 Laboratory Evaluation of Hereditary Hemochromatosis
Laboratory Evaluation of Hereditary Hemochromatosis
Introduction
Hemochromatosis, a syndrome caused by excessive iron absorption, was first described in the mid-1800s as a syndrome of "bronze diabetes and pigmentary cirrhosis." It is believed that the term hemochromatosis was coined by von Recklinghausen in 1889. A genetic origin of this metabolic problem was first suggested in the 1930s. In 1977, Simon et al reported the association between an HLA class I-like molecule and the presumed hemochromatosis gene on chromosome 6p, establishing the genetic basis of what is now referred to as hereditary hemochromatosis (HH).[1]
Although one of the most common genetic disorders in the United States, affecting over 1 million people, hereditary hemochromatosis (HH) is frequently an incidental discovery during routine laboratory iron measurements or the diagnostic workup of other conditions. However, increasing awareness of HH has also contributed to early detection. Early diagnosis allows for intervention before tissue damage occurs due to excessive iron deposition. Iron can deposit in the liver, pancreas, heart, joints, and other endocrine organs if left untreated.
Current classifications for HH identify 4 classes or types, with 5individual molecular subtypes, based on the age of onset, underlying genetic mutation, and mode of inheritance.[2]
Type I HH is considered the classic form of the disorder, and the onset of symptoms begins in adulthood. Loss-of-function mutations in the HFE (Hereditary Fe [iron]) gene are present in approximately 70% of patients diagnosed with HH. This form of HH disproportionately affects males, although females can also be affected.
Type 2 HH is frequently called "juvenile" hemochromatosis, as symptoms begin in childhood, and the disease is typically more clinically severe. Unlike Type I HH, there is no preference for a particular sex. Type 2 HH has 2 subtypes, 2a and 2b. Type 2a HH is caused by mutations in the gene initially known as hemojuvelin (HJV) and is now referred to as HFE2. Type 2b HH is due to hepcidin antimicrobial peptide (HAMP) gene mutations.
Type 3 HH has a typical onset at approximately 30 years of age and is caused by mutations in the transferring-receptor 2 (TFR2) gene.
Type 4 HH, also known as ferroportin disease, is caused by mutations in the SLC4OA1 gene. Ferroportin is an iron transmembrane transport protein, and Type 4 HH is the only known form of hemochromatosis that can be inherited in an autosomal dominant fashion. The onset of type 4 HH typically occurs in mid-adulthood.
Etiology and Epidemiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology and Epidemiology
Epidemiology
In the United States, about 1 in 300 non-Hispanic White people has HH, with lower rates among people of other races and ethnicities. Approximately 9% to 10% of males with HH will develop liver disease.[Centers for Disease Control, Hereditary Hemochromatosis] Types 1, 2, and 3 HH are transmitted via an autosomal recessive fashion; Type 4 HH is predominately an autosomal dominant disease.
HH is the most common inherited disorder among people of northern European ancestry. The United States, Europe, and Australia have a similar disease prevalence of one case per 200 to 400 people, with the highest prevalence in those of Irish and Scandinavian ancestry. The HH mutations are rare in people of Asian, African, Hispanic, and Pacific Islander ancestry. Despite the high prevalence of the gene mutation, there is a low and variable clinical penetrance. Up to 25% of people with C282Y homozygosity are clinically asymptomatic.[3][4]
Etiology
Over 100 known mutations in the HFE gene can cause Type 1 HH. The HFE protein regulates the production of hepcidin, a protein produced by the liver that regulates iron homeostasis. The most common genetic mutation in Type 1 HH is the p.Cys282Tyr or C282Y variant, which occurs from a single nucleotide change (845 G-A), substituting cysteine with tyrosine at amino acid 282. This mutation prevents the altered HFE protein from inserting into the cell's surface membrane. The second most common mutation in Type 1 HH replaces the amino acid histidine with the amino acid aspartic acid at position 63 (written as p.His63Asp or H63D).
Patients with Type 1 HH have two mutated copies of the HFE gene. Homozygous C282Y and heterozygous C282Y/H63D mutations of the HFE gene on chromosome 6 are responsible for up to 95% of Type 1 HH cases.[3]
Interestingly, the fact that the C282Y homozygosity was mainly detected in patients of Northern European ancestry and is not seen in other regions of the world, including Asia and Africa, led to the recognition of a heterogenous genetic basis of HH and the discovery of other variants involving the non-HFE genes, particularly the HAMP and HJV genes associated with severe early-onset juvenile forms of HH.[1]
While the C282Y variant was initially described as the causative mutation of HH, several other non-HFE genes, including variants of the hepcidin (HAMP), hemojuvelin (HJV), ferroportin/solute carrier family 40 member 1 (SLC40A1), and second receptor for transferrin (TFR2) genes have been identified in other types of HH.[5][6][7]
Pathophysiology
The clinical symptoms of HH occur due to genetic defects in the proteins of iron metabolism, resulting in iron overload and deposition in the liver, pancreas, myocardium, joints, and multiple endocrine organs, including the pituitary, thyroid, parathyroid, and adrenal glands. Humans are unable to actively excrete iron; regulation of intestinal iron absorption is the primary method of maintaining iron homeostasis. The total body iron pool in healthy adults ranges from 3 to 4 gm; approximately 0.5 grams is stored in the liver.[8] In severe untreated HH, total body iron stores may be more than 50 grams, one-third of which is stored in hepatocytes. Excessive iron appears directly toxic to tissues, although cells that are not irreversibly injured will recover function as the iron is removed. The characteristic findings of severe, untreated HH are micronodular cirrhosis, diabetes, and abnormal skin pigmentation.
Normal iron homeostasis is a complex mechanism that is incompletely understood. Humans must absorb iron from their diet; iron is used in multiple physiological processes, including DNA biosynthesis, oxygen transport, and cellular energy generation.[8] Hepcidin, the protein product of HAMP, is a circulating peptide hormone that is a negative regulator of iron absorption by enterocytes. When circulating levels of hepcidin are high, iron absorption by the gut is reduced. Ferroportin is an iron transmembrane transport protein encoded by SLC4OA1 that transports iron from the enterocyte into the vasculature. Ferroportin also transports iron out of reticuloendothelial cells. The proteolytic degradation of ferroportin is facilitated by hepcidin.
The HFE, HJV, and TFR2 gene products are hepatocyte membrane proteins that sense iron. In the presence of normal or excessive iron levels, these gene products activate hepcidin production, reducing enterocyte absorption and promoting the degradation of ferroportin.[3][8]
Mutations in HFE, HFE2, HAMP, HJV, and TFR2 significantly impair and reduce hepcidin synthesis, ultimately increasing intestinal iron absorption and iron deposition in tissues. The gain-of-function mutation of SLC40A1 impairs hepcidin-ferroportin binding. HFE-related mutations include C282Y, H63D, and S65C; together, these account for over 90% of cases of HH, most of which are due to C282Y homozygosity.[9][10][11]
The liver is most severely affected by untreated HH. Whatever the underlying genetic defect, excess iron deposition within hepatocytes results in lipid peroxidation via iron-catalyzed free radical reactions, stimulation of collagen formation by stellate cells, and DNA damage by reactive oxygen species. Ferroptosis is a recently discovered form of nonapoptotic cell death mediated by ROS and lipid peroxidation induction.[8] The risk of hepatocellular carcinoma in patients with HH is increased 200-fold.
Iron uptake into pancreatic beta-cells leads to impaired insulin synthesis and release, and liver fibrosis leads to high levels of circulating insulin and insulin resistance.[12] Iron deposition in the pancreas can result in hyperglycemia; HH should be suspected if there is skin hyperpigmentation, joint pain, hypogonadism, or features of liver disease, in addition to persistent hyperglycemia.[13] The pathogenesis of diabetes in HH is considered multifactorial; both insulin deficiency and resistance may contribute. Iron overload can impair insulin secretion and glucose tolerance early in HH before cirrhosis occurs.[14]
The myocardium is also affected by iron deposition. Iron deposition in HH occurs initially within the subepicardial cardiac myocytes; as the disease progresses, deposition occurs throughout the myocardium. The resulting myocardial hypertrophy results in diastolic dysfunction. If iron deposition continues, cardiomyopathy, systolic dysfunction, and arrhythmias ensue.
Deposition of iron in the cells of the anterior pituitary gland may result in reduced production of luteinizing hormone and follicle-stimulating hormone.[15] In non-HFE forms of HH, iron deposition starts early, is more severe, and deposits in the pituitary, especially the gonadotropes, although other lineages are affected.[16]
Joint pain affects up to 75% of patients with HH even before the diagnosis is made. Overt clinical symptoms related to joint involvement usually appear in the fifth decade of life but may appear as early as the third decade.[17] Hemochromatosis-associated pseudogout (chondrocalcinosis) is not common, but in those with severe chondrocalcinosis, the frequency of association may justify screening for hemochromatosis, especially in younger male patients.[18] Inhibition of the pyrophosphatases and synovial iron sequestration are likely mechanisms causing damage to the articular cartilage.[19]
Specimen Requirements and Procedure
Total serum iron and total iron-binding capacity (TIBC) assays can be used in conjunction with ferritin assays to aid in diagnosing iron deficiency or overload. No significant differences in apparent concentration have been reported between serum and plasma samples for total iron determination.
An early-morning fasting sample is likely optimal for analysis because normal diurnal variation may decrease the iron concentration by 30% during the day.[20] Peak iron concentrations occur in the early- to late-morning hours. The specimen should be centrifuged as soon as possible after collection.[21] Storage of whole blood without separation of serum or plasma renders specimens unsuitable for analysis. Serum iron is stable for up to 1 week at 4 to 8 °C and indefinitely when frozen at -20°C.[22] Hemolyzed samples must be rejected since erythrocytes contain iron; hemolysis falsely increases apparent iron concentration. Response to hemolysis is both heterogeneous and unpredictable, and therefore no reliable corrective measures can be made.[23]
Serum is the recommended sample type for a ferritin assay. No patient preparation is required. Blood should be drawn in plain red-top tubes or serum gel tubes.[24] Specimens are stable for at least 1 week when refrigerated at 4 °C and 6 months at -20 °C. Frozen specimens should not be thawed at 37 °C, and repeated freezing and thawing should be avoided, as well as violent mixing, which may denature ferritin.[22]
Diagnostic Tests
The serum ferritin concentration and serum transferrin saturation (TSAT) are increased in patients with HH. Further testing should be considered if the ferritin levels exceed 200 µg/L in menstruating females or 300 µg/L in males or nonmenstruating females, or if the TSAT is >45%.[1] TIBC values >450 mcg/dL (80.55 mmol/L) can also help diagnose pathological iron accumulation.[25] The negative predictive value for iron overload is 97% when serum ferritin is normal and TSAT <45%.[26]
Ferritin is an acute phase reactant often elevated in periods of inflammation or cases of malignancy; therefore, hyperferritinemia is a nonspecific finding that must be interpreted within the clinical context.[24] A persistently increased TSAT level >45% is a more reliable indicator of HH. Other findings that elicit suspicion of HH include imaging evidence of iron overload in the liver on MRI or iron deposits in hepatocytes on a liver biopsy.[27]
The absence of acquired risk factors for hepcidin deficiency, like alcohol misuse or end-stage liver disease, favors the possibility of HH.[28] Secondary causes of iron overload must be excluded; these include iatrogenic iron overload in the setting of regular blood transfusions, reduced hepcidin production due to hepatic dysfunction, chronic renal failure, adult-onset Still disease, hemophagocytic lymphohistiocytosis, hereditary hyperferritinemia, and disorders of ineffective erythropoiesis such as the thalassemias, other hemolytic anemias, and myelodysplastic syndromes.[29]
Genetic Testing
If serum ferritin levels are persistently elevated and the TSAT is >45%, the next diagnostic step is genetic testing; HFE genotyping is recommended.[30][31] Homozygous C282Y and compound heterozygous C282Y/H63D mutations of the HFE gene are responsible for up to 95% of all HH cases; Types 2 through 4 HH comprise the small percentage of remaining cases and are caused by non-HFE mutations.[4] The incidence of C282Y or H63D compound heterozygosity is less than 5%.[32]
Since mutations in HJV, HAMP, TFR2, or the ferroportin gene are far less common, genetic testing for non-HFE hemochromatosis may be considered in cases where other causes of hyperferritinemia have been ruled out, there is a family history of iron overload or hepatic iron overload can be demonstrated on MRI or liver biopsy. HJV, HAMP, and TFR2 mutations are usually seen in early-onset disease with severe clinical manifestations and are recessive, while the ferroportin mutations are dominant.[33][34][35]
Interestingly, while C282Y homozygosity is strongly associated with an increased risk of cirrhosis, there is a significant variance in phenotype due to incomplete penetrance. However, a significant positive statistical association exists between abdominal pain and cirrhosis in hemochromatosis probands with this homozygous mutation.[4][36]
Testing for Liver Involvement
Liver biopsy with histochemical, semi-quantitative assessment, or chemical determination of iron content was previously considered the gold standard of diagnosis in HH. However, in current clinical practice, the diagnosis is made via genetic testing for specific mutations with the presence of increased serum ferritin and TSAT.[37] A diagnostic liver biopsy is indicated when the diagnosis is unclear, as in certain types of non-HFE hemochromatosis, dysmetabolic iron overload syndrome, nonalcoholic fatty liver disease (NAFLD), and some types of alcoholic liver disease presenting with elevated ferritin and moderate iron overload. A liver biopsy can also assess the degree of fibrosis and detect hepatocellular carcinoma.[38]
Histopathological evaluation of the liver in HH reveals characteristic intracytoplasmic deposits of golden-yellow hemosiderin granules within hepatocytes. These deposits are initially located in the periportal region (see Image. Micrograph of Hemochromatosis) but eventually are found throughout the lobule, including within Kupffer cells and biliary epithelium. These hemosiderin granules will appear blue with the Prussian blue stain (Image 3 - Hemochromatosis; Prussian Blue Stain). Parenchymal architecture is preserved in early-stage HH; the onset of cirrhosis distorts normal architecture.
Noninvasive evaluation techniques such as magnetic resonance imaging (MRI) are now preferred to invasive liver biopsy when quantifying hepatic iron deposition.[39][40] MRI for noninvasive hepatic iron quantification also improves the diagnostic yield of next-generation sequencing (NGS) in patients with hyperferritinemia.[41]
Other noninvasive techniques for detecting liver fibrosis in patients with HH include the aspartate aminotransferase-to-platelet ratio index (APRI), fibrosis-4 Index (Fib4), Hepascore, and hepatic transient elastography (FibroScan).[42][43][44] The APRI cut-off value >0.44 and Fib4 cut-off value >1.1 have shown good diagnostic utility in correctly identifying liver biopsy-diagnosed advanced hepatic fibrosis in 85% and 80% of cases, respectively.[45] Hepascore incorporates the clinical variables of sex and age with blood-based markers, including bilirubin, gamma-glutamyl transferase (GGT), hyaluronic acid, and alpha2-macroglobulin, to detect hepatic fibrosis. Transient elastography (FibroScan) utilizes ultrasound to assess the degree of hepatic fibrosis and can be performed in an outpatient setting. Hepascore and TE are likely most reliable when serum ferritin levels are >1000 μg/L.[46]
Testing for Cardiac Involvement
It has been suggested that serial assessment of serum non–transferrin-bound iron levels might guide the initiation of treatment to avoid cardiac and other organ damage. However, standardization of this protocol has proven difficult.[47]
Electrocardiographic changes in HH are usually nonspecific and recognized late in the course of the disease. However, standard echocardiography can reveal cardiac abnormalities even in the early stages of HH. Two-dimensional speckle tracking echocardiography (2D STE) and 3-dimensional (3D) real-time echocardiography are novel techniques that seem more precise and distinctive in detecting the subtle cardiac abnormalities of early-stage HH.[25]
Cardiac MRI has indisputable value when diagnosing the cardiac abnormalities caused by HH. A reduction of T2 relaxation <20 ms is considered a reliable indicator of iron overload. However, the implementation of cardiac MRI in everyday practice is limited by the cost and availability of this diagnostic method.[48][49] Cardiac biopsy is rarely performed in the diagnosis and evaluation of HH. A cardiac biopsy may be indicated in cases where the diagnosis is unclear, and other causes of heart failure or infiltrative diseases need to be ruled out.[25]
Testing for Pancreatic Involvement
Pancreatic iron deposition can result in hyperglycemia, and HH is associated with an increased risk of developing type 2 diabetes. Fasting serum glucose levels, glucose tolerance tests, or hemoglobin A1c concentrations can be used to screen for and confirm a diagnosis of type 2 diabetes. Less commonly, HH may result in chronic pancreatitis; MRI can effectively assess iron deposition in the pancreas.[50]
Testing Hormone Levels to Evaluate Hypogonadism
Hypogonadism is the second most common endocrine abnormality in HH, with a frequency ranging from 10% to 100%.[51] Assessment of serum FSH, LH, testosterone, and occasionally gonadotropin-releasing hormone (GnRH) is advised for coexisting infertility or reproductive system disorders. Brain MRI can detect pituitary iron deposits.[52] There is evidence to suggest that screening for common endocrinopathies in patients with HH and evidence of pituitary iron deposition is beneficial, even in the absence of symptoms.[53] Additionally, if the serum ferritin is ≥300 μg/L, screening is beneficial regardless of clinical manifestations.[54] In premenopausal females, hypogonadism is evidenced by secondary amenorrhea and decreased FSH and LH serum levels.[55]
Testing for Musculoskeletal Involvement
Joint pain precedes a HH diagnosis in up to 75% of patients. Plain radiography of the affected joints can be used initially to know the extent of arthritis. A rheumatological scoring system, based on x-rays of joints, has been validated and used in hemochromatosis patients with arthritis. The presence of osteopenia and osteoporosis can be analyzed by dual-energy x-ray absorptiometry (DEXA scan) based on bone density measurement.[56] The trabecular bone score (TBS) based on the spatial grayscale analysis of DEXA images enables the evaluation of bone microarchitecture.[19]
Testing Procedures
Iron profiles can be performed via clinical chemistry and immunological assays.[57]
Real-time polymerase chain reaction (PCR) assay and fluorescence resonance energy transfer (FRET) hybridization probes can differentiate between wildtype and mutant alleles of HFE. Finding two copies of the HFE gene with the C282Y mutation confirms classic HH. Patients who are heterozygous for this mutation are reflexed to H63D analysis.
Histopathology and immunohistochemistry can be employed to confirm tissue involvement in HH. Radiological techniques are also utilized to assess cardiac, musculoskeletal, endocrine, liver, and other tissues for ongoing disease progress and degree of damage.[58]
Interfering Factors
Accurately diagnosing HH requires ruling out other disease processes with a similar presentation. Additionally, serum ferritin is an acute phase reactant and cannot be used in isolation to diagnose HH.[33] Secondary causes of iron overload such as ineffective erythropoiesis, repeated blood transfusions, reduced hepcidin production due to acute or chronic liver injury, chronic kidney injury, adult-onset Still disease, hemophagocytic lymphohistiocytosis, aceruloplasminemia, atransferrinemia, and hereditary hyperferritinemia must be ruled out. Dietary iron overloads, such as Bantu siderosis, and an oral or injectable iron overdose should also be ruled out.[59]
The presence of heterophilic antibodies can interfere with ferritin immunoassay procedures; ferritin values discordant with the clinical status may require further investigation.[60] In-vivo interferences such as ethanol, iron salts, and oral contraceptives may cause elevations of serum ferritin. Ferritin levels are decreased by exogenous erythropoietin.[61] Immunonephelometric analytical methods for ferritin are adversely affected by increased turbidity or lipemia.[62]
Clinical Significance
HH is a prevalent genetic disorder with low and variable penetrance; up to 25% of patients with Type 1 HH are clinically asymptomatic. The characteristic laboratory findings of elevated serum ferritin and transferrin saturation or elevated liver transaminases are frequently found incidentally during routine laboratory screening or evaluations for other common disorders. In these circumstances, further diagnostic testing is required to arrive at a specific diagnosis. Primary HH must be differentiated from secondary causes of iron overload.
Primary HH is frequently managed via serial phlebotomy. Secondary hemochromatosis is managed with iron chelation and treatment of the underlying cause of iron overload.
Type 1 HH is the most common form of the disorder and is inherited in an autosomal recessive fashion. Once a diagnosis of HH is made, screening and genetic testing should be offered to first- and second-degree relatives of the proband.[63] Additionally, if the proband desires to achieve pregnancy, their partner should be offered screening and genetic testing. Antenatal counseling should be offered to asymptomatic carriers in addition to patients with symptomatic HH; the risks of transmitting the mutation to potential offspring should be explained.[64]
Quality Control and Lab Safety
For optimum precision and accuracy of the test results, timely calibration and quality checks via internal & external quality control systems should be done per the laboratory's quality policy. Strict compliance with standard operating procedures and international guidelines becomes essential to enhance the accuracy of diagnosis.[65]
The analytical examination process's quality control (QC) monitors a measurement procedure to verify that it meets performance specifications appropriate for patient care or that an error condition must be corrected.[66] For non-waived tests, laboratory regulations require, at the minimum, analysis of at least two levels of quality control materials once every 24 hours. If necessary, laboratories can assay QC samples more frequently to ensure accurate results. Quality control samples should be assayed after calibration or maintenance of an analyzer to verify the correct method performance.[67] To minimize QC when performing tests for which manufacturers' recommendations are less than those required by the regulatory agency, the labs can develop an individualized quality control plan that involves performing a risk assessment of potential sources of error in all phases of testing and putting in place a QC plan to reduce the likelihood of errors.[68]
The design of a QC plan must consider the analytical performance capability of a measurement procedure and the risk of harm to a patient if an erroneous laboratory test result is used for a clinical care decision. An erroneous laboratory test result is a hazardous condition that may or may not cause harm to a patient, depending on what action or inaction a clinical care provider takes based on the erroneous result.[69]
The acceptable range and rules for interpreting QC results are based on the probability of detecting a significant analytical error condition with an acceptably low false-alert rate.[68] The desired process control performance characteristics must be established for each measurement before selecting the appropriate QC rules.[70] Westgard multi-rules are usually used to evaluate the quality control runs. If a run is declared out of control, investigate the system (instrument, standards, controls, etc.) to determine the cause of the problem. Do not perform any analysis until the issue has been resolved.[71]
Changing reagent lots can have an unexpected impact on QC results. Careful reagent lot crossover evaluation of QC target values is necessary. The matrix-related interaction between a QC material and a reagent can change with a different reagent lot; QC results may not be a reliable indicator of a measurement procedure's performance for patient samples after a reagent lot change.[72] It is necessary to use clinical patient samples to verify the consistency of results between old and new lots of reagents because of the unpredictability of a matrix-related bias being present for QC materials.[73]
The laboratory must participate in the external quality control or proficiency testing (PT) program; it is a regulatory requirement published by the Centers for Medicare and Medicaid Services (CMS) in the Clinical Laboratory Improvement Amendments (CLIA) regulations.[65] It is helpful to ensure the accuracy and reliability of the laboratory concerning other laboratories performing the same or comparable assays. Required participation and scored CMS and voluntary accreditation organizations monitor results. The PT plan should be included as an aspect of the quality assessment (QA) plan and the overall quality program of the laboratory.[74]
Consider all specimens, control materials, and calibrator materials as potentially infectious. Exercise the usual precautions required for handling all laboratory reagents. Disposal of all waste material should be in accordance with local guidelines. Wear gloves, a lab coat, and safety glasses when handling human blood specimens. Place all plastic tips, sample cups, and gloves that come into contact with blood in a biohazard waste container.[75] Discard all disposable glassware into sharps waste containers. Protect all work surfaces with disposable absorbent bench top paper, discarded into biohazard waste containers weekly or whenever blood contamination occurs. Wipe all work surfaces weekly.[76]
Enhancing Healthcare Team Outcomes
Improving outcomes for patients with HH involves a comprehensive multidisciplinary approach that addresses various aspects of the condition. Strategies to increase awareness and early detection of HH include educating healthcare professionals, particularly primary care providers, to recognize the signs, symptoms, and characteristic laboratory findings of HH. Promoting genetic testing for probands and close family members is imperative for promptly initiating treatment to improve morbidity rates. Plans for monitoring disease progression require collaboration among healthcare professionals across multiple disciplines, including gastroenterology, cardiology, rheumatology, and endocrinology. Patient education must include information on the potential complications of iron overload, the benefits of therapeutic phlebotomy, and the significance of regular monitoring.
Media
(Click Image to Enlarge)
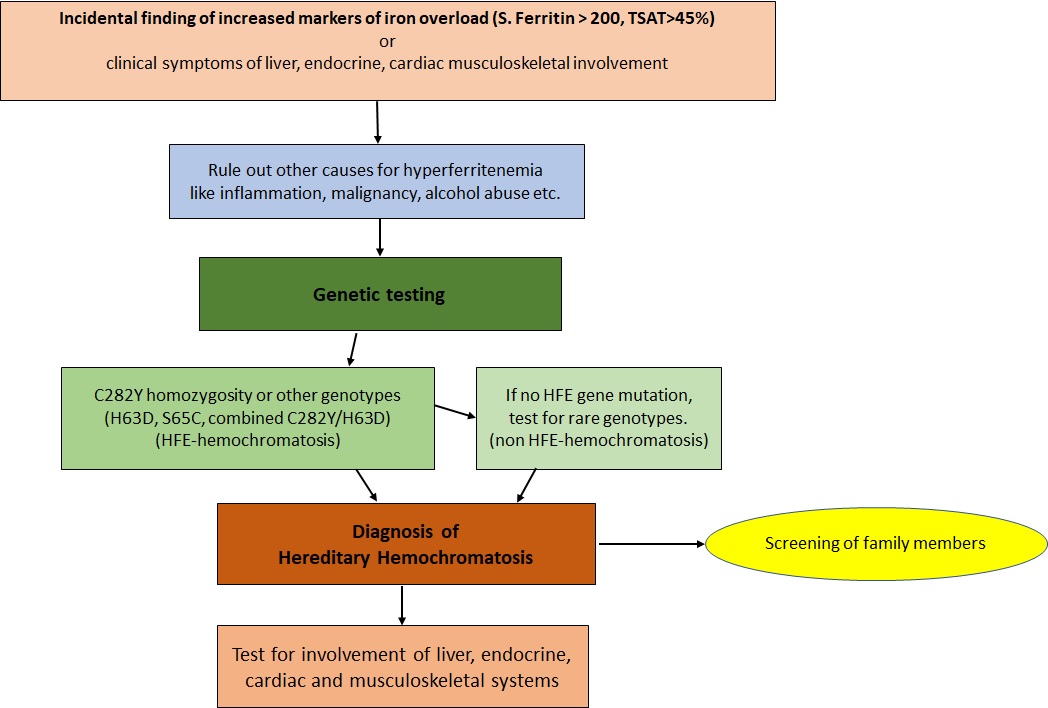
Flow chart for investigation of Hereditary Hemochromatosis Contributed by Dr. Jeevan Divakaran
(Click Image to Enlarge)
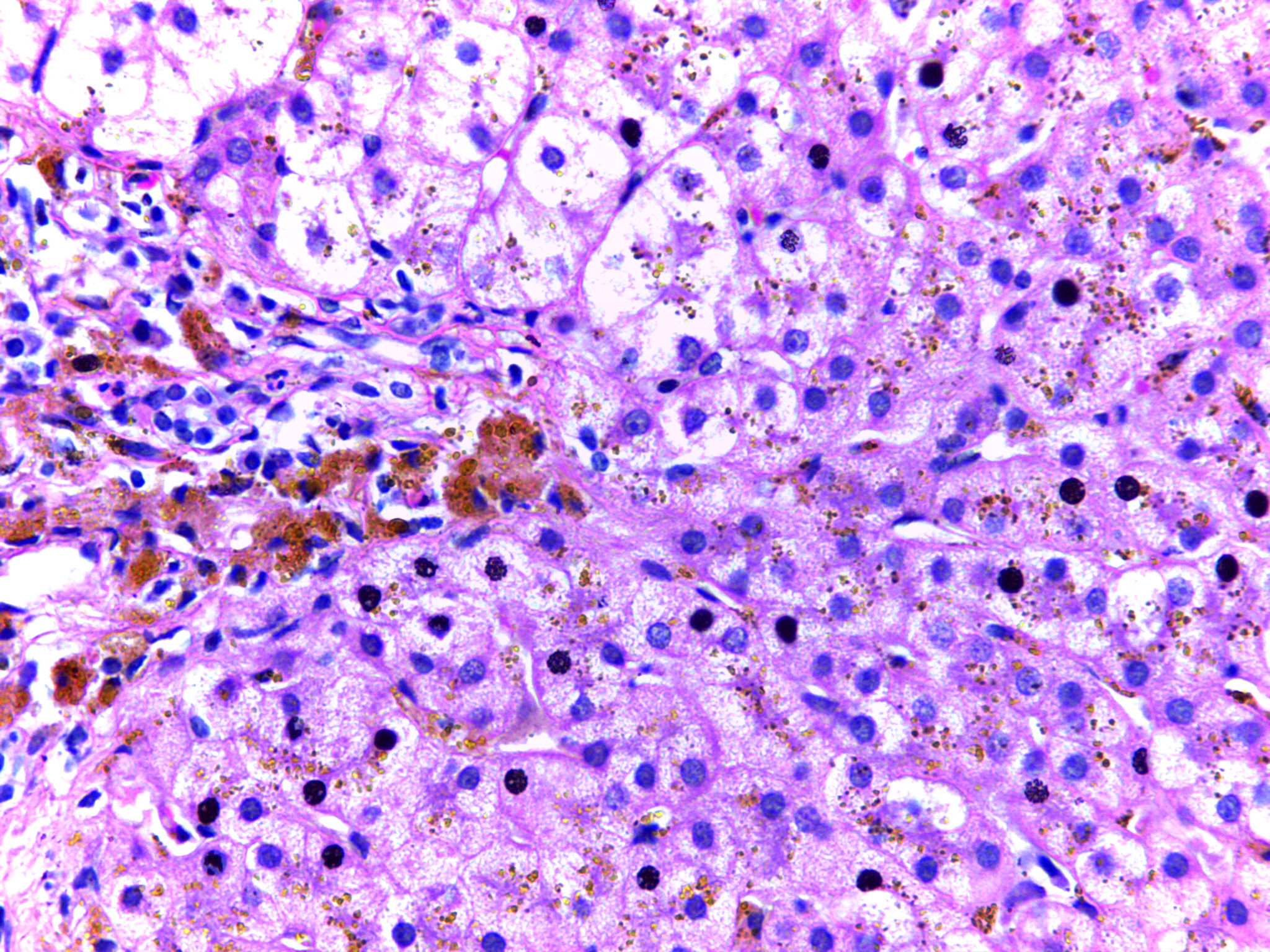
Image 1: Micrograph of hemochromatosis liver showing hepatocytes with coarse golden yellow granules of hemosiderin within the cytoplasm. These granules stain with Prussian blue stain. Contributed By Calicut Medical College - DEPARTMENT OF PATHOLOGY, CALICUT MEDICAL COLLEGE, CC BY-SA 4.0, https://commons.wikimedia.org/w/index.php?curid=44955377
(Click Image to Enlarge)
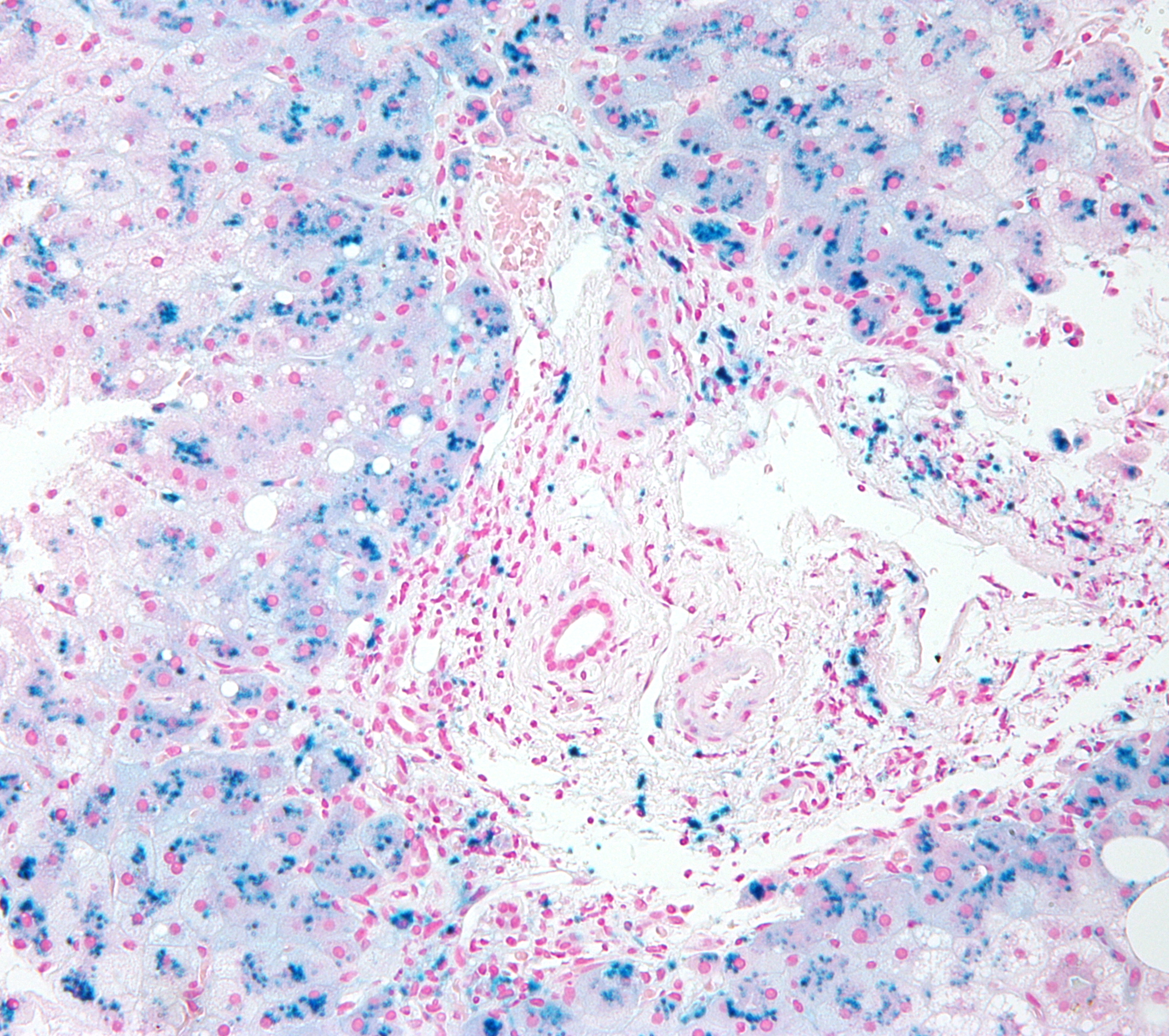
High magnification micrograph of hemosiderosis. Liver biopsy. Iron stain (Prussian blue). Contributed By Nephron - Own work, CC BY-SA 3.0, https://commons.wikimedia.org/w/index.php?curid=7765904
References
Girelli D, Busti F, Brissot P, Cabantchik I, Muckenthaler MU, Porto G. Hemochromatosis classification: update and recommendations by the BIOIRON Society. Blood. 2022 May 19:139(20):3018-3029. doi: 10.1182/blood.2021011338. Epub [PubMed PMID: 34601591]
Gerhard GS, Paynton BV, DiStefano JK. Identification of Genes for Hereditary Hemochromatosis. Methods in molecular biology (Clifton, N.J.). 2018:1706():353-365. doi: 10.1007/978-1-4939-7471-9_19. Epub [PubMed PMID: 29423808]
Kane SF, Roberts C, Paulus R. Hereditary Hemochromatosis: Rapid Evidence Review. American family physician. 2021 Sep 1:104(3):263-270 [PubMed PMID: 34523883]
Sandnes M, Vorland M, Ulvik RJ, Reikvam H. HFE Genotype, Ferritin Levels and Transferrin Saturation in Patients with Suspected Hereditary Hemochromatosis. Genes. 2021 Jul 28:12(8):. doi: 10.3390/genes12081162. Epub 2021 Jul 28 [PubMed PMID: 34440336]
Lanzara C, Roetto A, Daraio F, Rivard S, Ficarella R, Simard H, Cox TM, Cazzola M, Piperno A, Gimenez-Roqueplo AP, Grammatico P, Volinia S, Gasparini P, Camaschella C. Spectrum of hemojuvelin gene mutations in 1q-linked juvenile hemochromatosis. Blood. 2004 Jun 1:103(11):4317-21 [PubMed PMID: 14982873]
Level 2 (mid-level) evidenceMontosi G, Donovan A, Totaro A, Garuti C, Pignatti E, Cassanelli S, Trenor CC, Gasparini P, Andrews NC, Pietrangelo A. Autosomal-dominant hemochromatosis is associated with a mutation in the ferroportin (SLC11A3) gene. The Journal of clinical investigation. 2001 Aug:108(4):619-23 [PubMed PMID: 11518736]
Level 3 (low-level) evidenceTang S, Bai L, Gao Y, Hou W, Song W, Liu H, Hu Z, Duan Z, Zhang L, Zheng S. A Novel Mutation of Transferrin Receptor 2 in a Chinese Pedigree With Type 3 Hemochromatosis: A Case Report. Frontiers in genetics. 2022:13():836431. doi: 10.3389/fgene.2022.836431. Epub 2022 Apr 8 [PubMed PMID: 35464850]
Level 3 (low-level) evidenceRoemhild K, von Maltzahn F, Weiskirchen R, Knüchel R, von Stillfried S, Lammers T. Iron metabolism: pathophysiology and pharmacology. Trends in pharmacological sciences. 2021 Aug:42(8):640-656. doi: 10.1016/j.tips.2021.05.001. Epub 2021 Jun 2 [PubMed PMID: 34090703]
Bardou-Jacquet E, Ben Ali Z, Beaumont-Epinette MP, Loreal O, Jouanolle AM, Brissot P. Non-HFE hemochromatosis: pathophysiological and diagnostic aspects. Clinics and research in hepatology and gastroenterology. 2014 Apr:38(2):143-54. doi: 10.1016/j.clinre.2013.11.003. Epub 2013 Dec 8 [PubMed PMID: 24321703]
Golfeyz S, Lewis S, Weisberg IS. Hemochromatosis: pathophysiology, evaluation, and management of hepatic iron overload with a focus on MRI. Expert review of gastroenterology & hepatology. 2018 Aug:12(8):767-778. doi: 10.1080/17474124.2018.1496016. Epub 2018 Jul 19 [PubMed PMID: 29966105]
Piperno A, Pelucchi S, Mariani R. Inherited iron overload disorders. Translational gastroenterology and hepatology. 2020:5():25. doi: 10.21037/tgh.2019.11.15. Epub 2020 Apr 5 [PubMed PMID: 32258529]
Barton JC, Acton RT. Diabetes in HFE Hemochromatosis. Journal of diabetes research. 2017:2017():9826930. doi: 10.1155/2017/9826930. Epub 2017 Feb 26 [PubMed PMID: 28331855]
Lee YN, Huda MS. Uncommon forms of diabetes. Clinical medicine (London, England). 2021 Jul:21(4):e337-e341. doi: 10.7861/clinmed.2021-0369. Epub [PubMed PMID: 35192474]
Hramiak IM, Finegood DT, Adams PC. Factors affecting glucose tolerance in hereditary hemochromatosis. Clinical and investigative medicine. Medecine clinique et experimentale. 1997 Apr:20(2):110-8 [PubMed PMID: 9088667]
Bergeron C, Kovacs K. Pituitary siderosis. A histologic, immunocytologic, and ultrastructural study. The American journal of pathology. 1978 Nov:93(2):295-309 [PubMed PMID: 362939]
Level 3 (low-level) evidenceSantiago de Sousa Azulay R, Magalhães M, Tavares MDG, Dualibe R, Barbosa L, Sá Gaspar S, Faria AM, Nascimento GC, Damianse SDSP, Rocha VCC, Gomes MB, Dos Santos Faria M. Novel Mutation in the Hemojuvelin Gene (HJV) in a Patient with Juvenile Hemochromatosis Presenting with Insulin-dependent Diabetes Mellitus, Secondary Hypothyroidism and Hypogonadism. The American journal of case reports. 2020 Apr 24:21():e923108. doi: 10.12659/AJCR.923108. Epub 2020 Apr 24 [PubMed PMID: 32327622]
Level 3 (low-level) evidenceBanaszkiewicz K, Sikorska K, Panas D, Sworczak K. The Role of the Trabecular Bone Score in the Assessment of Osteoarticular Disorders in Patients with HFE-Hemochromatosis: A Single-Center Study from Poland. Genes. 2021 Aug 25:12(9):. doi: 10.3390/genes12091304. Epub 2021 Aug 25 [PubMed PMID: 34573286]
Timms AE, Sathananthan R, Bradbury L, Athanasou NA, Wordsworth BP, Brown MA. Genetic testing for haemochromatosis in patients with chondrocalcinosis. Annals of the rheumatic diseases. 2002 Aug:61(8):745-7 [PubMed PMID: 12117686]
Dallos T, Sahinbegovic E, Aigner E, Axmann R, Schöniger-Hekele M, Karonitsch T, Stamm T, Farkas M, Karger T, Cavallaro A, Stölzel U, Keysser G, Datz C, Schett G, Manger B, Zwerina J. Validation of a radiographic scoring system for haemochromatosis arthropathy. Annals of the rheumatic diseases. 2010 Dec:69(12):2145-51. doi: 10.1136/ard.2009.122119. Epub 2010 Aug 2 [PubMed PMID: 20679473]
Level 2 (mid-level) evidencePfeiffer CM, Looker AC. Laboratory methodologies for indicators of iron status: strengths, limitations, and analytical challenges. The American journal of clinical nutrition. 2017 Dec:106(Suppl 6):1606S-1614S. doi: 10.3945/ajcn.117.155887. Epub 2017 Oct 25 [PubMed PMID: 29070545]
Dale JC, Burritt MF, Zinsmeister AR. Diurnal variation of serum iron, iron-binding capacity, transferrin saturation, and ferritin levels. American journal of clinical pathology. 2002 May:117(5):802-8 [PubMed PMID: 12090432]
Monden S, Tanaka M, Takahashi H. [Pre-test blood samples at clinical laboratories--effects of temperature and duration of blood sample storage on measurements]. Rinsho byori. The Japanese journal of clinical pathology. 2008 Mar:56(3):239-42 [PubMed PMID: 18411808]
Lippi G, Salvagno GL, Montagnana M, Brocco G, Guidi GC. Influence of hemolysis on routine clinical chemistry testing. Clinical chemistry and laboratory medicine. 2006:44(3):311-6 [PubMed PMID: 16519604]
Daru J, Colman K, Stanworth SJ, De La Salle B, Wood EM, Pasricha SR. Serum ferritin as an indicator of iron status: what do we need to know? The American journal of clinical nutrition. 2017 Dec:106(Suppl 6):1634S-1639S. doi: 10.3945/ajcn.117.155960. Epub 2017 Oct 25 [PubMed PMID: 29070560]
Daniłowicz-Szymanowicz L, Świątczak M, Sikorska K, Starzyński RR, Raczak A, Lipiński P. Pathogenesis, Diagnosis, and Clinical Implications of Hereditary Hemochromatosis-The Cardiological Point of View. Diagnostics (Basel, Switzerland). 2021 Jul 16:11(7):. doi: 10.3390/diagnostics11071279. Epub 2021 Jul 16 [PubMed PMID: 34359361]
Bassett ML, Halliday JW, Ferris RA, Powell LW. Diagnosis of hemochromatosis in young subjects: predictive accuracy of biochemical screening tests. Gastroenterology. 1984 Sep:87(3):628-33 [PubMed PMID: 6745616]
Moyer TP, Highsmith WE, Smyrk TC, Gross JB Jr. Hereditary hemochromatosis: laboratory evaluation. Clinica chimica acta; international journal of clinical chemistry. 2011 Aug 17:412(17-18):1485-92. doi: 10.1016/j.cca.2011.04.007. Epub 2011 Apr 13 [PubMed PMID: 21510925]
Hagve TA, Asberg A, Ulvik R, Borch-Iohnsen B, Thorstensen K. [Hemochromatosis--from an underdiagnosed curiosity to a common disease]. Tidsskrift for den Norske laegeforening : tidsskrift for praktisk medicin, ny raekke. 2009 Apr 30:129(9):863-6. doi: 10.4045/tidsskr.08.0084. Epub [PubMed PMID: 19415085]
Brissot P, Troadec MB, Loréal O, Brissot E. Pathophysiology and classification of iron overload diseases; update 2018. Transfusion clinique et biologique : journal de la Societe francaise de transfusion sanguine. 2019 Feb:26(1):80-88. doi: 10.1016/j.tracli.2018.08.006. Epub 2018 Aug 15 [PubMed PMID: 30173950]
Bacon BR, Adams PC, Kowdley KV, Powell LW, Tavill AS, American Association for the Study of Liver Diseases. Diagnosis and management of hemochromatosis: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology (Baltimore, Md.). 2011 Jul:54(1):328-43. doi: 10.1002/hep.24330. Epub [PubMed PMID: 21452290]
Level 1 (high-level) evidenceEuropean Association for the Study of the Liver. Electronic address: easloffice@easloffice.eu, European Association for the Study of the Liver. EASL Clinical Practice Guidelines on haemochromatosis. Journal of hepatology. 2022 Aug:77(2):479-502. doi: 10.1016/j.jhep.2022.03.033. Epub 2022 Jun 1 [PubMed PMID: 35662478]
Level 1 (high-level) evidenceWalsh A, Dixon JL, Ramm GA, Hewett DG, Lincoln DJ, Anderson GJ, Subramaniam VN, Dodemaide J, Cavanaugh JA, Bassett ML, Powell LW. The clinical relevance of compound heterozygosity for the C282Y and H63D substitutions in hemochromatosis. Clinical gastroenterology and hepatology : the official clinical practice journal of the American Gastroenterological Association. 2006 Nov:4(11):1403-10 [PubMed PMID: 16979952]
Hsu CC, Senussi NH, Fertrin KY, Kowdley KV. Iron overload disorders. Hepatology communications. 2022 Aug:6(8):1842-1854. doi: 10.1002/hep4.2012. Epub 2022 Jun 14 [PubMed PMID: 35699322]
Barton JC, Lafreniere SA, Leiendecker-Foster C, Li H, Acton RT, Press RD, Eckfeldt JH. HFE, SLC40A1, HAMP, HJV, TFR2, and FTL mutations detected by denaturing high-performance liquid chromatography after iron phenotyping and HFE C282Y and H63D genotyping in 785 HEIRS Study participants. American journal of hematology. 2009 Nov:84(11):710-4. doi: 10.1002/ajh.21524. Epub [PubMed PMID: 19787796]
Level 2 (mid-level) evidenceWang Y, Du Y, Liu G, Guo S, Hou B, Jiang X, Han B, Chang Y, Nie G. Identification of novel mutations in HFE, HFE2, TfR2, and SLC40A1 genes in Chinese patients affected by hereditary hemochromatosis. International journal of hematology. 2017 Apr:105(4):521-525. doi: 10.1007/s12185-016-2150-8. Epub 2016 Nov 28 [PubMed PMID: 27896572]
Barton JC, Barton JC, Patel N, McLaren GD. Abdominal pain and cirrhosis at diagnosis of hemochromatosis: Analysis of 219 referred probands with HFE p.C282Y homozygosity and a literature review. PloS one. 2021:16(12):e0261690. doi: 10.1371/journal.pone.0261690. Epub 2021 Dec 21 [PubMed PMID: 34932603]
Pappachan JM, Babu S, Krishnan B, Ravindran NC. Non-alcoholic Fatty Liver Disease: A Clinical Update. Journal of clinical and translational hepatology. 2017 Dec 28:5(4):384-393. doi: 10.14218/JCTH.2017.00013. Epub 2017 Jul 26 [PubMed PMID: 29226105]
Milman NT, Schioedt FV, Junker AE, Magnussen K. Diagnosis and Treatment of Genetic HFE-Hemochromatosis: The Danish Aspect. Gastroenterology research. 2019 Oct:12(5):221-232. doi: 10.14740/gr1206. Epub 2019 Oct 4 [PubMed PMID: 31636772]
Henninger B, Alustiza J, Garbowski M, Gandon Y. Practical guide to quantification of hepatic iron with MRI. European radiology. 2020 Jan:30(1):383-393. doi: 10.1007/s00330-019-06380-9. Epub 2019 Aug 7 [PubMed PMID: 31392478]
França M, Carvalho JG. MR imaging assessment and quantification of liver iron. Abdominal radiology (New York). 2020 Nov:45(11):3400-3412. doi: 10.1007/s00261-020-02574-8. Epub [PubMed PMID: 32435848]
Viveiros A, Schaefer B, Panzer M, Henninger B, Plaikner M, Kremser C, Franke A, Franzenburg S, Hoeppner MP, Stauder R, Janecke A, Tilg H, Zoller H. MRI-Based Iron Phenotyping and Patient Selection for Next-Generation Sequencing of Non-Homeostatic Iron Regulator Hemochromatosis Genes. Hepatology (Baltimore, Md.). 2021 Nov:74(5):2424-2435. doi: 10.1002/hep.31982. Epub 2021 Jul 13 [PubMed PMID: 34048062]
Lin ZH, Xin YN, Dong QJ, Wang Q, Jiang XJ, Zhan SH, Sun Y, Xuan SY. Performance of the aspartate aminotransferase-to-platelet ratio index for the staging of hepatitis C-related fibrosis: an updated meta-analysis. Hepatology (Baltimore, Md.). 2011 Mar:53(3):726-36. doi: 10.1002/hep.24105. Epub 2011 Feb 11 [PubMed PMID: 21319189]
Level 1 (high-level) evidenceVallet-Pichard A, Mallet V, Nalpas B, Verkarre V, Nalpas A, Dhalluin-Venier V, Fontaine H, Pol S. FIB-4: an inexpensive and accurate marker of fibrosis in HCV infection. comparison with liver biopsy and fibrotest. Hepatology (Baltimore, Md.). 2007 Jul:46(1):32-6 [PubMed PMID: 17567829]
Adams LA, Bulsara M, Rossi E, DeBoer B, Speers D, George J, Kench J, Farrell G, McCaughan GW, Jeffrey GP. Hepascore: an accurate validated predictor of liver fibrosis in chronic hepatitis C infection. Clinical chemistry. 2005 Oct:51(10):1867-73 [PubMed PMID: 16055434]
Level 2 (mid-level) evidenceChin J, Powell LW, Ramm LE, Hartel GF, Olynyk JK, Ramm GA. Utility of Serum Biomarker Indices for Staging of Hepatic Fibrosis Before and After Venesection in Patients With Hemochromatosis Caused by Variants in HFE. Clinical gastroenterology and hepatology : the official clinical practice journal of the American Gastroenterological Association. 2021 Jul:19(7):1459-1468.e5. doi: 10.1016/j.cgh.2020.07.052. Epub 2020 Aug 1 [PubMed PMID: 32745684]
Ong SY, Khoo T, Nicoll AJ, Gurrin L, Worland T, Pateria P, Ramm LE, Testro A, Anderson GJ, Skoien R, Powell LW, Ramm GA, Olynyk JK, Delatycki MB. Utility and limitations of Hepascore and transient elastography to detect advanced hepatic fibrosis in HFE hemochromatosis. Scientific reports. 2021 Jul 19:11(1):14654. doi: 10.1038/s41598-021-94083-x. Epub 2021 Jul 19 [PubMed PMID: 34282177]
Jacobs EM, Hendriks JC, van Tits BL, Evans PJ, Breuer W, Liu DY, Jansen EH, Jauhiainen K, Sturm B, Porter JB, Scheiber-Mojdehkar B, von Bonsdorff L, Cabantchik ZI, Hider RC, Swinkels DW. Results of an international round robin for the quantification of serum non-transferrin-bound iron: Need for defining standardization and a clinically relevant isoform. Analytical biochemistry. 2005 Jun 15:341(2):241-50 [PubMed PMID: 15907869]
Shizukuda Y, Tripodi DJ, Zalos G, Bolan CD, Yau YY, Leitman SF, Waclawiw MA, Rosing DR. Incidence of cardiac arrhythmias in asymptomatic hereditary hemochromatosis subjects with C282Y homozygosity. The American journal of cardiology. 2012 Mar 15:109(6):856-60. doi: 10.1016/j.amjcard.2011.11.011. Epub 2011 Dec 22 [PubMed PMID: 22196777]
Level 2 (mid-level) evidenceAnderson LJ. Assessment of iron overload with T2* magnetic resonance imaging. Progress in cardiovascular diseases. 2011 Nov-Dec:54(3):287-94. doi: 10.1016/j.pcad.2011.07.004. Epub [PubMed PMID: 22014495]
Andersson R, Tingstedt B, Xia J. Pathogenesis of chronic pancreatitis: a comprehensive update and a look into the future. Scandinavian journal of gastroenterology. 2009:44(6):661-3. doi: 10.1080/00365520902718739. Epub [PubMed PMID: 19199163]
McDermott JH, Walsh CH. Hypogonadism in hereditary hemochromatosis. The Journal of clinical endocrinology and metabolism. 2005 Apr:90(4):2451-5 [PubMed PMID: 15657376]
Zafar AM, Zuberi L, Khan AH, Ahsan H. Utility of MRI in assessment of pituitary iron overload. JPMA. The Journal of the Pakistan Medical Association. 2007 Sep:57(9):475-7 [PubMed PMID: 18072648]
Level 3 (low-level) evidenceSmit SL, Peters TMA, Gisbertz IAM, Moolenaar W, Hendriks Y, Vincent HH, Houtsma D, Loosveld OJL, van Herwaarden AE, Rennings AJM, Swinkels DW. Variable workup calls for guideline development for type 2A hereditary haemochromatosis. The Netherlands journal of medicine. 2018 Oct:76(8):365-373 [PubMed PMID: 30362946]
El Osta R, Grandpre N, Monnin N, Hubert J, Koscinski I. Hypogonadotropic hypogonadism in men with hereditary hemochromatosis. Basic and clinical andrology. 2017:27():13. doi: 10.1186/s12610-017-0057-8. Epub 2017 Jul 8 [PubMed PMID: 28694969]
Kelly TM, Edwards CQ, Meikle AW, Kushner JP. Hypogonadism in hemochromatosis: reversal with iron depletion. Annals of internal medicine. 1984 Nov:101(5):629-32 [PubMed PMID: 6435491]
Bazzocchi A, Ponti F, Albisinni U, Battista G, Guglielmi G. DXA: Technical aspects and application. European journal of radiology. 2016 Aug:85(8):1481-92. doi: 10.1016/j.ejrad.2016.04.004. Epub 2016 Apr 14 [PubMed PMID: 27157852]
Worwood M. The laboratory assessment of iron status--an update. Clinica chimica acta; international journal of clinical chemistry. 1997 Mar 18:259(1-2):3-23 [PubMed PMID: 9086290]
Alvarenga AM, Brissot P, Santos PCJL. Haemochromatosis revisited. World journal of hepatology. 2022 Nov 27:14(11):1931-1939. doi: 10.4254/wjh.v14.i11.1931. Epub [PubMed PMID: 36483608]
Gattermann N, Muckenthaler MU, Kulozik AE, Metzgeroth G, Hastka J. The Evaluation of Iron Deficiency and Iron Overload. Deutsches Arzteblatt international. 2021 Dec 10:118(49):847-856. doi: 10.3238/arztebl.m2021.0290. Epub [PubMed PMID: 34755596]
Grouzmann E, Dayer N, Bain C, Neville CM, McCusker MD, Wang RY. Eliminating heterophilic antibody interference for ferritin detection using Olympus F(ab')2 based reagent. Clinical laboratory. 2008:54(9-10):355-7 [PubMed PMID: 19097493]
Level 3 (low-level) evidenceTate J, Ward G. Interferences in immunoassay. The Clinical biochemist. Reviews. 2004 May:25(2):105-20 [PubMed PMID: 18458713]
Dignass A, Farrag K, Stein J. Limitations of Serum Ferritin in Diagnosing Iron Deficiency in Inflammatory Conditions. International journal of chronic diseases. 2018:2018():9394060. doi: 10.1155/2018/9394060. Epub 2018 Mar 18 [PubMed PMID: 29744352]
Santos PCJL, Krieger JE, Pereira AC. Molecular diagnostic and pathogenesis of hereditary hemochromatosis. International journal of molecular sciences. 2012:13(2):1497-1511. doi: 10.3390/ijms13021497. Epub 2012 Feb 1 [PubMed PMID: 22408404]
Guggenbuhl P, Brissot P, Loréal O. Miscellaneous non-inflammatory musculoskeletal conditions. Haemochromatosis: the bone and the joint. Best practice & research. Clinical rheumatology. 2011 Oct:25(5):649-64. doi: 10.1016/j.berh.2011.10.014. Epub [PubMed PMID: 22142745]
Badrick T. Integrating quality control and external quality assurance. Clinical biochemistry. 2021 Sep:95():15-27. doi: 10.1016/j.clinbiochem.2021.05.003. Epub 2021 May 7 [PubMed PMID: 33965412]
Level 2 (mid-level) evidenceHowanitz PJ, Howanitz JH. Quality control for the clinical laboratory. Clinics in laboratory medicine. 1983 Sep:3(3):541-51 [PubMed PMID: 6357609]
Level 2 (mid-level) evidenceKinns H, Pitkin S, Housley D, Freedman DB. Internal quality control: best practice. Journal of clinical pathology. 2013 Dec:66(12):1027-32. doi: 10.1136/jclinpath-2013-201661. Epub 2013 Sep 26 [PubMed PMID: 24072731]
Level 2 (mid-level) evidenceKearney E. Internal quality control. Methods in molecular biology (Clifton, N.J.). 2013:1065():277-89. doi: 10.1007/978-1-62703-616-0_18. Epub [PubMed PMID: 23996371]
Level 2 (mid-level) evidenceWestgard JO. A Total Quality-Control Plan with Right-Sized Statistical Quality-Control. Clinics in laboratory medicine. 2017 Mar:37(1):137-150. doi: 10.1016/j.cll.2016.09.011. Epub 2016 Dec 20 [PubMed PMID: 28153361]
Level 2 (mid-level) evidenceWestgard JO. Internal quality control: planning and implementation strategies. Annals of clinical biochemistry. 2003 Nov:40(Pt 6):593-611 [PubMed PMID: 14629798]
Level 2 (mid-level) evidenceBayat H. Selecting multi-rule quality control procedures based on patient risk. Clinical chemistry and laboratory medicine. 2017 Oct 26:55(11):1702-1708. doi: 10.1515/cclm-2016-1077. Epub [PubMed PMID: 28236626]
Level 2 (mid-level) evidenceTopic E, Nikolac N, Panteghini M, Theodorsson E, Salvagno GL, Miler M, Simundic AM, Infusino I, Nordin G, Westgard S. How to assess the quality of your analytical method? Clinical chemistry and laboratory medicine. 2015 Oct:53(11):1707-18. doi: 10.1515/cclm-2015-0869. Epub [PubMed PMID: 26408611]
Level 2 (mid-level) evidenceDalenberg DA, Schryver PG, Klee GG. Analytical performance specifications: relating laboratory performance to quality required for intended clinical use. Clinics in laboratory medicine. 2013 Mar:33(1):55-73. doi: 10.1016/j.cll.2012.11.005. Epub 2012 Dec 20 [PubMed PMID: 23331729]
Level 2 (mid-level) evidenceCenters for Disease Control and Prevention (CDC) (2) Centers for Medicare & Medicaid Services (CMS), HHS. Medicare, Medicaid, and CLIA programs; laboratory requirements relating to quality systems and certain personnel qualifications. Final rule. Federal register. 2003 Jan 24:68(16):3639-714 [PubMed PMID: 12545998]
Level 2 (mid-level) evidenceRojo-Molinero E, Alados JC, de la Pedrosa EG, Leiva J, Pérez JL. [Safety in the Microbiology laboratory]. Enfermedades infecciosas y microbiologia clinica. 2015 Jun-Jul:33(6):404-10. doi: 10.1016/j.eimc.2014.06.014. Epub 2014 Nov 8 [PubMed PMID: 25444041]
Level 3 (low-level) evidenceLo J. Biological safety in the medical laboratory. Hong Kong medical journal = Xianggang yi xue za zhi. 2015 Jun:21(3):200. doi: 10.12809/hkmj154581. Epub [PubMed PMID: 26045068]