Clinical genetics. 2021 Jan:99(1):29-41. doi: 10.1111/cge.13792. Epub 2020 Jun 29 [PubMed PMID: 32506467]
McGrath JA. Recently Identified Forms of Epidermolysis Bullosa. Annals of dermatology. 2015 Dec:27(6):658-66. doi: 10.5021/ad.2015.27.6.658. Epub 2015 Dec 7 [PubMed PMID: 26719633]
Mallipeddi R, Keane FM, McGrath JA, Mayou BJ, Eady RA. Increased risk of squamous cell carcinoma in junctional epidermolysis bullosa. Journal of the European Academy of Dermatology and Venereology : JEADV. 2004 Sep:18(5):521-6 [PubMed PMID: 15324385]
Level 3 (low-level) evidenceKirkham J, Robinson C, Strafford SM, Shore RC, Bonass WA, Brookes SJ, Wright JT. The chemical composition of tooth enamel in junctional epidermolysis bullosa. Archives of oral biology. 2000 May:45(5):377-86 [PubMed PMID: 10739859]
Chung HJ, Uitto J. Type VII collagen: the anchoring fibril protein at fault in dystrophic epidermolysis bullosa. Dermatologic clinics. 2010 Jan:28(1):93-105. doi: 10.1016/j.det.2009.10.011. Epub [PubMed PMID: 19945621]
Baardman R, Bremer J, Diercks GFH, Jan SZ, Lemmink HH, Bolling MC, Van den Akker PC. Single glycine deletion in COL7A1 acting as glycine substitution in dystrophic epidermolysis bullosa. Journal of the European Academy of Dermatology and Venereology : JEADV. 2021 Sep:35(9):e597-e600. doi: 10.1111/jdv.17328. Epub 2021 May 13 [PubMed PMID: 33914976]
Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, Pfendner EG, Lucky AW. Dystrophic Epidermolysis Bullosa. GeneReviews(®). 1993:(): [PubMed PMID: 20301481]
Breitenbach J, Gruber C, Klausegger A, Trost A, Bogner B, Reitsamer H, Bauer JW. Pseudosyndactyly - an inflammatory and fibrotic wound healing disorder in recessive dystrophic epidermolysis bullosa. Journal der Deutschen Dermatologischen Gesellschaft = Journal of the German Society of Dermatology : JDDG. 2015 Dec:13(12):1257-66. doi: 10.1111/ddg.12839. Epub [PubMed PMID: 26612796]
Tang JY, Marinkovich MP, Lucas E, Gorell E, Chiou A, Lu Y, Gillon J, Patel D, Rudin D. A systematic literature review of the disease burden in patients with recessive dystrophic epidermolysis bullosa. Orphanet journal of rare diseases. 2021 Apr 13:16(1):175. doi: 10.1186/s13023-021-01811-7. Epub 2021 Apr 13 [PubMed PMID: 33849616]
Level 1 (high-level) evidenceMaier K, He Y, Wölfle U, Esser PR, Brummer T, Schempp C, Bruckner-Tuderman L, Has C. UV-B-induced cutaneous inflammation and prospects for antioxidant treatment in Kindler syndrome. Human molecular genetics. 2016 Dec 15:25(24):5339-5352. doi: 10.1093/hmg/ddw350. Epub [PubMed PMID: 27798104]
Guerrero-Aspizua S, Conti CJ, Escamez MJ, Castiglia D, Zambruno G, Youssefian L, Vahidnezhad H, Requena L, Itin P, Tadini G, Yordanova I, Martin L, Uitto J, Has C, Del Rio M. Assessment of the risk and characterization of non-melanoma skin cancer in Kindler syndrome: study of a series of 91 patients. Orphanet journal of rare diseases. 2019 Jul 24:14(1):183. doi: 10.1186/s13023-019-1158-6. Epub 2019 Jul 24 [PubMed PMID: 31340837]
Hon KL, Chu S, Leung AKC. Epidermolysis Bullosa: Pediatric Perspectives. Current pediatric reviews. 2022:18(3):182-190. doi: 10.2174/1573396317666210525161252. Epub [PubMed PMID: 34036913]
Level 3 (low-level) evidenceHorn HM, Tidman MJ. The clinical spectrum of dystrophic epidermolysis bullosa. The British journal of dermatology. 2002 Feb:146(2):267-74 [PubMed PMID: 11903238]
Mariath LM, Santin JT, Schuler-Faccini L, Kiszewski AE. Inherited epidermolysis bullosa: update on the clinical and genetic aspects. Anais brasileiros de dermatologia. 2020 Sep-Oct:95(5):551-569. doi: 10.1016/j.abd.2020.05.001. Epub 2020 Jul 8 [PubMed PMID: 32732072]
Uitto J, Pulkkinen L, McLean WH. Epidermolysis bullosa: a spectrum of clinical phenotypes explained by molecular heterogeneity. Molecular medicine today. 1997 Oct:3(10):457-65 [PubMed PMID: 9358473]
Intong LR, Murrell DF. How to take skin biopsies for epidermolysis bullosa. Dermatologic clinics. 2010 Apr:28(2):197-200, vii. doi: 10.1016/j.det.2009.12.002. Epub [PubMed PMID: 20447481]
Eady RA, Dopping-Hepenstal PJ. Transmission electron microscopy for the diagnosis of epidermolysis bullosa. Dermatologic clinics. 2010 Apr:28(2):211-22, vii. doi: 10.1016/j.det.2009.12.001. Epub [PubMed PMID: 20447483]
Rao R, Shetty VM. Utility of Immunofluorescence Antigen Mapping in Hereditary Epidermolysis Bullosa. Indian journal of dermatology. 2021 Jul-Aug:66(4):360-365. doi: 10.4103/ijd.IJD_131_20. Epub [PubMed PMID: 34759393]
Tenedini E, Artuso L, Bernardis I, Artusi V, Percesepe A, De Rosa L, Contin R, Manfredini R, Pellacani G, Giannetti A, Pagani J, De Luca M, Tagliafico E. Amplicon-based next-generation sequencing: an effective approach for the molecular diagnosis of epidermolysis bullosa. The British journal of dermatology. 2015 Sep:173(3):731-8. doi: 10.1111/bjd.13858. Epub 2015 Jul 29 [PubMed PMID: 25913354]
Vahidnezhad H, Youssefian L, Saeidian AH, Touati A, Sotoudeh S, Abiri M, Barzegar M, Aghazadeh N, Mahmoudi H, Norouz-Zadeh S, Hamid M, Zahabiyon M, Bagherian H, Zeinali S, Fortina P, Uitto J. Multigene Next-Generation Sequencing Panel Identifies Pathogenic Variants in Patients with Unknown Subtype of Epidermolysis Bullosa: Subclassification with Prognostic Implications. The Journal of investigative dermatology. 2017 Dec:137(12):2649-2652. doi: 10.1016/j.jid.2017.07.830. Epub 2017 Aug 19 [PubMed PMID: 28830826]
Chamcheu JC, Navsaria H, Pihl-Lundin I, Liovic M, Vahlquist A, Törmä H. Chemical chaperones protect epidermolysis bullosa simplex keratinocytes from heat stress-induced keratin aggregation: involvement of heat shock proteins and MAP kinases. The Journal of investigative dermatology. 2011 Aug:131(8):1684-91. doi: 10.1038/jid.2011.93. Epub 2011 Apr 14 [PubMed PMID: 21490615]
Denyer JE. Wound management for children with epidermolysis bullosa. Dermatologic clinics. 2010 Apr:28(2):257-64, viii-ix. doi: 10.1016/j.det.2010.01.002. Epub [PubMed PMID: 20447488]
Mellerio JE. Infection and colonization in epidermolysis bullosa. Dermatologic clinics. 2010 Apr:28(2):267-9, ix. doi: 10.1016/j.det.2010.01.004. Epub [PubMed PMID: 20447490]
Margulies S, Marion T, Saikaly SK. Use of Potent Topical Corticosteroids (TCS) for Hypergranulation Tissue (HGT) in Pediatric Patients. Cureus. 2022 Aug:14(8):e28304. doi: 10.7759/cureus.28304. Epub 2022 Aug 23 [PubMed PMID: 36158443]
Bonamonte D, Filoni A, De Marco A, Lospalluti L, Nacchiero E, Ronghi V, Colagrande A, Giudice G, Cazzato G. Squamous Cell Carcinoma in Patients with Inherited Epidermolysis Bullosa: Review of Current Literature. Cells. 2022 Apr 17:11(8):. doi: 10.3390/cells11081365. Epub 2022 Apr 17 [PubMed PMID: 35456044]
Goldschneider KR, Lucky AW. Pain management in epidermolysis bullosa. Dermatologic clinics. 2010 Apr:28(2):273-82, ix. doi: 10.1016/j.det.2010.01.008. Epub [PubMed PMID: 20447492]
von Bischhoffshausen S, Ivulic D, Alvarez P, Schuffeneger VC, Idiaquez J, Fuentes C, Morande P, Fuentes I, Palisson F, Bennett DLH, Calvo M. Recessive dystrophic epidermolysis bullosa results in painful small fibre neuropathy. Brain : a journal of neurology. 2017 May 1:140(5):1238-1251. doi: 10.1093/brain/awx069. Epub [PubMed PMID: 28369282]
Guide SV, Gonzalez ME, Bağcı IS, Agostini B, Chen H, Feeney G, Steimer M, Kapadia B, Sridhar K, Quesada Sanchez L, Gonzalez F, Van Ligten M, Parry TJ, Chitra S, Kammerman LA, Krishnan S, Marinkovich MP. Trial of Beremagene Geperpavec (B-VEC) for Dystrophic Epidermolysis Bullosa. The New England journal of medicine. 2022 Dec 15:387(24):2211-2219. doi: 10.1056/NEJMoa2206663. Epub [PubMed PMID: 36516090]
Haft MA, Park HH, Lee SS, Sprague JM, Paller AS, Cotton CH, Thyssen JP, Eichenfield LF. Diagnosis and Management of Pediatric Chronic Hand Eczema: The PeDRA CACHES Survey. Paediatric drugs. 2023 Jul:25(4):459-466. doi: 10.1007/s40272-023-00574-x. Epub 2023 May 24 [PubMed PMID: 37225932]
Level 3 (low-level) evidenceJensen JD, Resnick SD. Porphyria in childhood. Seminars in dermatology. 1995 Mar:14(1):33-9 [PubMed PMID: 7742238]
Hammers CM, Stanley JR. Mechanisms of Disease: Pemphigus and Bullous Pemphigoid. Annual review of pathology. 2016 May 23:11():175-97. doi: 10.1146/annurev-pathol-012615-044313. Epub 2016 Feb 22 [PubMed PMID: 26907530]
Odonwodo A, Vashisht P. Bullous Systemic Lupus Erythematosus. StatPearls. 2023 Jan:(): [PubMed PMID: 32491377]
Xie F, Lehman JS. Bullous Tinea Pedis. Mayo Clinic proceedings. 2022 Jul:97(7):1396-1397. doi: 10.1016/j.mayocp.2022.05.007. Epub [PubMed PMID: 35787867]
Schiller S, Seebode C, Hennies HC, Giehl K, Emmert S. Palmoplantar keratoderma (PPK): acquired and genetic causes of a not so rare disease. Journal der Deutschen Dermatologischen Gesellschaft = Journal of the German Society of Dermatology : JDDG. 2014 Sep:12(9):781-8. doi: 10.1111/ddg.12418. Epub [PubMed PMID: 25176457]
Kim JH, Kim SC. Epidermolysis bullosa acquisita. Journal of the European Academy of Dermatology and Venereology : JEADV. 2013 Oct:27(10):1204-13. doi: 10.1111/jdv.12096. Epub 2013 Feb 1 [PubMed PMID: 23368767]
Level 3 (low-level) evidenceTaj FT, Sajjan VV, Singh D. Ehlers-Danlos syndrome. Indian dermatology online journal. 2014 Nov:5(Suppl 1):S68-70. doi: 10.4103/2229-5178.144554. Epub [PubMed PMID: 25506578]
Level 3 (low-level) evidenceCammarata-Scalisi F, Fusco F, Ursini MV. Incontinentia Pigmenti. Actas dermo-sifiliograficas. 2019 May:110(4):273-278. doi: 10.1016/j.ad.2018.10.004. Epub 2019 Jan 17 [PubMed PMID: 30660327]
Has C, South A, Uitto J. Molecular Therapeutics in Development for Epidermolysis Bullosa: Update 2020. Molecular diagnosis & therapy. 2020 Jun:24(3):299-309. doi: 10.1007/s40291-020-00466-7. Epub [PubMed PMID: 32328988]
Marinkovich MP, Tang JY. Gene Therapy for Epidermolysis Bullosa. The Journal of investigative dermatology. 2019 Jun:139(6):1221-1226. doi: 10.1016/j.jid.2018.11.036. Epub 2019 May 5 [PubMed PMID: 31068252]
Kiritsi D, Garcia M, Brander R, Has C, Meijer R, Jose Escámez M, Kohlhase J, van den Akker PC, Scheffer H, Jonkman MF, Del Rio M, Bruckner-Tuderman L, Pasmooij AMG. Mechanisms of natural gene therapy in dystrophic epidermolysis bullosa. The Journal of investigative dermatology. 2014 Aug:134(8):2097-2104. doi: 10.1038/jid.2014.118. Epub 2014 Feb 27 [PubMed PMID: 24577406]
Bonafont J, Mencía Á, García M, Torres R, Rodríguez S, Carretero M, Chacón-Solano E, Modamio-Høybjør S, Marinas L, León C, Escamez MJ, Hausser I, Del Río M, Murillas R, Larcher F. Clinically Relevant Correction of Recessive Dystrophic Epidermolysis Bullosa by Dual sgRNA CRISPR/Cas9-Mediated Gene Editing. Molecular therapy : the journal of the American Society of Gene Therapy. 2019 May 8:27(5):986-998. doi: 10.1016/j.ymthe.2019.03.007. Epub 2019 Mar 15 [PubMed PMID: 30930113]
Shinkuma S, Guo Z, Christiano AM. Site-specific genome editing for correction of induced pluripotent stem cells derived from dominant dystrophic epidermolysis bullosa. Proceedings of the National Academy of Sciences of the United States of America. 2016 May 17:113(20):5676-81. doi: 10.1073/pnas.1512028113. Epub 2016 May 3 [PubMed PMID: 27143720]
Kwong A, Cogan J, Hou Y, Antaya R, Hao M, Kim G, Lincoln V, Chen Q, Woodley DT, Chen M. Gentamicin Induces Laminin 332 and Improves Wound Healing in Junctional Epidermolysis Bullosa Patients with Nonsense Mutations. Molecular therapy : the journal of the American Society of Gene Therapy. 2020 May 6:28(5):1327-1338. doi: 10.1016/j.ymthe.2020.03.006. Epub 2020 Mar 17 [PubMed PMID: 32222156]
Mahajan R, Manjunath S, Madakshira MG, De D, Handa S, Chatterjee D, Radotra BD. Topical Gentamicin 0.1% Promotes Collagen 7 Expression in Recessive Dystrophic Epidermolysis Bullosa. Indian dermatology online journal. 2022 Jul-Aug:13(4):480-483. doi: 10.4103/idoj.idoj_554_21. Epub 2022 Jun 24 [PubMed PMID: 36262564]
Pourani MR, Vahidnezhad H, Mansouri P, Youssefian L, Rakhshan A, Hajimoradi B, Abdollahimajd F, Uitto J. Losartan treatment improves recessive dystrophic epidermolysis bullosa: A case series. Dermatologic therapy. 2022 Jul:35(7):e15515. doi: 10.1111/dth.15515. Epub 2022 Apr 29 [PubMed PMID: 35420725]
Level 2 (mid-level) evidenceAfra TP, Razmi TM, Dogra S. Apremilast in Psoriasis and Beyond: Big Hopes on a Small Molecule. Indian dermatology online journal. 2019 Jan-Feb:10(1):1-12. doi: 10.4103/idoj.IDOJ_437_18. Epub [PubMed PMID: 30775293]
Schwieger-Briel A, Ott H, Kiritsi D, Laszczyk-Lauer M, Bodemer C. Mechanism of Oleogel-S10: A triterpene preparation for the treatment of epidermolysis bullosa. Dermatologic therapy. 2019 Jul:32(4):e12983. doi: 10.1111/dth.12983. Epub 2019 Jul 2 [PubMed PMID: 31168940]
Kern JS, Sprecher E, Fernandez MF, Schauer F, Bodemer C, Cunningham T, Löwe S, Davis C, Sumeray M, Bruckner AL, Murrell DF, EASE investigators. Efficacy and safety of Oleogel-S10 (birch triterpenes) for epidermolysis bullosa: results from the phase III randomized double-blind phase of the EASE study. The British journal of dermatology. 2023 Jan 23:188(1):12-21. doi: 10.1093/bjd/ljac001. Epub [PubMed PMID: 36689495]
Level 1 (high-level) evidenceWoodley DT, Keene DR, Atha T, Huang Y, Lipman K, Li W, Chen M. Injection of recombinant human type VII collagen restores collagen function in dystrophic epidermolysis bullosa. Nature medicine. 2004 Jul:10(7):693-5 [PubMed PMID: 15195089]
Level 3 (low-level) evidenceAikawa E, Fujita R, Kikuchi Y, Kaneda Y, Tamai K. Systemic high-mobility group box 1 administration suppresses skin inflammation by inducing an accumulation of PDGFRα(+) mesenchymal cells from bone marrow. Scientific reports. 2015 Jun 5:5():11008. doi: 10.1038/srep11008. Epub 2015 Jun 5 [PubMed PMID: 26046579]
Kim M, Li M, Intong LR, Tran K, Melbourne W, Marucci D, Bucci J, de Souza P, Mallesara G, Murrell DF. Use of cetuximab as an adjuvant agent to radiotherapy and surgery in recessive dystrophic epidermolysis bullosa with squamous cell carcinoma. The British journal of dermatology. 2013 Jul:169(1):208-10. doi: 10.1111/bjd.12272. Epub [PubMed PMID: 23398414]
Level 3 (low-level) evidenceAtanasova VS, Pourreyron C, Farshchian M, Lawler M, Brown CA 4th, Watt SA, Wright S, Warkala M, Guttmann-Gruber C, Hofbauer JP, Fuentes I, Prisco M, Rashidghamat E, Has C, Salas-Alanis JC, Palisson F, Hovnanian A, McGrath JA, Mellerio JE, Bauer JW, South AP. Identification of Rigosertib for the Treatment of Recessive Dystrophic Epidermolysis Bullosa-Associated Squamous Cell Carcinoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2019 Jun 1:25(11):3384-3391. doi: 10.1158/1078-0432.CCR-18-2661. Epub 2019 Mar 7 [PubMed PMID: 30846478]
Hunjan MK, Bardhan A, Zuzarte L, Balacco DL, Harper N, Heagerty A. Are existing validated severity scores for epidermolysis bullosa reflective of the burden of disease in patients with epidermolysis bullosa simplex? Clinical and experimental dermatology. 2023 Jun 5:48(6):706-707. doi: 10.1093/ced/llad069. Epub [PubMed PMID: 36972307]
Lucky AW, Whalen J, Rowe S, Marathe KS, Gorell E. Diagnosis and Care of the Newborn with Epidermolysis Bullosa. NeoReviews. 2021 Jul:22(7):e438-e451. doi: 10.1542/neo.22-7-e438. Epub 2021 Jul 1 [PubMed PMID: 34210808]
Yang CS, Kroshinksy D, Cummings BM. Neonatal junctional epidermolysis bullosa: treatment conundrums and ethical decision making. American journal of clinical dermatology. 2014 Oct:15(5):445-50. doi: 10.1007/s40257-014-0091-7. Epub [PubMed PMID: 25117154]
Robertson SJ, Orrin E, Lakhan MK, O'Sullivan G, Felton J, Robson A, Greenblatt DT, Bernardis C, McGrath JA, Martinez AE, Mellerio JE. Cutaneous Squamous Cell Carcinoma in Epidermolysis Bullosa: a 28-year Retrospective Study. Acta dermato-venereologica. 2021 Aug 24:101(8):adv00523. doi: 10.2340/00015555-3875. Epub 2021 Aug 24 [PubMed PMID: 34230977]
Wright JT. Oral manifestations in the epidermolysis bullosa spectrum. Dermatologic clinics. 2010 Jan:28(1):159-64. doi: 10.1016/j.det.2009.10.022. Epub [PubMed PMID: 19945630]
Polizzi A, Santonocito S, Patini R, Quinzi V, Mummolo S, Leonardi R, Bianchi A, Isola G. Oral Alterations in Heritable Epidermolysis Bullosa: A Clinical Study and Literature Review. BioMed research international. 2022:2022():6493156. doi: 10.1155/2022/6493156. Epub 2022 May 31 [PubMed PMID: 35686231]
Fantauzzi RS, Maia MO, Cunha FC, Simões RV, Gonçalves DU, Maia AF. Otorhinolaryngological and esophageal manifestations of epidermolysis bullosa. Brazilian journal of otorhinolaryngology. 2008 Sep-Oct:74(5):657-661. doi: 10.1016/S1808-8694(15)31373-2. Epub [PubMed PMID: 19082345]
Level 2 (mid-level) evidenceAdam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, Lucky AW, Gorell E. Epidermolysis Bullosa with Pyloric Atresia. GeneReviews(®). 1993:(): [PubMed PMID: 20301336]
Salera S, Tadini G, Rossetti D, Grassi FS, Marchisio P, Agostoni C, Giavoli C, Rodari G, Guez S. A nutrition-based approach to epidermolysis bullosa: Causes, assessments, requirements and management. Clinical nutrition (Edinburgh, Scotland). 2020 Feb:39(2):343-352. doi: 10.1016/j.clnu.2019.02.023. Epub 2019 Feb 21 [PubMed PMID: 30857908]
Reimer A, Hess M, Schwieger-Briel A, Kiritsi D, Schauer F, Schumann H, Bruckner-Tuderman L, Has C. Natural history of growth and anaemia in children with epidermolysis bullosa: a retrospective cohort study. The British journal of dermatology. 2020 Jun:182(6):1437-1448. doi: 10.1111/bjd.18475. Epub 2019 Nov 27 [PubMed PMID: 31487386]
Level 2 (mid-level) evidenceFreeman EB, Köglmeier J, Martinez AE, Mellerio JE, Haynes L, Sebire NJ, Lindley KJ, Shah N. Gastrointestinal complications of epidermolysis bullosa in children. The British journal of dermatology. 2008 Jun:158(6):1308-14. doi: 10.1111/j.1365-2133.2008.08507.x. Epub 2008 Mar 20 [PubMed PMID: 18363753]
Martinez AE, Mellerio JE. Osteopenia and osteoporosis in epidermolysis bullosa. Dermatologic clinics. 2010 Apr:28(2):353-5, xi. doi: 10.1016/j.det.2010.01.006. Epub [PubMed PMID: 20447502]
Vozdvizhensky SI, Albanova VI. Surgical treatment of contracture and syndactyly of children with epidermolysis bullosa. British journal of plastic surgery. 1993 Jun:46(4):314-6 [PubMed PMID: 8392421]
Khan MT, O'Sullivan M, Faitli B, Mellerio JE, Fawkes R, Wood M, Hubbard LD, Harris AG, Iacobaccio L, Vlahovic T, James L, Brains L, Fitzpatrick M, Mayre-Chilton K. Foot care in epidermolysis bullosa: evidence-based guideline. The British journal of dermatology. 2020 Mar:182(3):593-604. doi: 10.1111/bjd.18381. Epub 2019 Oct 23 [PubMed PMID: 31397882]
Alvarez VC, Penttilä ST, Salutto VL, Udd B, Mazia CG. Epidermolysis bullosa simplex with muscular dystrophy associated with PLEC deletion mutation. Neurology. Genetics. 2016 Dec:2(6):e109 [PubMed PMID: 27766310]
Figueira EC, Murrell DF, Coroneo MT. Ophthalmic involvement in inherited epidermolysis bullosa. Dermatologic clinics. 2010 Jan:28(1):143-52. doi: 10.1016/j.det.2009.10.021. Epub [PubMed PMID: 19945628]
Fine JD, Johnson LB, Weiner M, Stein A, Cash S, Deleoz J, Devries DT, Suchindran C. Eye involvement in inherited epidermolysis bullosa: experience of the National Epidermolysis Bullosa Registry. American journal of ophthalmology. 2004 Aug:138(2):254-62 [PubMed PMID: 15289135]
Level 2 (mid-level) evidenceCicek N, Yildiz N, Asadov R, Yucelten AD, Tugtepe H, Alpay H. Kidney and Urinary Tract Involvement in Epidermolysis Bullosa: Is Routine Follow-Up Necessary? Dermatology practical & conceptual. 2021 May:11(3):e2021051. doi: 10.5826/dpc.1103a51. Epub 2021 May 20 [PubMed PMID: 34123558]
Steinkampf MP, Reilly SD, Ackerman GE. Vaginal agglutination and hematometra associated with epidermolysis bullosa. Obstetrics and gynecology. 1987 Mar:69(3 Pt 2):519-21 [PubMed PMID: 3808539]
Level 3 (low-level) evidenceMałecki M, Domański M, Ciechanowski K. End-stage kidney disease in patient with epidermolysis bullosa - what are the treatment options? - case report. BMC nephrology. 2017 Jun 14:18(1):193. doi: 10.1186/s12882-017-0606-6. Epub 2017 Jun 14 [PubMed PMID: 28615054]
Level 3 (low-level) evidenceFine JD, Hall M, Weiner M, Li KP, Suchindran C. The risk of cardiomyopathy in inherited epidermolysis bullosa. The British journal of dermatology. 2008 Sep:159(3):677-82. doi: 10.1111/j.1365-2133.2008.08697.x. Epub 2008 Jul 4 [PubMed PMID: 18616785]
He Y, Balasubramanian M, Humphreys N, Waruiru C, Brauner M, Kohlhase J, O'Reilly R, Has C. Intronic ITGA3 Mutation Impacts Splicing Regulation and Causes Interstitial Lung Disease, Nephrotic Syndrome, and Epidermolysis Bullosa. The Journal of investigative dermatology. 2016 May:136(5):1056-1059. doi: 10.1016/j.jid.2015.11.031. Epub 2016 Feb 15 [PubMed PMID: 26854491]
Lyos AT, Levy ML, Malpica A, Sulek M. Laryngeal involvement in epidermolysis bullosa. The Annals of otology, rhinology, and laryngology. 1994 Jul:103(7):542-6 [PubMed PMID: 8024217]
Level 3 (low-level) evidenceSoon K, Mason R, Martinez AE, Mellerio JE. The psychological functioning of children with epidermolysis bullosa and its relationship with specific aspects of disease. The British journal of dermatology. 2020 Mar:182(3):789-790. doi: 10.1111/bjd.18592. Epub 2019 Nov 25 [PubMed PMID: 31587254]
Bruckner AL, Losow M, Wisk J, Patel N, Reha A, Lagast H, Gault J, Gershkowitz J, Kopelan B, Hund M, Murrell DF. The challenges of living with and managing epidermolysis bullosa: insights from patients and caregivers. Orphanet journal of rare diseases. 2020 Jan 3:15(1):1. doi: 10.1186/s13023-019-1279-y. Epub 2020 Jan 3 [PubMed PMID: 31900176]
Pfendner EG, Nakano A, Pulkkinen L, Christiano AM, Uitto J. Prenatal diagnosis for epidermolysis bullosa: a study of 144 consecutive pregnancies at risk. Prenatal diagnosis. 2003 Jun:23(6):447-56 [PubMed PMID: 12813757]
Sybert VP. Genetic counseling in epidermolysis bullosa. Dermatologic clinics. 2010 Apr:28(2):239-43, viii. doi: 10.1016/j.det.2009.12.004. Epub [PubMed PMID: 20447486]
Fassihi H, McGrath JA. Prenatal diagnosis of epidermolysis bullosa. Dermatologic clinics. 2010 Apr:28(2):231-7, viii. doi: 10.1016/j.det.2010.02.001. Epub [PubMed PMID: 20447485]
Fassihi H, Liu L, Renwick PJ, Braude PR, McGrath JA. Development and successful clinical application of preimplantation genetic haplotyping for Herlitz junctional epidermolysis bullosa. The British journal of dermatology. 2010 Jun:162(6):1330-6. doi: 10.1111/j.1365-2133.2010.09701.x. Epub 2010 Feb 15 [PubMed PMID: 20163412]
Patel PM, Jones VA, Murray TN, Amber KT. A Review Comparing International Guidelines for the Management of Bullous Pemphigoid, Pemphigoid Gestationis, Mucous Membrane Pemphigoid, and Epidermolysis Bullosa Acquisita. American journal of clinical dermatology. 2020 Aug:21(4):557-565. doi: 10.1007/s40257-020-00513-3. Epub [PubMed PMID: 32180161]
Retrosi C, Diociaiuti A, De Ranieri C, Corbeddu M, Carnevale C, Giancristoforo S, Marchili MR, Salvatori G, Atti MLCD, El Hachem M, Raponi M. Multidisciplinary care for patients with epidermolysis bullosa from birth to adolescence: experience of one Italian reference center. Italian journal of pediatrics. 2022 Apr 12:48(1):58. doi: 10.1186/s13052-022-01252-3. Epub 2022 Apr 12 [PubMed PMID: 35414096]