Introduction
Amyloidosis is a heterogeneous group of disorders characterized by the deposition of an abnormal protein known as amyloid in various parts of the body, including the eyes and periocular structures.[1] Amyloid, an insoluble fibrillary protein resulting from a dynamic misfolding process, aggregates in the intracellular and extracellular spaces of the body, leading to progressive organ damage.
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
Amyloidosis is classified into systemic or localized forms.[1] This disease may occur as a primary condition or secondary to poorly controlled inflammatory diseases, such as chronic infections, autoimmune conditions, familial Mediterranean fever, and plasma cell dyscrasia, such as multiple myeloma.[2][3] Based on the amyloid protein type, clinicians refer to the systemic disease as AL (amyloid immunoglobulin light chain), AA (amyloid A protein), or ATTR (amyloid transport protein transthyretin).[1]
Epidemiology
AL amyloidosis is the most common form of systemic amyloidosis in the United States, with approximately 78% of new cases annually. The hereditary form of ATTR amyloidosis is a less common systemic condition, accounting for 10% to 20% of diagnosed cases in tertiary centers, and is more prevalent in men. AA amyloidosis is a rare acquired condition secondary to chronic inflammation, with an annual incidence of 6%.[4]
Amyloidosis may involve the eye or ocular adnexa with high phenotypic variability. Ocular amyloidosis is frequently attributed to localized AL amyloidosis, accounting for 78.6% of cases, followed by systemic AL amyloidosis at 16.2% and multiple myeloma-associated AL amyloidosis at 10.7%.[2] Approximately 11.8% of patients with AL amyloidosis have ocular involvement, and in 5% of cases, ocular involvement is the initial presentation.[5]
Ocular manifestations in AL amyloidosis are mostly extraocular, involving the orbit, conjunctiva, temporal artery, and extraocular muscles.[5] In hereditary forms of amyloidosis, such as non-transthyretin familial amyloidosis, the cornea is primarily affected, representing 20% of cases, while vitreous involvement occurs in 10% of cases in ATTR amyloidosis, with the retina being affected only rarely.[5][6] Ocular involvement in secondary amyloidosis is rare.[5] Ocular orbital amyloidosis affects those who are between 45 and 65 years with no sex predilection.[7] In transthyretin amyloidosis, women experience ocular symptoms more frequently, representing 46% of cases, compared to men at 15%.[5]
Pathophysiology
In response to extracellular stimuli like low pH, oxidation, and high temperature, proteins synthesized in the endoplasmic reticulum undergo unfolding and misfolding. Consequently, these proteins form monomers that aggregate to create amyloid fibrils, which deposit across the body in intra and extracellular tissues. In hereditary amyloidosis, misfolding occurs due to a mutation in the protein structure that stimulates amyloid fibril formation. Amyloid fibrils cause tissue damage by various mechanisms such as alteration of tissue architecture, interaction with cell surface receptors, inflammation, oxidative stress, and activation of apoptosis. The amyloid may be deposited locally at the synthesis or distant sites, leading to tissue damage.[8] The deposit may manifest as neuropathy or vascular obstruction and increased vascular fragility. The mass effect of amyloid deposition may lead to structural distortion. Deposition in the vitreous cavity leads to media opacification.[9]
Histopathology
The confirmatory test for amyloidosis is the presence of apple-green birefringence on Congo red staining on the biopsy specimen. Other features of amyloidosis are homogeneous granular, filamentous eosinophilic appearance in hematoxylin and eosin staining, metachromasia on crystal violet staining, and ultraviolet fluorescence on thioflavin T staining.[10]
History and Physical
Various clinical presentations are observed based on the part of the eye affected.
Orbital Amyloidosis
Orbital involvement is more common in systemic AL amyloidosis or can occur as a part of localized amyloidosis, a rare condition that affects the head and neck. The latter may present in 2 forms. The most common clinical picture is a progressive proptosis with limitation of extraocular movements due to bilateral nodular infiltration of extraocular muscles and orbital adnexa. Rarely, this condition may present as an amyloidoma involving the anterior orbital area close to the lacrimal glands.[11][12] Most commonly, amyloidosis occurs in the fourth to sixth decades of life and affects both sexes equally.[7] Amyloidosis may cause restrictive exophthalmoplegia, which can be mistaken for ocular thyroidopathy, and an accurate diagnosis may require orbital imaging and thyroid function tests.
Optic nerve involvement in amyloidosis usually results from amyloid masses in the orbital apex or due to dural infiltration leading to compressive optic neuropathy. Neuropathic amyloidosis usually spares the optic nerve.[13] Amyloidosis uncommonly involves the lacrimal gland and may present as a hard, mobile mass in the upper temporal orbit that rarely attaches to the periocular bones. Amyloidosis may also be associated with eccentric proptosis. Bilateral involvement is common in systemic amyloidosis. An accurate diagnosis may require excluding similar conditions such as vascular malformations, dacryoadenitis, or lacrimal gland neoplasms.[14] AL amyloidosis may present a clinical picture similar to temporal arteritis with bilateral vision loss due to amyloid light chain infiltration of the temporal artery. A temporal artery biopsy confirms the diagnosis of AL amyloid.[15] Patients with AL amyloidosis may also develop spontaneous bilateral purplish discoloration around the eyelids and the surrounding area. This symptom is usually a late sign caused by increased vascular fragility resulting from the deposition of misfolded light-chain fragments.[2]
Conjunctival amyloidosis can affect healthy people of any age or gender and may cause nodular or diffuse deposits on the bulbar or tarsal conjunctiva. The tarsal amyloid deposits on the upper eyelids can lead to ptosis, while bulbar amyloidosis commonly affects the superior fornices. Amyloid deposits may be unilateral or bilateral and have a firm, waxy appearance with color varying from yellow, pink, or salmon patches. In some cases, recurrent subconjunctival hemorrhages may occur due to increased capillary fragility caused by amyloid deposits. Other conditions, such as lymphoproliferative deposits and conjunctival inflammatory granuloma, must be ruled out. Excision biopsy and a characteristic histopathology result can confirm the diagnosis.[16] Conjunctival amyloidosis may be an early indication of systemic amyloidosis; hence, systemic evaluation and follow-up are necessary.
Corneal Amyloidosis
Corneal involvement in amyloidosis may occur as a primary localized condition or as a part of the systemic disease. Primary localized corneal amyloidosis has 2 distinct inherited forms: gelatinous drop-like corneal dystrophy and lattice corneal dystrophy types I and III. Meretoja syndrome is the main form of primary systemic amyloidosis that presents with lattice corneal dystrophy type II.[17] Secondary localized corneal amyloidosis is present in various ocular diseases such as trichiasis, trachoma, sarcoidosis, interstitial keratitis, phlyctenular keratitis, uveitis, chronic post-traumatic inflammation, glaucoma, or keratoconus. However, secondary systemic amyloidosis typically spares the cornea.
Gelatinous drop-like corneal dystrophy is a rare autosomal recessive condition that mainly affects the Asian population, particularly Japanese individuals. Gelatinous drop-like corneal dystrophy usually presents within the first and second decades and is a slowly progressive condition. Nodular amyloid deposits initially involve the subepithelial part of the cornea and later coalesce to form mulberry-like opacities extending to the stroma. Nodular amyloid deposits lead to photophobia, foreign body sensation, and impaired vision. Despite attempting various surgical modalities, recurrence remains a significant challenge, leading to a poor prognosis.[18]
Lattice dystrophy type I, also known as Biber-Haab-Dimmer corneal dystrophy, is a bilateral disease with autosomal dominant inheritance, albeit asymmetrical. Biber-Haab-Dimmer corneal dystrophy most commonly affects people in the western world, with few reports from Japan. At the end of the first decade of life, Biber-Haab-Dimmer corneal dystrophy usually presents with ocular irritation due to recurrent corneal erosions and decreased vision. These opacities usually appear denser anteriorly and centrally but typically spare the peripheral cornea. The progressive condition leads to significant visual impairment by the fourth to sixth decade.[19] Lattice dystrophy type III is an autosomal recessive disease that presents after the fourth decade of life, leading to visual impairment by the seventh decade of life.[19]
Lattice corneal dystrophy type II, or the gelsolin type, is an autosomal dominant form of systemic amyloidosis, previously called familial amyloidotic polyneuropathy type IV, Finnish type. Lattice corneal dystrophy type II is a rare condition initially reported in Finland. A few cases have also been detected worldwide in Europe, North America, and India.[2][20] This condition is part of Meretoja syndrome, which includes the typical triad of lattice corneal dystrophy, progressive bilateral cranial neuropathy characterized by facial palsy, mild sensory peripheral neuropathy, walking difficulties, dysarthria, drooling, loose skin resulting in a mask-like face, protruding lips, and dry skin. Ocular symptoms manifest after the third decade of life, primarily as dry eyes. Vision typically remains preserved until the seventh decade. The condition affects both sexes, although women exhibit an earlier onset and more severe involvement than men. Patients with this disease are at increased risk of open-angle glaucoma. Facial paralysis and skin laxity affect eyelid closure and cause ectropion, leading to exposure keratitis.[21]
Vitreoretinal Amyloidosis
Vitreous amyloidosis is a rare condition associated with familial amyloidotic polyneuropathy (FAP), which can occur in hereditary or sporadic non-hereditary forms. Mutations in the transthyretin gene cause FAP, the most common form of hereditary amyloidosis, transmitted in an autosomal dominant manner.[22] The liver mainly synthesizes transthyretin. However, transthyretin can also be produced by the brain's retinal pigment epithelium and choroid plexus. Ocular involvement is reported in 10% of patients with ATTR amyloidosis.[2] While FAP commonly occurs in Western populations, instances of vitreous amyloidosis with distinct TTR mutations also emerge in India and China.[23][24]
Vitreous opacities are usually bilateral but may be asymmetric. They can appear as cobwebs, sheets, crystalline needle-like structures, veils, or strings of pearls. Visual symptoms vary from glare, floaters, and mild to severe visual impairment depending on the density of opacities (see Image. Vitreous Amyloidosis). These vitreous opacities typically extend from the peripheral vitreous in a centripetal manner. Vitreous opacities abutting the posterior lens capsule may form footplates or pseudopodia lentis (see Image. Pseudopodia Lentis in Vitreous Amyloidosis).
In FAP, the incidence of vitreous opacity varies from 5.4% to 35%.[23] Vitreous opacities can be an isolated sign of amyloidosis or associated with other signs such as amyloid deposits on the iris, pupillary margin, choroidal infiltration, and glaucoma.[25] Retinal perivascular amyloid deposits can appear as focal plaques, tortuosity, beading, or vascular sheathing. Retinal hemorrhages and cotton wool spots may be seen in 20% of patients with hereditary TTR amyloidosis, leading to a higher incidence of retinal vein occlusions.[6] Retinal amyloid angiopathy and choroidal amyloid angiopathy have also been described.[26]
Evaluation
Although the clinical features may suggest ocular amyloidosis, the definitive diagnosis is through a biopsy of the lesion and a histopathology evaluation. Surgeons may take an excision biopsy from lesions of the conjunctiva, eyelid, lacrimal gland, or temporal artery. Additionally, vitreous samples obtained from vitrectomy can help diagnose the condition. In an orbital mass, a biopsy via the superior lateral sulcus may be necessary to confirm the diagnosis of amyloidoma and rule out other conditions, such as a lymphoproliferative disorder.[27] The histopathology examination shows the characteristic appearance, which helps to confirm the diagnosis of amyloidosis.[10]
Computed tomography scans are usually more informative than magnetic resonance imaging (MRI) in assessing periocular and orbital amyloidosis due to their higher sensitivity in detecting bone changes and calcifications in tissues. The mass effect may cause global displacement. Orbital amyloidosis may exhibit extraocular muscle enlargement, soft tissue infiltration or mass, and calcification. Extraocular muscle shows highly irregular nodular or fusiform enlargement with areas of calcification. Muscle involvement may be unilateral or bilateral, which usually spares the tendons.[13][28][29][30] However, MRI provides a more accurate anatomical definition of the extension of the lesion.[31]
In vitreoretinal amyloidosis, fundus fluorescein angiography may show retinal vascular occlusion and focal or diffuse leakage that is more prominent in the posterior pole than the retinal periphery.[32] Optical coherence tomography in eyes with veil-like vitreous opacities shows needle-shaped deposits on the retinal surface that extend to the vitreous cavity.[33] When amyloid has been found in a site-specific for localized amyloidosis, screening for amyloid in another site of the body, such as subcutaneous fat or bone marrow, with a tissue biopsy is recommended. Ancillary tests to rule out systemic involvement include a complete blood count, peripheral blood smear, liver and renal function tests, prothrombin time, beta-microglobulin assay, serum immunofixation electrophoresis with measurement of the free light chain, bone marrow aspiration, urine electrophoresis, chest x-ray, electrocardiogram, echocardiography, and ultrasonography of the abdomen.[27]
Treatment / Management
Upon diagnosing the disease, it is mandatory to assess for systemic involvement. Observation is advisable in asymptomatic or mildly symptomatic patients with localized amyloidosis. In extensive conjunctival infiltration, close follow-up is vital as surgical excision or other interventions are not effectiveineffective.[13] Surgical debulking is indicated for patients experiencing symptomatic diseases such as ocular motility disturbances, compressive optic neuropathy, and unacceptable cosmetic appearance. In patients with medical contraindications for surgery or with extensive infiltrative disease, radiotherapy with or without surgical debulking may be useful. Close follow-up of the patients is necessary forto the prevention ofprevent complications.[28] Cryotherapy may be effective for conjunctival amyloidosis as it disrupts the blood supply to the amyloid deposits. Surgical debulking before cryotherapy is advisable as it allows access to freezing to the deeper blood supply.[34]
Differential Diagnosis
Lacrimal gland involvement can mimic lymphoproliferative disorders, vascular malformations, dacryoadenitis, or lacrimal gland neoplasms.[14] A computed tomography scan helps assess bone invasion or calcification. A definitive diagnosis is made by biopsy and histopathology evaluation. Focal orbital amyloidosis mimics are pseudotumor, lymphoproliferative lesion, sarcoidosis, or cavernous hemangioma. Characteristic findings on computed tomography and MRI differentiate these conditions.[35][36][37]
Corneal amyloidosis may mimic other forms of corneal dystrophies.[38] Other differential diagnoses include various causes of monoclonal gammopathy, lecithin-cholesterol-acyltransferase deficiency, Fabry disease, cystinosis, tyrosine transaminase deficiency, systemic lysosomal storage diseases such as mucopolysaccharidoses, lipidoses, mucolipidoses, and several skin diseases known as X-linked ichthyosis, and keratosis follicularis. The main differentials of vitreoretinal amyloidosis are intermediate uveitis of infectious or non-infectious etiology, masquerades such as intra-ocular lymphoma, and multiple sclerosis.[39] After ruling out various causes for vitritis, histopathology of the vitrectomy samples helps to confirm the diagnosis.
Prognosis
Orbital amyloidosis
The prognosis of orbital amyloidosis depends on the extent of involvement and the area affected. Observing asymptomatic patients is an option, while surgical debulking of the mass may be necessary in symptomatic cases. Excision treatment may continue by cryotherapy to treat residual infiltrating tissue, but incomplete removal of the conjunctival infiltration may result in frequent recurrences.[40] Resection of the levator palpebrae muscle is rarely required, even in ptosis and globe displacement.[41] External beam radiotherapy may benefit patients where complete surgical debulking or excision cannot avoid or reduce the risk of recurrence.[27]
Corneal amyloidosis
Corneal dystrophy type I presents with visual symptoms by 10 years of age, leading to marked visual impairment by 40 to 60 years of age. In type III corneal dystrophy, visual symptoms manifest after 40 years of age, and vision is impaired after 60 years of age. Both conditions are progressive, and there is no therapy to prevent progressive visual impairment. Surgeons most commonly treat corneal opacities with deep anterior lamellar keratoplasty. However, recurrence and stromal graft rejection remain possible complications. Penetrating keratoplasty may also be of value in these cases. An overall graft survival rate of 100% at 12 years has been reported.[42]
There is no curative treatment for patients with Meretoja syndrome. Visual symptoms typically begin before 25 years of age, but individuals usually retain reasonably good visual acuity up to 65 years of age. Early intervention to control dry eye may help to alleviate symptoms. In patients with recurrent corneal ulcers and corneal scarring leading to corneal opacification, keratoplasty is necessary. However, the visual outcome after traditional penetrating keratoplasty is usually poor because of frequent postoperative complications related to dry eye and corneal anesthesia, persistent epithelial defects, and the ongoing deposition of amyloid in the transplanted cornea.[21]
In gelatinous drop-like corneal dystrophy, visual symptoms present before 10 years of age, with marked visual impairment at 10 to 30 years of age. Penetrating keratoplasty, deep lamellar keratoplasty, or superficial keratectomy may help to achieve visual rehabilitation, although early post-surgical relapses have been reported.[21][18]
Vitreoretinal amyloidosis
Pars plana vitrectomy usually clears media haze and significantly improves visual acuity in vitreoretinal amyloidosis. However, recurrence may occur in 25% of patients due to retrolental residual vitreous, persistent intraocular production of amyloid fibrils by the retinal pigment epithelium, or both.[23] Repeated vitrectomy may be necessary in these specific cases.
Complications
Based on the part of the eye affected, complications vary and include:
- Orbital or ocular adnexal amyloidosis.
- Ptosis, eyelid swelling, proptosis, lid or conjunctival masses, ophthalmoplegia, and paresis of accommodation.
- Amyloid deposition in trabecular meshwork can lead to glaucoma.
- Corneal amyloidosis.
- Keratoconjunctivitis sicca, corneal opacification, and severe visual loss.
- Vitreoretinal disorders.
- ATTR amyloidosis causes significant visual defects due to dense vitreous opacities.[2][9]
Deterrence and Patient Education
Educating patients diagnosed with ocular amyloidosis about the potential systemic implications of the disease is essential. Patients should consult with an internist and other physicians as needed. Emphasize the importance of regular follow-up appointments with both physicians and ophthalmologists.
Enhancing Healthcare Team Outcomes
Ocular amyloidosis can occur as a localized condition or as part of systemic amyloidosis. AL amyloidosis and familial non-TTR or TTR amyloidosis may be associated with ocular complications. Ocular features may be the initial presentation or appear after the systemic condition diagnosis. AL amyloidosis is also known to cause severe neuropathy, myopathy, nephropathy, cardiomyopathy, or vasculopathy.
Once detecting ocular amyloidosis, healthcare providers should refer patients for a thorough evaluation by the internist. In addition, patients with unexplained ocular findings and a diagnosis of conditions such as Waldenstrom macroglobulinemia and multiple myeloma should undergo a systemic workup. Patients presenting with corneal lattice dystrophy also need systemic evaluation.[2]
Interprofessional management, including a team of internists, oncologists, hematologists, nephrologists, cardiologists, and neurologists working with ophthalmologists, helps optimal patient management. Early diagnosis is necessary for treatment options such as chemotherapy and bone marrow transplant to mitigate the severity of the condition.[43]
Media
(Click Image to Enlarge)
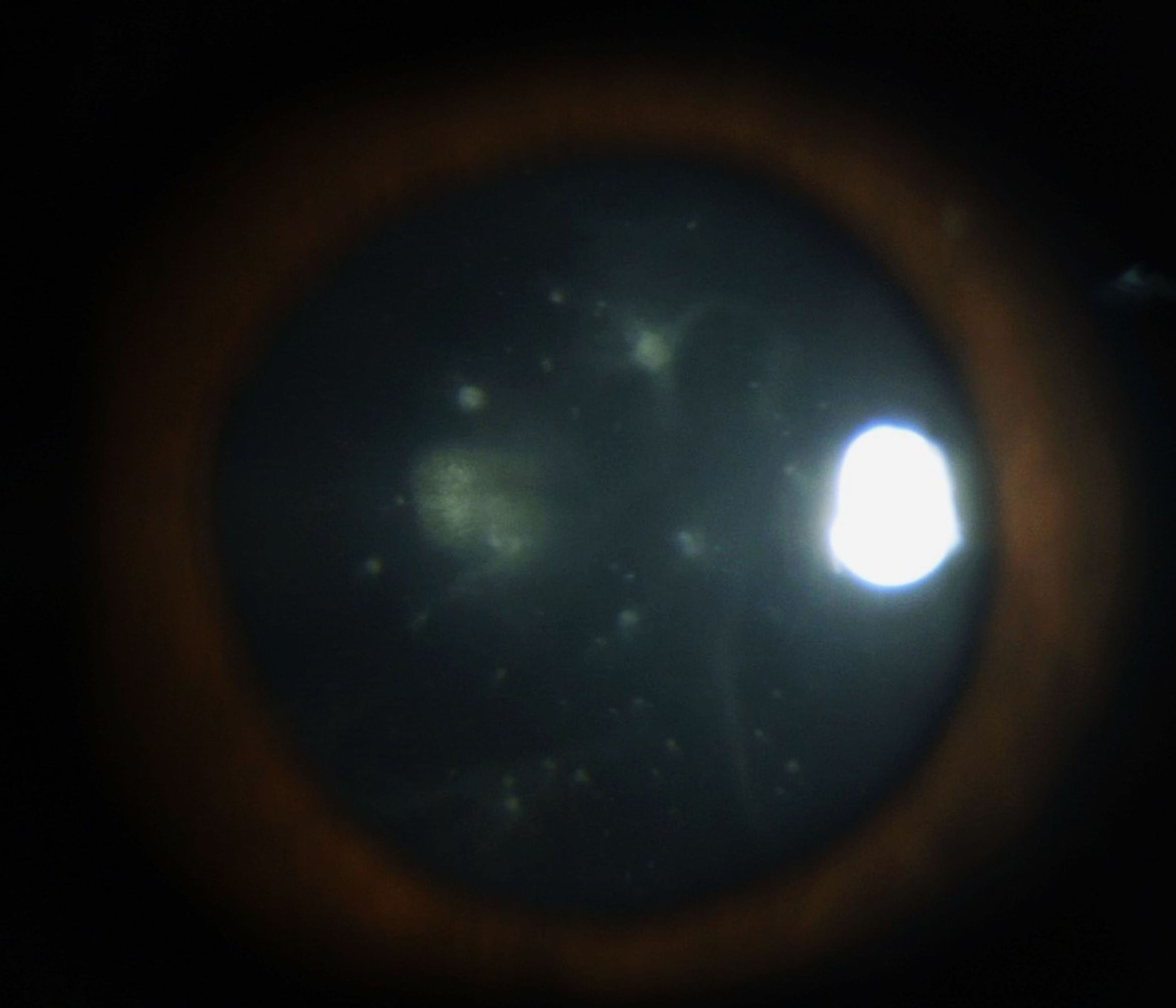
Pseudopodia Lentis in Vitreous Amyloidosis
Reproduced with kind permission from Elsevier. Source publication: Venkatesh P, Selvan H, Singh SB, et al. Vitreous Amyloidosis: Ocular, Systemic, and Genetic Insights. Ophthalmology. 2017;124(7):1014-1022. doi:10.1016/j.ophtha.2017.03.011
(Click Image to Enlarge)
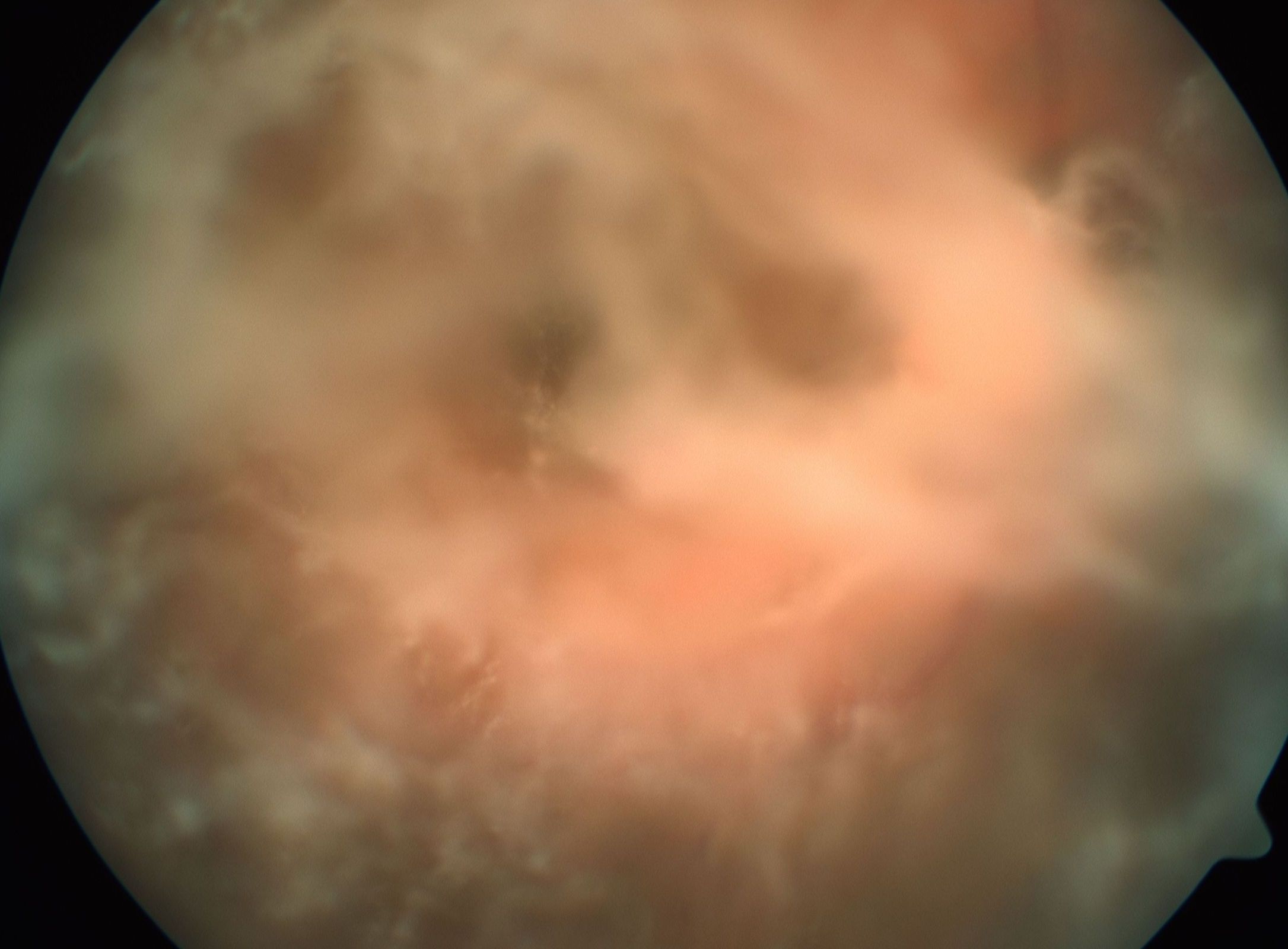
Vitreous Amyloidosis. Fundus color photo of an eye with vitreous amyloidosis
Reproduced with kind permission from Elsevier, Source publication: Venkatesh P, Selvan H, Singh SB, et al. Vitreous Amyloidosis: Ocular, Systemic, and Genetic Insights. Ophthalmology. 2017;124(7):1014-1022. doi:10.1016/j.ophtha.2017.03.011
References
Sipe JD, Benson MD, Buxbaum JN, Ikeda S, Merlini G, Saraiva MJ, Westermark P, Nomenclature Committee of the International Society of Amyloidosis. Amyloid fibril protein nomenclature: 2012 recommendations from the Nomenclature Committee of the International Society of Amyloidosis. Amyloid : the international journal of experimental and clinical investigation : the official journal of the International Society of Amyloidosis. 2012 Dec:19(4):167-70. doi: 10.3109/13506129.2012.734345. Epub 2012 Nov 1 [PubMed PMID: 23113696]
Dammacco R, Merlini G, Lisch W, Kivelä TT, Giancipoli E, Vacca A, Dammacco F. Amyloidosis and Ocular Involvement: an Overview. Seminars in ophthalmology. 2020 Jan 2:35(1):7-26. doi: 10.1080/08820538.2019.1687738. Epub 2019 Dec 12 [PubMed PMID: 31829761]
Level 3 (low-level) evidenceLachmann HJ, Goodman HJ, Gilbertson JA, Gallimore JR, Sabin CA, Gillmore JD, Hawkins PN. Natural history and outcome in systemic AA amyloidosis. The New England journal of medicine. 2007 Jun 7:356(23):2361-71 [PubMed PMID: 17554117]
Bustamante JG, Zaidi SRH. Amyloidosis. StatPearls. 2024 Jan:(): [PubMed PMID: 29261990]
Reynolds MM, Veverka KK, Gertz MA, Dispenzieri A, Zeldenrust SR, Leung N, Pulido JS. OCULAR MANIFESTATIONS OF SYSTEMIC AMYLOIDOSIS. Retina (Philadelphia, Pa.). 2018 Jul:38(7):1371-1376. doi: 10.1097/IAE.0000000000001901. Epub [PubMed PMID: 29068915]
Minnella AM, Rissotto R, Antoniazzi E, Di Girolamo M, Luigetti M, Maceroni M, Bacherini D, Falsini B, Rizzo S, Obici L. Ocular Involvement in Hereditary Amyloidosis. Genes. 2021 Jun 22:12(7):. doi: 10.3390/genes12070955. Epub 2021 Jun 22 [PubMed PMID: 34206500]
Banerjee P, Alam MS, Subramanian N, Kundu D, Koka K, Poonam NS, Mukherjee B. Orbital and adnexal amyloidosis: Thirty years experience at a tertiary eye care center. Indian journal of ophthalmology. 2021 May:69(5):1161-1166. doi: 10.4103/ijo.IJO_2528_20. Epub [PubMed PMID: 33913851]
Ikura H, Endo J, Kitakata H, Moriyama H, Sano M, Fukuda K. Molecular Mechanism of Pathogenesis and Treatment Strategies for AL Amyloidosis. International journal of molecular sciences. 2022 Jun 6:23(11):. doi: 10.3390/ijms23116336. Epub 2022 Jun 6 [PubMed PMID: 35683015]
Singh RB, Singhal S, Sinha S, Cho J, Nguyen AX, Dhingra LS, Kaur S, Sharma V, Agarwal A. Ocular complications of plasma cell dyscrasias. European journal of ophthalmology. 2023 Sep:33(5):1786-1800. doi: 10.1177/11206721231155974. Epub 2023 Feb 9 [PubMed PMID: 36760117]
Sipe JD, Benson MD, Buxbaum JN, Ikeda S, Merlini G, Saraiva MJ, Westermark P. Nomenclature 2014: Amyloid fibril proteins and clinical classification of the amyloidosis. Amyloid : the international journal of experimental and clinical investigation : the official journal of the International Society of Amyloidosis. 2014 Dec:21(4):221-4. doi: 10.3109/13506129.2014.964858. Epub 2014 Sep 29 [PubMed PMID: 25263598]
Murdoch IE, Sullivan TJ, Moseley I, Hawkins PN, Pepys MB, Tan SY, Garner A, Wright JE. Primary localised amyloidosis of the orbit. The British journal of ophthalmology. 1996 Dec:80(12):1083-6 [PubMed PMID: 9059275]
Jamshidi P, Levi J, Suarez MJ, Rivera R, Mahoney N, Eberhart CG, Rosenberg A, Rodriguez FJ. Clinicopathologic and Proteomic Analysis of Amyloidomas Involving the Ocular Surface and Adnexa. American journal of clinical pathology. 2022 Apr 1:157(4):620-627. doi: 10.1093/ajcp/aqab161. Epub [PubMed PMID: 34698334]
Taban M, Piva A, See RF, Sadun AA, Quiros PA. Review: orbital amyloidosis. Ophthalmic plastic and reconstructive surgery. 2004 Mar:20(2):162-5 [PubMed PMID: 15083087]
Segado Martínez M, Ruiz García G, Abenza Baeza S. Amyloidosis localized in lacrimal gland. Medicina clinica. 2022 Aug 12:159(3):e23-e24. doi: 10.1016/j.medcli.2022.02.027. Epub 2022 May 25 [PubMed PMID: 35643772]
Neri A, Rubino P, Macaluso C, Gandolfi SA. Light-chain amyloidosis mimicking giant cell arteritis in a bilateral anterior ischemic optic neuropathy case. BMC ophthalmology. 2013 Dec 20:13():82. doi: 10.1186/1471-2415-13-82. Epub 2013 Dec 20 [PubMed PMID: 24359546]
Level 3 (low-level) evidenceSuesskind D, Ziemssen F, Rohrbach JM. Conjunctival amyloidosis -- clinical and histopathologic features. Graefe's archive for clinical and experimental ophthalmology = Albrecht von Graefes Archiv fur klinische und experimentelle Ophthalmologie. 2015 Aug:253(8):1377-83. doi: 10.1007/s00417-015-2932-3. Epub 2015 Jan 27 [PubMed PMID: 25619666]
Carrwik C, Stenevi U. Lattice corneal dystrophy, gelsolin type (Meretoja's syndrome). Acta ophthalmologica. 2009 Nov:87(8):813-9. doi: 10.1111/j.1755-3768.2009.01686.x. Epub [PubMed PMID: 19832730]
Kaza H, Barik MR, Reddy MM, Mittal R, Das S. Gelatinous drop-like corneal dystrophy: a review. The British journal of ophthalmology. 2017 Jan:101(1):10-15. doi: 10.1136/bjophthalmol-2016-309555. Epub 2016 Dec 2 [PubMed PMID: 27913443]
Milovanova E, Gomon S, Rocha G. Classic lattice corneal dystrophy: a brief review and summary of treatment modalities. Graefe's archive for clinical and experimental ophthalmology = Albrecht von Graefes Archiv fur klinische und experimentelle Ophthalmologie. 2023 Nov 7:():. doi: 10.1007/s00417-023-06297-6. Epub 2023 Nov 7 [PubMed PMID: 37934291]
Maramattom BV, Chickabasaviah YT. A new Indian family affected by gelsolin amyloidosis. Neurology India. 2013 Nov-Dec:61(6):673-5. doi: 10.4103/0028-3886.125372. Epub [PubMed PMID: 24441349]
Casal I, Monteiro S, Abreu C, Neves M, Oliveira L, Beirão M. Meretoja's Syndrome: Lattice Corneal Dystrophy, Gelsolin Type. Case reports in medicine. 2017:2017():2843417. doi: 10.1155/2017/2843417. Epub 2017 Jan 31 [PubMed PMID: 28250773]
Adams D, Ando Y, Beirão JM, Coelho T, Gertz MA, Gillmore JD, Hawkins PN, Lousada I, Suhr OB, Merlini G. Expert consensus recommendations to improve diagnosis of ATTR amyloidosis with polyneuropathy. Journal of neurology. 2021 Jun:268(6):2109-2122. doi: 10.1007/s00415-019-09688-0. Epub 2020 Jan 6 [PubMed PMID: 31907599]
Level 3 (low-level) evidenceVenkatesh P, Selvan H, Singh SB, Gupta D, Kashyap S, Temkar S, Gogia V, Tripathy K, Chawla R, Vohra R. Vitreous Amyloidosis: Ocular, Systemic, and Genetic Insights. Ophthalmology. 2017 Jul:124(7):1014-1022. doi: 10.1016/j.ophtha.2017.03.011. Epub 2017 Apr 12 [PubMed PMID: 28412068]
Li Z, Du K, Chu X, Lv H, Zhang W, Wang Z, Yuan Y, Meng L. TTR Gly83Arg Mutation: Beyond Familial Vitreous Amyloidosis. Frontiers in neurology. 2021:12():821003. doi: 10.3389/fneur.2021.821003. Epub 2022 Feb 3 [PubMed PMID: 35185758]
Pathak-Ray V, Pulpa V, Blyth C. Vitreous amyloidosis and secondary glaucoma-a case report. Eye (London, England). 2002 Jul:16(4):492-4 [PubMed PMID: 12101461]
Level 3 (low-level) evidenceMano F, Dispenzieri A, Kusaka S, Pavesio C, Khalid H, Keane PA, Pulido JS. ASSOCIATION BETWEEN CHOROIDAL CHARACTERISTICS AND SYSTEMIC SEVERITY IN AMYLOIDOSIS. Retina (Philadelphia, Pa.). 2021 May 1:41(5):1037-1046. doi: 10.1097/IAE.0000000000002961. Epub [PubMed PMID: 32826787]
Mora-Horna ER, Rojas-Padilla R, López VG, Guzmán MJ, Ceriotto A, Salcedo G. Ocular adnexal and orbital amyloidosis: a case series and literature review. International ophthalmology. 2016 Apr:36(2):281-98. doi: 10.1007/s10792-015-0138-7. Epub 2015 Oct 14 [PubMed PMID: 26466598]
Level 2 (mid-level) evidenceLeibovitch I, Selva D, Goldberg RA, Sullivan TJ, Saeed P, Davis G, McCann JD, McNab A, Rootman J. Periocular and orbital amyloidosis: clinical characteristics, management, and outcome. Ophthalmology. 2006 Sep:113(9):1657-64 [PubMed PMID: 16828514]
Hamidi Asl K, Liepnieks JJ, Nunery WR, Yazaki M, Benson MD. Kappa III immunoglobulin light chain origin of localized orbital amyloidosis. Amyloid : the international journal of experimental and clinical investigation : the official journal of the International Society of Amyloidosis. 2004 Sep:11(3):179-83 [PubMed PMID: 15523920]
Yerli H, Aydin E, Avci S, Haberal N, Oto S. Focal Amyloidosis of the Orbit Presenting as a Mass: MRI and CT Features. Iranian journal of radiology : a quarterly journal published by the Iranian Radiological Society. 2011 Dec:8(4):241-4. doi: 10.5812/iranjradiol.4555. Epub 2011 Dec 25 [PubMed PMID: 23329948]
Menetti F, Bartolomei I, Ambrosini-Spaltro A, Salvi F, Agati R, Leonardi M. Amyloidoma Involving the Orbit, Meckel's Cave and Infratemporal Fossa: 3T MRI Findings. The neuroradiology journal. 2009 Mar 23:22(1):41-7 [PubMed PMID: 24206952]
Kakihara S, Hirano T, Kitahara J, Matsuda Y, Imai A, Miyahara T, Murata T. OCULAR ANGIOGRAPHIC FEATURES IN JAPANESE PATIENTS WITH VAL30MET HEREDITARY TRANSTHYRETIN AMYLOIDOSIS. Retina (Philadelphia, Pa.). 2022 Jan 1:42(1):210-215. doi: 10.1097/IAE.0000000000003291. Epub [PubMed PMID: 34483312]
Kakihara S, Hirano T, Matsuda Y, Takano D, Imai A, Miyahara T, Murata T. Deposits on Retinal Surface Seen on OCT in Ocular Amyloidosis. Ophthalmology. Retina. 2021 Oct:5(10):1005-1008. doi: 10.1016/j.oret.2020.12.028. Epub 2021 Jan 7 [PubMed PMID: 33422693]
Fraunfelder FW. Liquid nitrogen cryotherapy for conjunctival amyloidosis. Archives of ophthalmology (Chicago, Ill. : 1960). 2009 May:127(5):645-8. doi: 10.1001/archophthalmol.2008.535. Epub [PubMed PMID: 19433714]
Asao C, Korogi Y, Hotta A, Shimomura O, Kitajima M, Negi A, Takahashi M. Orbital pseudotumors: value of short inversion time inversion-recovery MR imaging. Radiology. 1997 Jan:202(1):55-9 [PubMed PMID: 8988192]
Simon EM, Zoarski GH, Rothman MI, Numaguchi Y, Zagardo MT, Mathis JM. Systemic sarcoidosis with bilateral orbital involvement: MR findings. AJNR. American journal of neuroradiology. 1998 Feb:19(2):336-7 [PubMed PMID: 9504490]
Thorn-Kany M, Arrué P, Delisle MB, Lacroix F, Lagarrigue J, Manelfe C. Cavernous hemangiomas of the orbit: MR imaging. Journal of neuroradiology = Journal de neuroradiologie. 1999 Jun:26(2):79-86 [PubMed PMID: 10444931]
Klintworth GK. Corneal dystrophies. Orphanet journal of rare diseases. 2009 Feb 23:4():7. doi: 10.1186/1750-1172-4-7. Epub 2009 Feb 23 [PubMed PMID: 19236704]
Touhami S, Leclercq M, Stanescu-Segall D, Touitou V, Bodaghi B. Differential Diagnosis of Vitritis in Adult Patients. Ocular immunology and inflammation. 2021 May 19:29(4):786-795. doi: 10.1080/09273948.2021.1898001. Epub 2021 May 18 [PubMed PMID: 34003716]
Prager AJ, Habib LA, Gambogi T, Busam KJ, Marr BP. Long-Term Follow-Up of 4 Patients with Conjunctival Amyloidosis. Ocular oncology and pathology. 2018 Sep:4(5):313-317. doi: 10.1159/000485918. Epub 2018 Feb 13 [PubMed PMID: 30320104]
Eneh AA, Farmer J, Kratky V. Primary localized orbital amyloid: case report and literature review; 2004-2015. Canadian journal of ophthalmology. Journal canadien d'ophtalmologie. 2016 Aug:51(4):e131-e136. doi: 10.1016/j.jcjo.2016.03.019. Epub 2016 Jul 1 [PubMed PMID: 27521683]
Level 3 (low-level) evidenceOno T, Ishiyama S, Hayashidera T, Mori Y, Nejima R, Miyata K, Amano S. Twelve-year follow-up of penetrating keratoplasty. Japanese journal of ophthalmology. 2017 Mar:61(2):131-136. doi: 10.1007/s10384-016-0489-2. Epub 2016 Nov 24 [PubMed PMID: 27885526]
Palladini G, Merlini G. Current treatment of AL amyloidosis. Haematologica. 2009 Aug:94(8):1044-8. doi: 10.3324/haematol.2009.008912. Epub [PubMed PMID: 19644136]