Introduction
Blepharophimosis syndrome (BPES) is a relatively rare genetic condition that primarily affects eyelid formation and results in a distinctive facial appearance present at birth.[1] This condition was first described by Komoto in 1921.[2] The condition is characterized by the existence of a reduced horizontal opening of the eyelid (blepharophimosis), drooping of the upper eyelid (ptosis), increased distance between the inner corners of the eyes (telecanthus), and an upward fold of skin near the inner corner of the eye, where the medial eyelid skin fold appears more prominent in the lower eyelid, extending across the canthus and tapering into the upper eyelid (epicanthus inversus).[3] These congenital features define the syndrome, hence the acronym BPES (see Image. Male Child with Blepharophimosis Syndrome).[4]
Blepharophimosis syndrome is an autosomal dominant disease affecting the eyelids and mid-face structures. These characteristics significantly impact the patient's vision and facial appearance. BPES can also be associated with additional systemic defects, specifically premature ovarian insufficiency in females, resulting in BPES type I.[5] Most cases are caused by mutations in FOXL2 on chromosome 3q23.
The structural defects observed in BPES are caused by the interruption of the development of the eyelid and related tissues during embryogenesis. The course of BPES is contingent upon the specific subtype and severity, resulting in varying outcomes. Some persons may encounter substantial visual impairment caused by ptosis, whereas others may be concerned with cosmetic issues. The condition has no propensity for dissemination but remains limited to the congenital structural defects. Nevertheless, if ptosis is not addressed, subsequent complications such as amblyopia may result, underscoring the importance of prompt detection and surgery.
There are 2 main types of BPES. Each type harbors the 4 classic clinical signs—blepharophimosis, ptosis, epicanthus inversus, and telecanthus. Type I is associated with premature ovarian insufficiency, whereas type II is characterized by the classic facial features alone. These features are associated with a high incidence of amblyopia if not correctly managed. Both types require surgical treatment early in life for normal vision development (see Image. Male Child After Surgery for Blepharophimosis Syndrome).
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
BPES is mainly caused by mutations in FOXL2, which is crucial for the development of eyelids and ovarian follicles. This gene encodes a forkhead family transcription factor that plays a vital role in controlling gene expression throughout embryonic development.[6] Pathogenic mutations dsirupt the normal functioning of FOXL2, resulting in the observable characteristics of BPES.
The genetic cause of BPES was discovered in 2001 through genetic sequencing of 7 families affected by BPES.[7] Pathogenic mutations in FOXL2 on chromosome 3q23 are responsible for BPES. FOXL2 encodes a forkhead transcription factor of 376 amino acids, which contain a tract of 14 alanine residues. To date, over 100 unique FOXL2 mutations have been identified in BPES families.[8] The exact function of FOXL2 is unknown.[9] Still, expression studies have shown that FOXL2 is expressed in the mesenchyme of developing fetal eyelids and the granulosa cells of fetal and adult ovaries.[7]
The 2 types of BPES are phenotypically indistinguishable but can be determined through genetic testing. Mutations that truncate FOXL2 before the polyalanine tract result in type I BPES. Mutations that expand the polyalanine tract result in type II BPES.[10] BPES can be caused by various types of mutations including missense, nonsense, or frameshift mutations.[11][12] These mutations can result in a loss-of-function or dominant-negative effect.[13] In individuals with type I BPES, the genetic abnormalities not only impact the formation of eyelids but also interfere with the functioning of the ovaries, resulting in premature ovarian insufficiency in affected females. Type II BPES is characterized by ocular manifestations without concurrent ovarian dysfunction.[14]
Epidemiology
BPES is a rare disorder, occurring in approximately 1 of 50,000 newborns.[15] Although type I BPES affects both males and females, the specific risk of early ovarian insufficiency is exclusive to females. The syndrome has been observed in several ethnic groups globally, without any notable geographical or ethnic inclination observed. Most cases of BPES are familial via an autosomal dominant pattern. However, spontaneous mutations may occur. Within affected families, BPES generally shows complete penetrance but exhibits varying expressivity, implying that the degree of eyelid abnormalities and related symptoms can greatly differ, even among individuals within the same family.
Up to 75% of affected individuals have a FOXL2 mutation.[15] Approximately 64% of cases occur in women.[16] Type I has 100% penetrance and is transmitted primarily through males due to females developing premature ovarian insufficiency and reduced fertility. Type II has 96.5% penetrance and is transmitted through males and females.[17][16] The incidence of strabismus in BPES is 20 to 27%.[18] Development of amblyopia from ptosis or strabismus is between 39% and 56%.[17] Refractive error is common and as high as 94%, with simple hyperopia being the most common etiology.[15]
Pathophysiology
The underlying mechanism of BPES revolves around the interference with typical eyelid and face formation caused by abnormalities in FOXL2. The eyelid abnormalities, such as blepharophimosis, ptosis, and epicanthus inversus, occur due to the hindered differentiation and migration of the mesenchymal tissues that shape the eyelids during embryonic development. The most frequently observed characteristics are constricted palpebral fissures, drooping eyelids, and telecanthus. Type I BPES is characterized by a FOXL2 mutation, which causes ovarian dysfunction and disrupts the process of folliculogenesis, leading to early ovarian insufficiency.
Eyelid morphogenesis is a complex process involving the coordinated movement of neural crest-derived periocular mesenchymal cells during embryonic development. These cells form eyelid structures, including the levator palpebrae superioris, smooth muscle, and tarsus.[19]
In congenital ptosis, the levator palpebrae superioris muscle is maldeveloped, resulting in a droopy upper eyelid. Genes associated with congenital ptosis include ZFH4 and FOXL2. Mutations in FOXL2 disrupt actin gene expression in the smooth muscle cells of periocular mesenchyma. This disruption results in severe hypoplasia of the levator palpebrae superioris and other craniofacial defects, causing blepharophimosis, ptosis, and epicanthus inversus.[19][20]
The FOXL2 transcription factor is crucial in sexual development and ovarian function. The expression of FOXL2 is the highest in granulosa cells of the ovary, and it plays a vital role in folliculogenesis, hormone signaling, cell regulation, proliferation, and apoptosis. In the absence of FOXL2, primary follicle formation in the ovaries is disrupted, and the number of follicles recruited during fetal ovarian development is depleted. The decreased number of follicles formed during ovarian development causes premature ovarian insufficiency and sequelae of reduced estrogen.[9][21]
Histopathology
In cases of BPES, histopathological analysis of tissue indicates the presence of undeveloped or dysplastic levator palpebrae muscles, which are responsible for raising the eyelids.[22] Abnormalities may also be observed in the connective tissue components of the eyelids, such as the tarsus and orbicularis oculi muscle.[23] In instances involving concurrent premature ovarian insufficiency, an examination of the ovaries by biopsy may uncover a diminished quantity of follicles and other indications that are in line with ovarian dysgenesis.[24] Nevertheless, histopathology is not commonly employed for diagnosing BPES, as the primary methods used are clinical and genetic.
History and Physical
The evaluation of a patient with BPES typically starts with the observations of ocular abnormalities shortly after birth. There may be a hereditary pattern of comparable facial characteristics or a history of early menopause in female relatives. The physical examination reveals the characteristic manifestations of the syndrome, including blepharophimosis (narrowed eyelid fissures), ptosis (drooping of the upper eyelids), telecanthus (increased distance between the inner corners of the eyes), and epicanthus inversus (an upward fold of skin near the inner corner of the eye). These findings are typically bilateral and symmetrical.
Females with type I BPES may show signs of premature ovarian insufficiency, such as irregular menstrual periods or secondary amenorrhea, during adolescence or early adulthood. The degree of ptosis might vary, with certain patients encountering substantial visual blockage requiring prompt surgical intervention.
The diagnosis of BPES is mainly clinical (see Image. Male Child with Blepharophimosis Syndrome). Physical examination findings include:
- Blepharophimosis: A horizontal shortening of the palpebral fissure. The average horizontal palpebral fissure measures 25 to 30 mm, but in BPES, it measures 20 to 22 mm.[25]
- Ptosis: Upper eyelid droopiness develops from bilateral dysplasia of the levator palpebral superioris muscle, resulting in poor levator function and bilateral ptosis.[26][27]
- Epicanthus inverus: A skin fold arising from the medial lower eyelid and running inward and upward to the upper eyelid. This feature is always bilateral; the caruncle and plica semilunaris beneath have a hypoplastic appearance.[28]
- Telecanthus: The bony walls of the orbit are unaffected in BPES. The medial canthal tendons are elongated, which causes a widened intercanthal distance with normal interpupillary distance.[29]
There are 2 clinical types of BPES.
- Type I BPES is characterized by blepharophimosis, ptosis, epicanthus inversus, telecanthus, and premature ovarian insufficiency.
- Type II BPES is characterized by the same classic 4 findings without premature ovarian insufficiency.
Numerous other coexisting malformations and compensatory responses have been reported. Craniofacial malformations include thin and short upper eyelid skin with a margin that has an S-shape, whereas the lower margin has a downward concavity, with or without slight ectropion.[30][31]
Abnormalities of the lacrimal drainage apparatus include lateral displacement of the lower punctum, canaliculi stenosis, and elongation of the horizontal canaliculi.[32][33] Other ophthalmic manifestations include strabismus, microphthalmos, and optic disc coloboma.[34] Facial abnormalities include wide and flattened nasal bridges and anteverted ears.[16]
Individuals compensate for ptosis by adopting a chin-up head position and constantly contracting their frontalis muscles. The eyebrows can become more prominent and arched, giving patients a continually surprised look.[2]
Evaluation
The assessment of BPES involves a combination of clinical examination and genetic testing. Diagnosis is typically made by observing distinctive facial characteristics and confirmed through genetic testing that identifies pathogenic mutations in FOXL2.[35] FOXL2 sequencing is considered the most reliable method for diagnosis.[36] Procedures such as chromosomal microarray analysis or karyotyping might be used to detect significant deletions or chromosomal rearrangements affecting 3q23.[37]
In addition to genetic testing, a thorough ophthalmologic evaluation is essential, involving the measurement of palpebral fissure length, evaluation of ptosis severity, and examination of levator palpebrae muscle functionality. Visual acuity should also be evaluated to identify any associated conditions, such as amblyopia. Endocrine assessment may be required in females, especially those with type I BPES, to determine ovarian function.[38]
Individuals with the characteristic features of BPES can undergo gene-targeted testing to detect mutations in FOXL2.[39] If no variant is detected, comprehensive genomic testing can be conducted to detect deletions or duplications in the FOXL2 region. In some cases, cytogenetic testing is completed to further evaluate for balanced translocations or other interruptions of the FOXL2 region.[8][40]
Female individuals with type I BPES develop primary ovarian insufficiency, manifesting as pubertal delay, primary amenorrhea, secondary amenorrhea, or oligomenorrhea. Endocrinologic laboratory testing can aid in diagnosis, with elevated levels of follicle-stimulating and luteinizing hormones and decreased serum concentrations of estrogen and progesterone. Transvaginal ultrasound with antral follicular count can assess ovarian reserve and aid discussions regarding poor fertility.[41]
Treatment / Management
The management of BPES involves multiple disciplines, including ophthalmology, genetics, and, in the case of type I BPES, endocrinology. The main objectives of treatment are to enhance visual function and address aesthetic considerations. Treatment goals include surgical eyelid repair to promote normal visual development, improve cosmesis, alleviate neck strain from chin-up posture, and address primary ovarian insufficiency and infertility in females with type I BPES. Initial management involves a pediatric ophthalmologist assessing for amblyopia, strabismus, and refractive error. Referral to an oculoplastic surgeon should be made to evaluate the degree of ptosis, blepharophimosis, and epicanthus inversus (see Image. Male Child After Surgery for Blepharophimosis Syndrome).[42](B3)
Surgical correction is the primary treatment for eyelid abnormalities. Ptosis correction is frequently necessary to prevent or treat amblyopia. Frontalis sling surgery, a procedure that uses a suspensory device to raise the eyelid, is commonly employed. Medial canthoplasty is a surgical procedure that can repair telecanthus by repositioning the inner corners of the eyelids to a more typical position. Traditionally, eyelid correction was performed as a 2-stage surgery at 3 to 5 years of age. A medial canthoplasty using the Mustarde technique or double-opposing z-plasty to repair epicanthus inversus and telecanthus is followed by frontalis suspension for ptosis repair 6 to 12 months later. Performing ptosis correction and medial canthoplasty simultaneously could lead to tension between tightened vertical and horizontal tissues, potentially reducing the effectiveness of the procedures if done separately.[42][43](B2)
Some surgeons argue that medial canthus reconstruction can worsen ptosis and should be addressed first. Other surgeons recommend repairing ptosis first due to the high incidence of amblyopia and then performing medial canthus repair at a later age when the face has grown. Recent reports have described successful outcomes with single-stage surgery combining medial canthoplasty and ptosis repair for a select group of individuals. Additional studies are required to assess the outcomes of single-stage versus two-stage repair.[44](B2)
In general, the following surgical recommendations are made:
- If the central visual axis is unobstructed, surgical repair can wait until 3 to 5 years.
- If the central visual axis is obscured, but the vertical interpalpebral fissure height is more than 2 mm, either single-stage or two-stage repair is associated with satisfactory outcomes.[45]
- If the central visual axis is obscured, and the vertical interpalpebral fissure height is less than 2 mm, a two-stage repair should be pursued as early as possible, involving ptosis repair first to prevent amblyopia, followed by medial canthoplasty.[17] (B2)
Females who inherit BPES from the paternal parent are more likely to have type I disease and should be referred to a clinical geneticist to discuss the nature of the disease, its mode of inheritance, and the fertility implications of the genetic disorder.[46] Women should be informed of the risk of premature ovarian insufficiency and referred to a pediatric or reproductive endocrinologist and gynecologist to monitor ovarian status. Hormone replacement therapy is recommended to address early ovarian insufficiency and to prevent the long-term consequences of estrogen deprivation, such as osteoporosis and cardiovascular disease. Young women may consider fertility preservation alternatives, such as oocyte or embryo cryopreservation, before the decrease of ovarian function.[47][48] The American Society for Reproductive Medicine and the International Menopause Society recommend estrogen replacement therapy for women with primary ovarian insufficiency.[49] Hormone replacement therapy under the guidance of an endocrinologist and gynecologist is reasonable for maintaining normal bone mineral density. The options for women who wish to pursue parenthood include adoption, foster parenthood, embryo donation, egg donation, and cryopreservation.[50][46](B2)
Genetic counseling is a process that involves providing individuals and families with information about genetic conditions, inheritance patterns, and the risks of passing on genetic disorders. The goal is to assist individuals in making informed decisions about family planning.[51] Affected families are advised to undergo genetic counseling to discuss inheritance patterns, the likelihood of passing on the condition to their children, and available reproductive choices. Families with known FOXL2 mutations can access prenatal genetic testing and preimplantation genetic diagnosis.[52]
Differential Diagnosis
Most patients with BPES have a clear family history, but de novo mutations are possible.[53] If eyelid findings are present without a clear family history, it is important to consider other conditions that include ptosis and blepharophimosis as major characteristic features—NR2F2-associated 46, XX sex reversal 5, the Say-Barber-Biesecker variant of Ohdo syndrome, congenital ptosis, ptosis with external ophthalmoplegia, Noonan syndrome, Marden-Walker syndrome, Schwartz Jampel syndrome, Waardenburg syndrome, Williams syndrome, trisomy 18, cerebro-oculo-facial-digital syndrome, Dubowitz syndrome, and Smith-Lemli-Opitz syndrome.[54][55]
Conditions such as congenital ptosis [56] caused by isolated levator muscle failure must be distinguished from BPES, in which ptosis is a component of a more comprehensive syndrome. Waardenburg syndrome [57] is defined by the presence of dystopia canthorum (abnormally wide-set eyes), which can sometimes be mistaken for BPES. Nevertheless, Waardenburg syndrome encompasses hearing impairment and abnormalities in pigmentation, whereas these characteristics are not present in BPES. Individuals with Noonan syndrome [58] may exhibit ptosis (drooping of the upper eyelid) and hypertelorism (increased distance between the eyes), along with other characteristics such as below average height, congenital cardiac abnormalities, and delays in development. Apert syndrome [59] can be distinguished from BPES by the presence of syndactyly (fusion of fingers and toes), severe skull malformations, and craniofacial anomalies.
Staging
BPES does not adhere to a conventional stage scheme, as shown in oncological conditions. Nevertheless, it is possible to categorize it according to the kind (type I or type II) and the severity of symptoms, specifically in terms of their effect on vision and ovarian function. The classification assists in directing the care strategy, especially in females, where timely recognition of type I BPES is crucial for treating premature ovarian insufficiency.[60]
Prognosis
The outlook for individuals with BPES is typically favorable with proper care. Timely surgical intervention for ptosis can effectively avoid the development of amblyopia and enhance visual results (see Image. Male Child After Surgery for Blepharophimosis Syndrome). Cosmetic surgery has the potential to significantly improve the physical appearance and psychological well-being of individuals who undergo it.[61] The reproductive prognosis of females with type I BPES depends on the timing and severity of ovarian insufficiency. Early identification and intervention can alleviate certain detrimental consequences linked to early ovarian insufficiency. Patients require a multidisciplinary approach and coordination of care across many specialties. The prognosis is excellent when medical and surgical management is addressed early in life. Depending on the severity of the disorder, surgical treatment can require multiple stages, but outcomes are very successful. Patients with BPES have an average lifespan.
Complications
The primary complications of BPES result from untreated ptosis, which can cause amblyopia by blocking the visual axis during crucial stages of visual development. Possible surgical consequences may involve the occurrence of either under- or overcorrection of the condition. Ptosis, asymmetry, and scarring are present. Type I BPES is associated with problems such as infertility, osteoporosis, and cardiovascular disease, all resulting from prolonged estrogen deficiency due to premature ovarian insufficiency. Females with typical facial features and concomitant infertility may also experience psychological challenges, such as low self-esteem and social difficulties. Complications can occur after surgical repair. Early popular techniques, such as the Mustarde technique and z-epicanthoplasty, had major incidences of severe scarring. Other techniques, such as the Y-to-V technique and medial epicanthoplasty with skin redraping, have been developed to improve cosmetic outcomes.[62]
Many organic and inorganic materials are used for frontalis sling suspension to treat congenital ptosis surgically. Alloplastic materials include braided polyester sutures, polypropylene sutures, expanded polytetrafluorethylene, and silastic bands. Placing foreign material in the tissue can result in granulomatous formation, infection, or extrusion.[63]
Homografts, such as autogenous fascia lata, are associated with longer recovery time and a second potential site of infection. Overcorrection of congenital ptosis can result in lagophthalmos and cause corneal complications.
Deterrence and Patient Education
Education for individuals and families affected by BPES should prioritize the significance of early identification and intervention to mitigate visual problems. Educating parents about the signs of amblyopia and the importance of regular ophthalmologic monitoring is essential. For females with type I BPES, it is crucial to provide early information to about the risks of premature ovarian insufficiency and the available alternatives for fertility preservation. Genetic counseling is essential for delivering precise information regarding the pattern of inheritance, potential dangers to future offspring, and the range of available reproductive options.
Families must understand the need for lifelong monitoring of vision into adulthood. Surgery is almost always necessary early in life to prevent amblyopia, and it may be required again later in adulthood for cosmetic reconstruction if desired.
Families and patients should know that multiple methods exist to lift the eyelid and repair ptosis. The operation should be personalized to the patient's needs. When considering any procedure, a do-nothing option should be explained to the family and patient. Adolescence can be a challenging time for female patients, and support from a multidisciplinary team of healthcare providers is necessary. Genetic testing also involves different approaches. Females should discuss infertility and family planning with a genetic counselor.
Pearls and Other Issues
BPES should be considered a possible diagnosis in infants who display the distinctive combination of blepharophimosis, ptosis, telecanthus, and epicanthus inversus. The diagnosis can be confirmed through genetic testing for FOXL2 mutations. Prompt surgical surgery for ptosis is essential to prevent the development of amblyopia. Regular ophthalmologic surveillance is essential to assess the progress of visual development and determine whether further surgical interventions are required. Genetic counseling and reproductive alternatives, such as prenatal diagnosis and preimplantation genetic diagnosis, should be considered in families with a hereditary predisposition to BPES.
Enhancing Healthcare Team Outcomes
Management of BPES is complex and requires a multidisciplinary approach, including a pediatric ophthalmologist, oculoplastic surgeon, pediatrician, endocrinologist, gynecologist, nurses, and genetic counselor. Ophthalmologists play a critical role in educating fellow clinicians and healthcare professionals about the risk of amblyopia and its visual implications if unrecognized.
Genetic information is complex, and genetic counselors are essential in educating families and patients on test results. The decision to pursue genetic testing is very personal, particularly for female patients. FOXL2 gene mutation analysis is primarily used to identify future fertility status. Genetic counselors can lead one-on-one counseling to families regarding unexpected test results and discussions regarding reproductive choices. The Clinical Genetics Society recommends postponing genetic testing for disorders with purely reproductive implications until the individual has reached an age or level of maturity where they can fully understand the impact of the test and make an informed decision about whether to proceed with testing.[64]
Endocrinologists and gynecologists are critical in the interprofessional approach to treating these patients. Individuals with type I BPES should be referred to an endocrinologist to address ovarian insufficiency and administer suitable hormonal therapy. Regular interprofessional meetings can facilitate the coordination of care and enhance patient outcomes. Low estrogen levels at a young age increase the risk of osteoporosis, heart disease, and depression. Women with premature ovarian failure cannot get pregnant naturally but can carry a pregnancy with in vitro fertilization with donor oocytes. Patients should be referred to a gynecologist or infertility specialist to discuss reproductive treatments. Ensuring patients understand reproductive biology and therapeutic options can help them cope with this emotionally distressing condition.
Involving patients and their families in decision-making is crucial, particularly when it comes to surgical alternatives and genetic counseling. Providing the family with information about the ailment and treatment alternatives facilitates the ability to make well-informed decisions and improves compliance with treatment regimens.
BPES raises ethical concerns related to fertility preservation and the possibility of doing preimplantation genetic diagnosis in families with identified FOXL2 mutations. These discussions should be approached with sensitivity while respecting the patient's right to make decisions about their health and considering their cultural views.
By providing comprehensive care that addresses the physical and psychological elements of BPES, healthcare professionals can significantly enhance the quality of life for individuals affected by this condition. To uphold a superior level of care, it is advisable to engage in regular training and stay updated on the most recent guidelines and surgical methods.
Media
(Click Image to Enlarge)
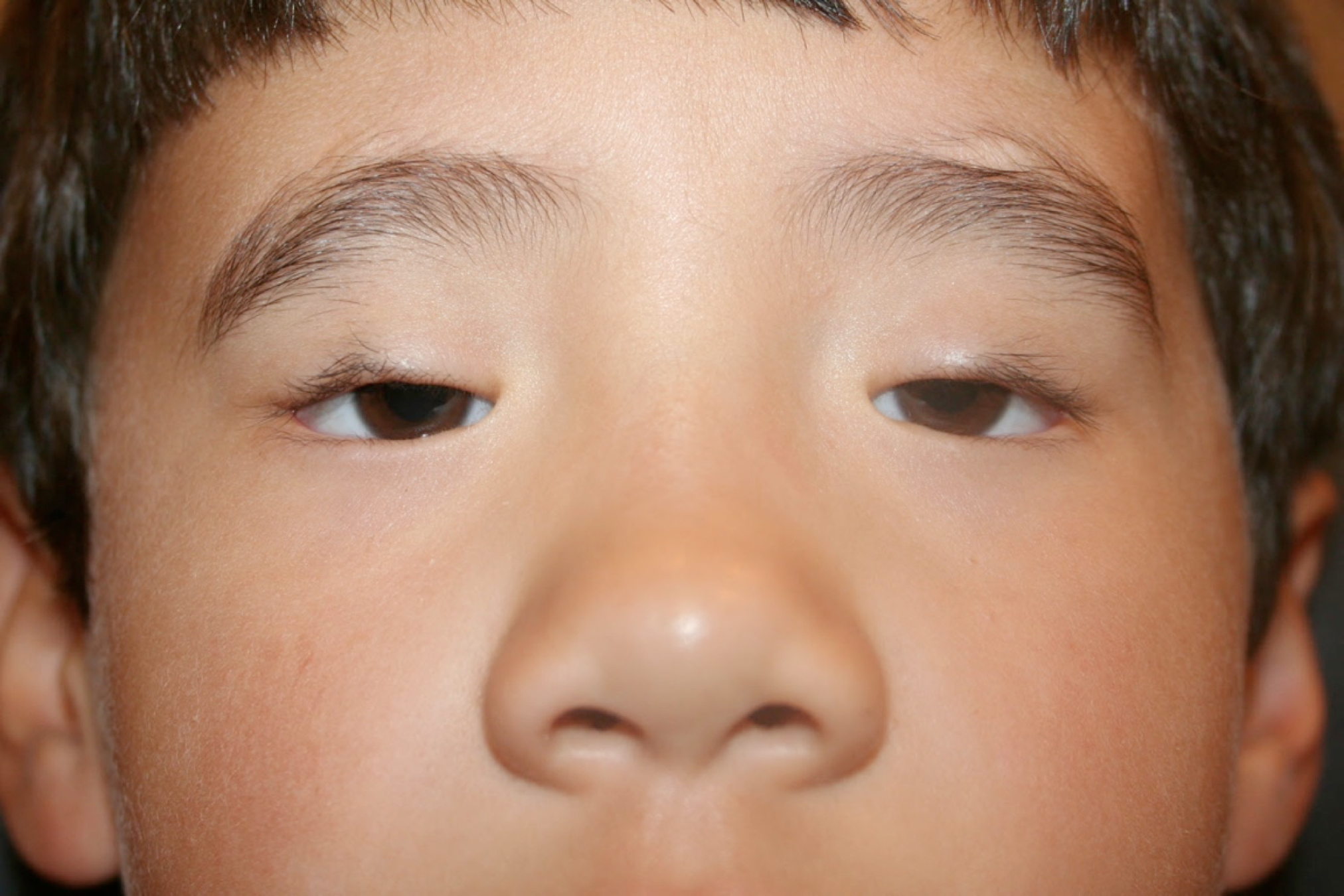
Male Child with Blepharophimosis Syndrome. The image shows the classic findings of blepharophimosis, ptosis, and epicanthus inverus in each eye, and telecanthus.
Contributed by A Harrison, MD
(Click Image to Enlarge)
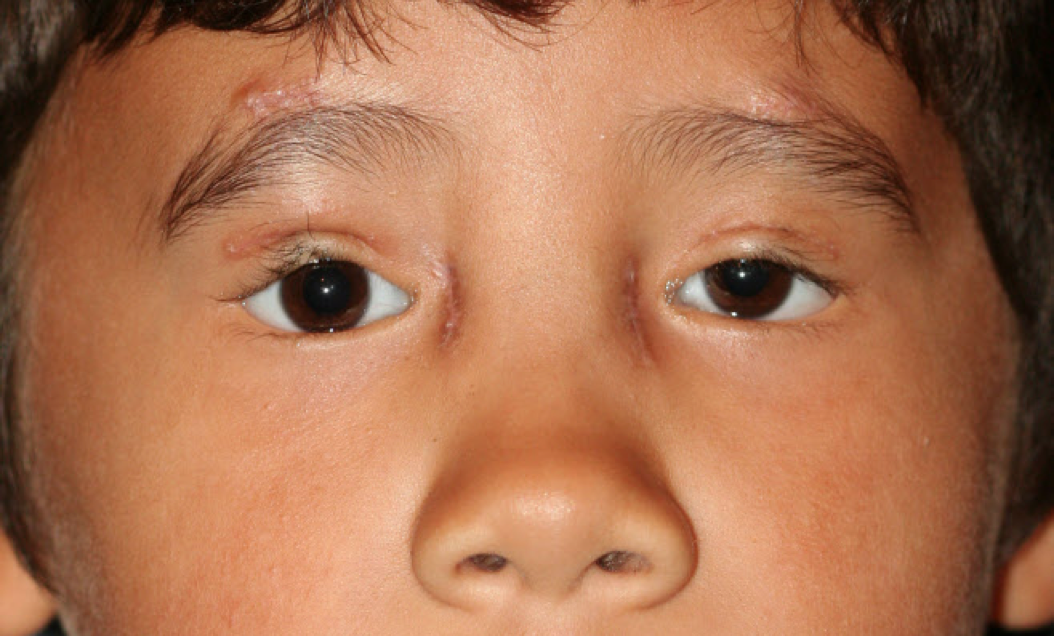
Male Child After Surgery for Blepharophimosis Syndrome. The image shows a male patient 3 weeks after surgical repair of blepharophimosis syndrome. Ptosis repair is performed by frontalis suspension, and telecanthus repair is performed by transnasal wiring.
Contributed by A Harrison, MD
References
Méjécase C, Nigam C, Moosajee M, Bladen JC. The Genetic and Clinical Features of FOXL2-Related Blepharophimosis, Ptosis and Epicanthus Inversus Syndrome. Genes. 2021 Mar 4:12(3):. doi: 10.3390/genes12030364. Epub 2021 Mar 4 [PubMed PMID: 33806295]
Level 3 (low-level) evidenceAllen CE, Rubin PA. Blepharophimosis-ptosis-epicanthus inversus syndrome (BPES): clinical manifestation and treatment. International ophthalmology clinics. 2008 Spring:48(2):15-23. doi: 10.1097/IIO.0b013e3181694eee. Epub [PubMed PMID: 18427257]
Gupta AK, Gupta DC, Khan SA, Razi SM. Blepharophimosis Ptosis Epicanthus Inversus Syndrome (BPES) Type 1 in an Indian Family. Journal of the ASEAN Federation of Endocrine Societies. 2017:32(1):68-71. doi: 10.15605/jafes.032.01.13. Epub 2017 May 9 [PubMed PMID: 33442089]
Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, Verdin H, Matton C, De Baere E. Blepharophimosis, Ptosis, and Epicanthus Inversus Syndrome. GeneReviews(®). 1993:(): [PubMed PMID: 20301614]
Yu Y, Ji M, Xu W, Zhang L, Qi M, Shu J. Confrontment and solution to gonadotropin resistance and low oocyte retrieval in in vitro fertilization for type I BPES: a case series with review of literature. Journal of ovarian research. 2021 Oct 28:14(1):143. doi: 10.1186/s13048-021-00900-2. Epub 2021 Oct 28 [PubMed PMID: 34711234]
Level 2 (mid-level) evidenceWang S, Ge S, Zhuang A. A Novel Forkhead Box L2 Missense Mutation, c.1068G}C, in a Chinese Family With Blepharophimosis/Ptosis/ Epicanthus Inversus Syndrome. The Journal of craniofacial surgery. 2022 May 1:33(3):e238-e240. doi: 10.1097/SCS.0000000000008042. Epub 2021 Aug 9 [PubMed PMID: 34374675]
Crisponi L, Deiana M, Loi A, Chiappe F, Uda M, Amati P, Bisceglia L, Zelante L, Nagaraja R, Porcu S, Ristaldi MS, Marzella R, Rocchi M, Nicolino M, Lienhardt-Roussie A, Nivelon A, Verloes A, Schlessinger D, Gasparini P, Bonneau D, Cao A, Pilia G. The putative forkhead transcription factor FOXL2 is mutated in blepharophimosis/ptosis/epicanthus inversus syndrome. Nature genetics. 2001 Feb:27(2):159-66 [PubMed PMID: 11175783]
Level 3 (low-level) evidenceBeysen D, De Paepe A, De Baere E. FOXL2 mutations and genomic rearrangements in BPES. Human mutation. 2009 Feb:30(2):158-69. doi: 10.1002/humu.20807. Epub [PubMed PMID: 18726931]
Level 3 (low-level) evidenceTucker EJ. The Genetics and Biology of FOXL2. Sexual development : genetics, molecular biology, evolution, endocrinology, embryology, and pathology of sex determination and differentiation. 2022:16(2-3):184-193. doi: 10.1159/000519836. Epub 2021 Nov 2 [PubMed PMID: 34727551]
De Baere E, Beysen D, Oley C, Lorenz B, Cocquet J, De Sutter P, Devriendt K, Dixon M, Fellous M, Fryns JP, Garza A, Jonsrud C, Koivisto PA, Krause A, Leroy BP, Meire F, Plomp A, Van Maldergem L, De Paepe A, Veitia R, Messiaen L. FOXL2 and BPES: mutational hotspots, phenotypic variability, and revision of the genotype-phenotype correlation. American journal of human genetics. 2003 Feb:72(2):478-87 [PubMed PMID: 12529855]
Lin ZB, Chen ZJ, Yang H, Ding XR, Li J, Pan AP, Sun HS, Yu AY, Chen SH. Expanded phenotypic spectrum of FOXL2 Variant c.672_701dup revealed by whole-exome sequencing in a rare blepharophimosis, ptosis, and epicanthus inversus syndrome family. BMC ophthalmology. 2023 Nov 7:23(1):446. doi: 10.1186/s12886-023-03189-5. Epub 2023 Nov 7 [PubMed PMID: 37932670]
Shen Q, Zhao X, Ji Y, Chai P. Deletion of cis-regulatory Element in FOXL2 Promoter in a Chinese Family of Type II Blepharophimosis-ptosis-epicanthus Inversus Syndrome with Polydactyly. The Journal of craniofacial surgery. 2024 Jan-Feb 01:35(1):e52-e56. doi: 10.1097/SCS.0000000000009801. Epub 2023 Nov 8 [PubMed PMID: 37938073]
Fuller PJ, Nguyen T, Alexiadis M, Chu S. FOXL2(C134W) : much ado about something!(†). The Journal of pathology. 2022 Jan:256(1):1-3. doi: 10.1002/path.5816. Epub 2021 Nov 18 [PubMed PMID: 34687235]
França MM, Mendonca BB. Genetics of ovarian insufficiency and defects of folliculogenesis. Best practice & research. Clinical endocrinology & metabolism. 2022 Jan:36(1):101594. doi: 10.1016/j.beem.2021.101594. Epub 2021 Oct 14 [PubMed PMID: 34794894]
Chawla B, Bhadange Y, Dada R, Kumar M, Sharma S, Bajaj MS, Pushker N, Chandra M, Ghose S. Clinical, radiologic, and genetic features in blepharophimosis, ptosis, and epicanthus inversus syndrome in the Indian population. Investigative ophthalmology & visual science. 2013 Apr 26:54(4):2985-91. doi: 10.1167/iovs.13-11794. Epub 2013 Apr 26 [PubMed PMID: 23513057]
Level 2 (mid-level) evidenceKrastinova D, Jasinski MA. Orbitoblepharophimosis syndrome: a 16-year perspective. Plastic and reconstructive surgery. 2003 Mar:111(3):987-99 [PubMed PMID: 12621168]
Level 3 (low-level) evidenceBeckingsale PS, Sullivan TJ, Wong VA, Oley C. Blepharophimosis: a recommendation for early surgery in patients with severe ptosis. Clinical & experimental ophthalmology. 2003 Apr:31(2):138-42 [PubMed PMID: 12648048]
Level 2 (mid-level) evidenceDawson EL, Hardy TG, Collin JR, Lee JP. The incidence of strabismus and refractive error in patients with blepharophimosis, ptosis and epicanthus inversus syndrome (BPES). Strabismus. 2003 Sep:11(3):173-7 [PubMed PMID: 14710475]
Level 2 (mid-level) evidenceLiu CY. Wakayama Symposium: Notch-FoxL2-α-SMA axis in eyelid levator muscle development and congenital blepharophimosis. The ocular surface. 2012 Oct:10(4):221-3. doi: 10.1016/j.jtos.2012.07.003. Epub 2012 Jul 25 [PubMed PMID: 23084143]
Heude É, Bellessort B, Fontaine A, Hamazaki M, Treier AC, Treier M, Levi G, Narboux-Nême N. Etiology of craniofacial malformations in mouse models of blepharophimosis, ptosis and epicanthus inversus syndrome. Human molecular genetics. 2015 Mar 15:24(6):1670-81. doi: 10.1093/hmg/ddu579. Epub 2014 Nov 21 [PubMed PMID: 25416281]
Level 3 (low-level) evidenceHarris SE, Chand AL, Winship IM, Gersak K, Aittomäki K, Shelling AN. Identification of novel mutations in FOXL2 associated with premature ovarian failure. Molecular human reproduction. 2002 Aug:8(8):729-33 [PubMed PMID: 12149404]
Yang XW, He WB, Gong F, Li W, Li XR, Zhong CG, Lu GX, Lin G, Du J, Tan YQ. Novel FOXL2 mutations cause blepharophimosis-ptosis-epicanthus inversus syndrome with premature ovarian insufficiency. Molecular genetics & genomic medicine. 2018 Mar:6(2):261-267. doi: 10.1002/mgg3.366. Epub 2018 Jan 29 [PubMed PMID: 29378385]
Xue M, Zheng J, Zhou Q, Hejtmancik JF, Wang Y, Li S. Novel FOXL2 mutations in two Chinese families with blepharophimosis-ptosis-epicanthus inversus syndrome. BMC medical genetics. 2015 Sep 1:16():73. doi: 10.1186/s12881-015-0217-7. Epub 2015 Sep 1 [PubMed PMID: 26323275]
Wen F, Ding Y, Wang M, Du J, Zhang S, Kee K. FOXL2 and NR5A1 induce human fibroblasts into steroidogenic ovarian granulosa-like cells. Cell proliferation. 2024 May:57(5):e13589. doi: 10.1111/cpr.13589. Epub 2024 Jan 8 [PubMed PMID: 38192172]
JOHNSON CC. SURGICAL REPAIR OF THE SYNDROME OF EPICANTHUS INVERSUS, BLEPHAROPHIMOSIS AND PTOSIS. Archives of ophthalmology (Chicago, Ill. : 1960). 1964 Apr:71():510-6 [PubMed PMID: 14109036]
Beaconsfield M, Walker JW, Collin JR. Visual development in the blepharophimosis syndrome. The British journal of ophthalmology. 1991 Dec:75(12):746-8 [PubMed PMID: 1768667]
KLEIN M. Hereditary bilateral ptosis and blepharophimosis associated with other developmental abnormalities of the outer eye. Proceedings of the Royal Society of Medicine. 1950 Dec:43(12):1025-6 [PubMed PMID: 14808191]
Kohn R, Romano PE. Blepharoptosis, blepharophimosis, epicanthus inversus, and telecanthus--a syndrome with no name. American journal of ophthalmology. 1971 Sep:72(3):625-32 [PubMed PMID: 5568616]
MUSTARDE JC. EPICANTHUS AND TELECANTHUS. British journal of plastic surgery. 1963 Oct:16():346-56 [PubMed PMID: 14077771]
JOHNSON CC. Operations for epicanthus and blepharophimosis; an evaluation and a method for shortening the medial canthal ligament. American journal of ophthalmology. 1956 Jan:41(1):71-9 [PubMed PMID: 13275546]
Lewis SR, Arons MS, Lynch JB, Blocker TG Jr. The congenital eyelid syndrome. Plastic and reconstructive surgery. 1967 Mar:39(3):271-7 [PubMed PMID: 5336149]
MUSTARDE JC. EPICANTHUS AND TELECANTHUS. International ophthalmology clinics. 1964 Jun:4():359-76 [PubMed PMID: 14199224]
Garden JW. Blepharophimosis, ptosis, epicanthus inversus and lacrimal stenosis. American journal of ophthalmology. 1969 Jan:67(1):153-4 [PubMed PMID: 5782860]
Choi KH, Kyung S, Oh SY. The factors influencing visual development in blepharophimosis-ptosis-epicanthus inversus syndrome. Journal of pediatric ophthalmology and strabismus. 2006 Sep-Oct:43(5):285-8. doi: 10.3928/01913913-20060901-03. Epub [PubMed PMID: 17022162]
Level 2 (mid-level) evidenceLandau Prat D, Nguyen BJ, Strong A, Katowitz WR, Katowitz JA. "Blepharophimosis-plus" syndromes: Frequency of systemic genetic disorders that also include blepharophimosis. Clinical & experimental ophthalmology. 2021 Jul:49(5):448-453. doi: 10.1111/ceo.13933. Epub 2021 Apr 29 [PubMed PMID: 33882191]
Bertini V, Valetto A, Baldinotti F, Azzarà A, Cambi F, Toschi B, Giacomina A, Gatti GL, Gana S, Caligo MA, Bertelloni S. Blepharophimosis, Ptosis, Epicanthus Inversus Syndrome: New Report with a 197-kb Deletion Upstream of FOXL2 and Review of the Literature. Molecular syndromology. 2019 May:10(3):147-153. doi: 10.1159/000497092. Epub 2019 Mar 20 [PubMed PMID: 31191203]
Zhuang J, Zeng S, Wang Y, Jiang Y. [Clinical and genetic analysis of a patient with 10q26.3 microdeletion in conjunct with 18q22.3q23 microduplication]. Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical genetics. 2022 Dec 10:39(12):1415-1418. doi: 10.3760/cma.j.cn511374-20211126-00943. Epub [PubMed PMID: 36453971]
Chen M, Jiang H, Zhang C. Selected Genetic Factors Associated with Primary Ovarian Insufficiency. International journal of molecular sciences. 2023 Feb 23:24(5):. doi: 10.3390/ijms24054423. Epub 2023 Feb 23 [PubMed PMID: 36901862]
Zeng L, Zhang Y, Lin L, Dong X, Li L. [Genetic testing and prenatal diagnosis for a family with 10q22.3q23.2 microdeletion]. Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical genetics. 2021 Aug 10:38(8):768-770. doi: 10.3760/cma.j.cn511374-20200608-00419. Epub [PubMed PMID: 34365621]
Yang Y, Yang C, Zhu Y, Chen H, Zhao R, He X, Tao L, Wang P, Zhou L, Zhao L, Tu M, Dong Z, Chen H, Xie Z. Intragenic and extragenic disruptions of FOXL2 mapped by whole genome low-coverage sequencing in two BPES families with chromosome reciprocal translocation. Genomics. 2014 Sep:104(3):170-6. doi: 10.1016/j.ygeno.2014.07.010. Epub 2014 Jul 30 [PubMed PMID: 25086333]
Huhtaniemi I, Hovatta O, La Marca A, Livera G, Monniaux D, Persani L, Heddar A, Jarzabek K, Laisk-Podar T, Salumets A, Tapanainen JS, Veitia RA, Visser JA, Wieacker P, Wolczynski S, Misrahi M. Advances in the Molecular Pathophysiology, Genetics, and Treatment of Primary Ovarian Insufficiency. Trends in endocrinology and metabolism: TEM. 2018 Jun:29(6):400-419. doi: 10.1016/j.tem.2018.03.010. Epub 2018 Apr 26 [PubMed PMID: 29706485]
Level 3 (low-level) evidenceKaracaoğlan N, Sahin U, Ercan U, Bozdogan N. One-stage repair of blepharophimosis: a new method. Plastic and reconstructive surgery. 1994 Jun:93(7):1406-9 [PubMed PMID: 8208806]
Level 3 (low-level) evidenceBhattacharjee K, Bhattacharjee H, Kuri G, Shah ZT, Deori N. Single stage surgery for Blepharophimosis syndrome. Indian journal of ophthalmology. 2012 May-Jun:60(3):195-201. doi: 10.4103/0301-4738.95870. Epub [PubMed PMID: 22569380]
Level 2 (mid-level) evidenceLiu H, Shao Y, Zhao Z, Zhang D. One-stage correction of blepharophimosis-ptosis-epicanthus inversus syndrome using a frontalis muscle transfer technique. Journal of plastic surgery and hand surgery. 2014 Feb:48(1):74-9. doi: 10.3109/2000656X.2013.819004. Epub 2013 Aug 23 [PubMed PMID: 23968369]
Level 2 (mid-level) evidenceWu SY, Ma L, Tsai YJ, Kuo JZ. One-stage correction for blepharophimosis syndrome. Eye (London, England). 2008 Mar:22(3):380-8 [PubMed PMID: 17115018]
Level 2 (mid-level) evidenceMastellari E, La Marca A. Genetic conditions impairing female fertility. Panminerva medica. 2020 Dec:62(4):260-267. doi: 10.23736/S0031-0808.20.04208-1. Epub 2020 Nov 13 [PubMed PMID: 33185415]
Bonus ML, Pothast R, Lamb JD, Feinberg EC, Bernardi LA. Planned oocyte cryopreservation in women with blepharophimosis-ptosis-epicanthus inversus syndrome: a case series. F&S reports. 2021 Sep:2(3):332-337. doi: 10.1016/j.xfre.2021.05.006. Epub 2021 May 27 [PubMed PMID: 34553160]
Level 2 (mid-level) evidenceMeng T, Zhang W, Zhang R, Li J, Gao Y, Qin Y, Jiao X. Ovarian Reserve and ART Outcomes in Blepharophimosis-Ptosis-Epicanthus Inversus Syndrome Patients With FOXL2 Mutations. Frontiers in endocrinology. 2022:13():829153. doi: 10.3389/fendo.2022.829153. Epub 2022 Apr 28 [PubMed PMID: 35574016]
Level 2 (mid-level) evidenceBaber RJ, Panay N, Fenton A, IMS Writing Group. 2016 IMS Recommendations on women's midlife health and menopause hormone therapy. Climacteric : the journal of the International Menopause Society. 2016 Apr:19(2):109-50. doi: 10.3109/13697137.2015.1129166. Epub 2016 Feb 12 [PubMed PMID: 26872610]
Nelson LM. Clinical practice. Primary ovarian insufficiency. The New England journal of medicine. 2009 Feb 5:360(6):606-14. doi: 10.1056/NEJMcp0808697. Epub [PubMed PMID: 19196677]
Rong WN, Ma MJ, Yang W, Yuan SQ, Sheng XL. Identification of a novel FOXL2 mutation in a fourth-generation Chinese family with blepharophimosis-ptosis-epicanthus inversus syndrome. International journal of ophthalmology. 2021:14(4):504-509. doi: 10.18240/ijo.2021.04.04. Epub 2021 Apr 18 [PubMed PMID: 33875939]
Cheng T, Yuan X, Yuan S, Zhu J, Tang S, Zhang Y. ITGB5 mutation discovered in a Chinese family with blepharophimosis-ptosis-epicanthus inversus syndrome. Open life sciences. 2021:16(1):1268-1277. doi: 10.1515/biol-2021-0129. Epub 2021 Dec 10 [PubMed PMID: 34966851]
Bashamboo A, Eozenou C, Jorgensen A, Bignon-Topalovic J, Siffroi JP, Hyon C, Tar A, Nagy P, Sólyom J, Halász Z, Paye-Jaouen A, Lambert S, Rodriguez-Buritica D, Bertalan R, Martinerie L, Rajpert-De Meyts E, Achermann JC, McElreavey K. Loss of Function of the Nuclear Receptor NR2F2, Encoding COUP-TF2, Causes Testis Development and Cardiac Defects in 46,XX Children. American journal of human genetics. 2018 Mar 1:102(3):487-493. doi: 10.1016/j.ajhg.2018.01.021. Epub 2018 Feb 22 [PubMed PMID: 29478779]
Clayton-Smith J, O'Sullivan J, Daly S, Bhaskar S, Day R, Anderson B, Voss AK, Thomas T, Biesecker LG, Smith P, Fryer A, Chandler KE, Kerr B, Tassabehji M, Lynch SA, Krajewska-Walasek M, McKee S, Smith J, Sweeney E, Mansour S, Mohammed S, Donnai D, Black G. Whole-exome-sequencing identifies mutations in histone acetyltransferase gene KAT6B in individuals with the Say-Barber-Biesecker variant of Ohdo syndrome. American journal of human genetics. 2011 Nov 11:89(5):675-81. doi: 10.1016/j.ajhg.2011.10.008. Epub [PubMed PMID: 22077973]
Level 3 (low-level) evidenceOley C, Baraitser M. Blepharophimosis, ptosis, epicanthus inversus syndrome (BPES syndrome). Journal of medical genetics. 1988 Jan:25(1):47-51 [PubMed PMID: 3270326]
Shukla UV, Patel BC. Congenital Ptosis. StatPearls. 2024 Jan:(): [PubMed PMID: 33760447]
Ahmed jan N, Mui RK, Masood S. Waardenburg Syndrome. StatPearls. 2024 Jan:(): [PubMed PMID: 32809714]
Saint-Laurent C, Mazeyrie L, Yart A, Edouard T. Novel therapeutic perspectives in Noonan syndrome and RASopathies. European journal of pediatrics. 2024 Mar:183(3):1011-1019. doi: 10.1007/s00431-023-05263-y. Epub 2023 Oct 21 [PubMed PMID: 37863846]
Level 3 (low-level) evidenceSingh N, Verma P, Bains R, Mutalikdesai J. Apert syndrome: craniofacial challenges and clinical implications. BMJ case reports. 2024 Jul 16:17(7):. pii: e260724. doi: 10.1136/bcr-2024-260724. Epub 2024 Jul 16 [PubMed PMID: 39013624]
Level 3 (low-level) evidenceNuovo S, Passeri M, Di Benedetto E, Calanchini M, Meldolesi I, Di Giacomo MC, Petruzzi D, Piemontese MR, Zelante L, Sangiuolo F, Novelli G, Fabbri A, Brancati F. Characterization of endocrine features and genotype-phenotypes correlations in blepharophimosis-ptosis-epicanthus inversus syndrome type 1. Journal of endocrinological investigation. 2016 Feb:39(2):227-33. doi: 10.1007/s40618-015-0334-3. Epub 2015 Jun 23 [PubMed PMID: 26100530]
Amer AA, Abdellah MM, Hassan NHF, Mounir A. Surgical outcome of epicanthus and telecanthus correction by C-U medial canthoplasty with lateral canthoplasty in treatment of Blepharophimosis syndrome. BMC ophthalmology. 2022 May 19:22(1):226. doi: 10.1186/s12886-022-02455-2. Epub 2022 May 19 [PubMed PMID: 35590300]
Sa HS, Lee JH, Woo KI, Kim YD. A new method of medial epicanthoplasty for patients with blepharophimosis-ptosis-epicanthus inversus syndrome. Ophthalmology. 2012 Nov:119(11):2402-7. doi: 10.1016/j.ophtha.2012.05.037. Epub 2012 Jul 24 [PubMed PMID: 22835816]
Level 3 (low-level) evidenceSavino G, Mandarà E, Calandriello L, Dickmann A, Petroni S. A Modified One-Stage Early Correction of Blepharophimosis Syndrome Using Tutopatch Slings. Orbit (Amsterdam, Netherlands). 2015:34(4):186-91. doi: 10.3109/01676830.2015.1015146. Epub 2015 Jun 4 [PubMed PMID: 26043072]
Fokstuen S, Antonarakis SE, Blouin JL. FOXL2-mutations in blepharophimosis-ptosis-epicanthus inversus syndrome (BPES); challenges for genetic counseling in female patients. American journal of medical genetics. Part A. 2003 Mar 1:117A(2):143-6 [PubMed PMID: 12567411]
Level 3 (low-level) evidence