Introduction
The term “macular dystrophy” is historically used for a group of heritable disorders that present with retinal abnormalities between the temporal vascular arcades. Stargardt disease type 1 (STGD1) is the most common cause of juvenile macular dystrophy.[1] German ophthalmologist Karl Stargardt first described the condition in 1909 in seven patients of two families who presented with macular dystrophy surrounded by yellow-white pisciform flecks.[2]
Stargardt disease is a genetic disorder that leads to the accumulation of lipofuscin, a metabolic waste product, in the retina. This condition is a heterogeneous disease with many clinical presentations, which vary vastly in the age of onset and the rate of progression. Patients present with progressive visual impairment, usually beginning in the first or second decades of life. The phenotypic heterogeneity makes the clinical diagnosis of STGD1 challenging. Reports from the Progression of Atrophy Secondary to Stargardt Disease (ProgStar) study have described the natural history of the disease with the help of various structural and functional investigative modalities.[3] Several therapeutic options are currently being evaluated. However, none have yet received approval from the United States Food and Drug Administration (US-FDA) as of May 2023.
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
STGD (Online Mendelian Inheritance in Man, OMIM #248200; juvenile macular degeneration; macular dystrophy with flecks, type 1; fundus flavimaculatus; or early-onset severe retinal dystrophy) is commonly inherited as an autosomal recessive trait due to missense and null mutations in the adenosine triphosphate-binding cassette transporter alpha 4 subunit (ABCA4) gene.[4] The ABCA4 gene is complex and consists of 50 exons mapped to the short arm of the human chromosome 1. While the former is associated with a milder, adult-onset, "foveal-sparing" disease, the latter can lead to severe and early-onset disease. There are few exceptions to this rule as a severe disease secondary to a few missense variants like p.Leu541Pro, p.Ala1038Val complex, and p.Arg1640Trp have been noted.[1] Other diseases associated with the mutation of the ABCA4 gene include cone-rod dystrophy 3 (OMIM #604116, autosomal recessive), retinitis pigmentosa 19 (OMIM #601718, autosomal recessive), and age-related macular degeneration 2 (OMIM #153800, autosomal dominant). ABCA4-associated retinopathies or ABCA4 retinopathies encompass a group of monogenic autosomal recessive diseases caused by varying amounts of decreased ABCA4 function.[5][6] The spectrum of phenotypes includes bull's eye maculopathy, cone-rod dystrophy, retinitis pigmentosa, rapid-onset chorioretinopathy,[7] and deep scleral exposure due to extensive chorioretinal damage.[6][8] It is thought that the progressive reduction of function of the ABCA4 gene produces Stargardt disease, cone-rod dystrophy, and retinitis pigmentosa, respectively.[5] Retinitis pigmentosa has the most severe phenotype with a flat electroretinogram and absent ABCA4 function.[5]
Stargardt-like diseases are retinal diseases with phenotypes similar to STGD1 but with an autosomal dominant inheritance.[9][10] Such diseases include STGD2, STGD3, and STGD4. STGD2 was erroneously linked to chromosome 13q34[11]; however, this phenotype occurs due to mutation in the ELOVL4 gene at 6q14.1 as in STGD3 (OMIM #600110, Stargardt disease type 3; macular dystrophy with flakes, type 3; autosomal dominant Stargardt-like macular dystrophy).[12] Patients with STGD3 do not usually show the silent or jet-black dark choroid as seen in STGD1; therefore, fluorescein angiograms may be darker than usual.[9][13][14] Variants in ELOVL4 are associated with autosomal dominant spinocerebellar ataxia, autosomal recessive disorder congenital ichthyosis, spastic quadriplegia, and impaired intellectual development (ISQMR).
STGD4 (OMIM #603786, Stargardt disease type 4) is an autosomal dominant disease caused by the prominin-1 gene (PROM1) mutation at chromosome 4p15.32.[10][15] The disease severity may increase in such cases if an additional mutation in ABCA4 is also present.[16] PROM1 mutations can also cause macular dystrophy, bull's eye maculopathy, cone-rod dystrophy, and retinitis pigmentosa.[6] WDR19 (IFT144 or NPHP13, chromosome 4p14), a gene classically associated with ciliopathies including Senior-Løken syndrome, Cranio-ectodermal dysplasia, nephronophthisis, short-rib thoracic dysplasia 5 with or without polydactyly, may also cause Stargardt disease-like phenotype.[17]
Other genes that can cause similar phenotypes include PRPH2 (multifocal pattern dystrophy simulating fundus flavimaculatus), CDH3, CERKL, CRX, BEST1, IMPG1, RDH12, and RPGR;[17] however, all these phenotypes do not show the typical triad of Stargardt disease.
Epidemiology
Although Stugardt disease is the most common cause of juvenile macular dystrophy, its prevalence in the United States is estimated to be 10 to 12.5 per 100,000 individuals.[2] Cornish et al. estimated the annual incidence of this condition in the United Kingdom to be around 0.110 to 0.128 per 100,000 individuals.[18] A study conducted in the Netherlands found the incidence of Stargardt disease to be 1.67 to 1.95 per 1,000,000 individuals per year.[19] The point prevalence of Stargardt disease was 1:22,000 to 1:19,000 in 2018 in the Netherlands.[19]
Pathophysiology
The ABCA4 gene encodes for the rim protein (RmP), a retinal adenosine triphosphate (ATP)-binding cassette protein. RmP is present on the membrane of the discs in the outer segment of the photoreceptors (rods and cones),[20] and participates in the visual cycle-related vitamin A metabolism. The light photons lead to isomerization of the 11-cis retinal to the all-trans retinal form. The all-trans-retinal form then enters the intradiscal space, where some of its molecules combine with a phospholipid (phosphatidylethanolamine, PE) ordinarily present in the disc membranes to form a complex called N-retinylidene-PE (N-RPE).
The RmP protein breaks the N-RPE complex and transports it from the intradiscal space. The absence of the RmP protein leads to the accumulation of the N-RPE complexes, leading to N-retinylidene-N-retinyl-PE (A2PE) formation by combining N-RPE and all-trans-retinal. A2PE hydrolyzes to form N-retinylidene-N-retinyl-ethanolamine (A2E). The A2E produced during these reactions leads to lipofuscin formation, accumulating in the retinal pigment epithelium (RPE) cells. The accumulation of lipofuscin leads to the formation of flecks. In vivo, study results have shown that patients with STGD1 have 2 to 5 times the amount of lipofuscin compared to the age-matched controls.[21][22]
Lipofuscin is resistant to lysosomal enzymatic degradation. High levels of A2E slow the phagocytic ability of the RPE cells, ultimately leading to RPE and photoreceptor cell death.[23][24]
Multimodal imaging studies have suggested that the degeneration of the photoreceptors’ inner and outer segments leads to hyper-reflective debris on the apical surface of the RPE cells, seen clinically as flecks. These flecks cause gradual disruption of the interdigitation zone (IZ), the ellipsoid zones (EZ), the external limiting membrane (ELM), and, eventually, the thinning of the outer nuclear layer (ONL).[25]
Histopathology
Light microscopy studies have shown the presence of abnormal RPE cells and loss of photoreceptors and RPE cells in eyes with STGD1. In addition, the RPE cells are heterogeneous in size and contain numerous PAS-positive intracytoplasmic material, lipofuscin.[26] Reactive Müller cell hypertrophy may be noted, which stains positively for glial fibrillary acid protein.[27] Cones are more affected than rod cells in all retinal locations, and the macula may show severe degeneration.[26]
History and Physical
Patients with STGD1 usually present with progressive, painless bilateral central vision loss. The presenting visual acuity ranges from 20/20 to 20/400. The other presenting symptoms include central scotoma, delayed dark adaptation, photosensitivity, photopsia, and abnormal color vision. Usually, both eyes are involved. Both males and females can be affected. The age of presentation is usually childhood, with a second peak at early adulthood.[2] The early-onset disease may cause severe vision loss compared to late-onset or adult-onset disease. A nonsense mutation usually causes the early-onset disease and severe loss of function of ABCA4, and late-onset "foveal-sparing" Stargardt disease (FS-STGD1) can be seen in patients with less reduction of ABCA4 function (missense mutation).[2]
There is a marked phenotypic heterogeneity of STGD1 due to a considerable number (more than 1200) of disease-causing mutations of varying severity and different types in ABCA4.[2][28] The phenotypes vary in age of onset, severity, visual acuity, multimodal imaging features, and electrophysiological characteristics. The typical triad associated with STGD1 includes flecks, macular atrophy, and peripapillary sparing.[29] Another crucial feature is jet-black dark choroid or silent choroid in the fluorescein angiogram.
Flecks are yellowish-white round or pisciform (fish-tail) shaped lesions of varying sizes located within the RPE. They tend to develop centrifugally from the macular region and may even progress beyond the vascular arcades to the mid-periphery but usually spare the far periphery. Around 30% of patients with childhood-onset disease may not show flecks, which may appear as the child grows.[30] This is a critical cause of misdiagnosis.
The macula is involved early in the disease course. The macular atrophy changes are usually progressive. However, its severity can be variable, ranging from a small region of central macular atrophy to atrophy beyond the vascular arcades. In a child with STGD1 with visual decline, the macular changes may be very subtle and missed on routine ophthalmoscopic examination. The severity of visual decline may not correlate with a normal-appearing retina. Multimodal imaging, including autofluorescence, optical coherence tomography, and electroretinography, helps to diagnose such cases. The macular manifestations include normal macula, vermillion fundus, abnormal foveal reflex, mild retinal pigment epithelial changes, pigmentary changes in the macula, foveal thinning, beaten bronze metal appearance, snail-slime appearance, bull's eye macula (BEM), dense pigmentation at the macula, macular atrophy, and even scleral show with very severe chorioretinal atrophy that may extend beyond the arcades.[8]
Peripapillary sparing (absence of flecks around the optic disc) is a diagnostic feature in STGD1 that is more prominently visible on fundus autofluorescence imaging. Pentosan polysulfate maculopathy, which is a close differential diagnosis of STGD1, may not spare the peripapillary region.[31]
BEM may be seen in STGD1. The differential diagnoses for BEM include hydroxychloroquine/chloroquine retinopathy, STGD1, cone dystrophy, cone-rod dystrophy, advanced dry age-related macular degeneration, central areolar choroidal dystrophy, benign concentric annular macular dystrophy, fenestrated sheen macular dystrophy, juvenile Batten disease, fucosidosis, ceroid lipofuscinosis, and clofazimine maculopathy. Flecks tend to appear later in eyes with this phenotype.
Chorioretinal degeneration is the most severe phenotype related to ABCA4 gene mutation. Choriocapillaris atrophy, widespread nummular pigmentary deposits, and bony spicule characterize ABCA4 gene mutation. Severe cases can show a visible sclera, vascular attenuation, and have peripapillary disease involvement. Florid retinitis pigmentosa with bony spicule pigmentation may be a severe form of ABCA4 retinopathy.[5]
Foveal-sparing Stargardt disease (FS-STGD1) is a milder phenotype that presents late and has an excellent visual prognosis with foveal sparing. The usual age of presentation is 35 to 45 years.[32] Such cases may be misdiagnosed as age-related macular degeneration. The central visual acuity is usually preserved (at least 20/40), though the rate of progression may vary due to the phenotypic variability associated with ABCA4.[33] On autofluorescence imaging, such patients have normal foveal autofluorescence surrounded by at least 180 degrees of retinal atrophy (hypoautofluorescence).[33] Factors leading to foveal sparing and relatively better prognosis include genetic factors (less severe missense mutation or hypomorphic mutation), environmental factors, the individual's cone density, and macular pigment density.[2]
Evaluation
The various imaging modalities have provided great insights into the anatomical and functional status of the retina. They are valuable for screening and early diagnosis of the disease. Lambertus et al reported that almost one-fourth of patients diagnosed with childhood-onset STGD1 had no clinically identifiable retinal lesions at presentation.[34] Patients can easily be misdiagnosed and subjected to multiple investigations like neuroimaging, lumbar punctures, and therapies like occlusion. Multimodal imaging can help in early and correct diagnosing, monitoring the disease progression, and evaluating the treatment modalities' efficacy. The emergence of novel ABCA4-specific molecular and genetic therapies can help patients with the early stages of the disease.
i) Color fundus photography (Figure 1. ) is helpful in the documentation and monitoring of the type of retinal changes and extent of the disease. The Fishman classification is based on color fundus photographs.[35] Stage 1 is characterized by macular pigmentary changes, which may range from faint or irregular pigment mottling to bronze-beaten appearance; a ring of pisciform flecks within 1 disc-diameter (DD) on all sides of the fovea; and a normal electroretinogram (ERG) and electrooculogram (EOG). The presence of pisciform flecks beyond 1 DD of the fovea and may extend beyond the vascular arcades and nasal to the optic nerve; a normal ERG and EOG except for occasional subnormal cone/rod response; and prolonged dark adaptation is representative of stage 2. Atrophic macular changes replacing the previously recognized flecks, subnormal EOG and ERG responses, and central and mid-peripheral and peripheral visual field defects characterize stage 3. Stage 4 occurs with extensive atrophy throughout the entire fundus, reduced ERG responses, and moderate to extensive peripheral field restriction.[35]
Noble and Carr classified STGD1 (Stargatdt disease and fundus flavimaculatus) based on fundus appearance into 4 groups:[36]
- Group 1: macular degeneration without flecks
- Group 2: perifoveal flecks with macular degeneration (Stargatdt disease)
- Group 3: diffuse flecks with macular degeneration (fundus flavimaculatus with macular degeneration)
- Group 4: diffuse flecks, absent macular degeneration (pure fundus flavimaculatus)
ii) Fundus fluorescein angiography:[14] The hallmark finding is the "dark choroid," which appears as a highlighted retinal blood vessel against a hypofluorescent choroid. The dark choroid is a diagnostic feature seen in at least 80% of patients. Results from a study found the "dark choroid sign" in 94% of patients with STGD1 and ABCA4 mutation[37] occurring due to blockage of background choroidal fluorescence—because of lipofuscin in the RPE cells. Flecks appear hypofluorescent due to the blocked transmission but may appear hyperfluorescent in the presence of atrophy. They can be seen on fundus fluorescin angiography (FFA) before they become clinically visible. The regions of RPE atrophy appear hyperfluorescent due to transmission defects, and a dark choroid ring may be seen.[37]
iii) Infrared imaging: Flecks typically appear hyperreflective; however, the resorbing flecks appear hyporeflective. The macular atrophy region also appears hyperreflective and is usually surrounded by a dark ring.[37]
iv) Optical coherence tomography: Optical coherence tomography (OCT) findings include thinning outer retinal layers and disruption of the EZ. The flecks appear as subretinal and intraretinal hyperreflective deposits.
Querques et al categorized flecks into 2 types.[38] While dome-shaped hyperreflective deposits characterize type 1 flecks on the inner part of the RPE, type 2 flecks represent small linear deposits in the ONL and spare the RPE. Voigt and colleagues have devised a newer classification describing 5 subtypes of flecks.[38] Class A flecks are seen as hyperreflective deposits at the level of the outer segment of the photoreceptors, RPE, interdigitation zone (IZ), and RPE-Brunch membrane complex. The other subtypes progress toward the inner retinal layers. Class B flecks reach the ELM, and Class C flecks break into the ONL. Class A to C represent the type 1 flecks per the previous classification. Class D flecks are only present within the ONL, while deposits in the outer retinal layers disappear; these only represent the type 2 flecks as per the previous classification. Class E flecks represent atypical drusen-like lesions.[38]
The other OCT changes noted previously to the fundoscopy changes are visible, including a subfoveal optical gap and thickening of the ELM or hyper-reflectivity at the base of ONL.[39][40][41] The disruption of the EZ and thinning of proximal ONL characterizes the subfoveal optical gap.
Noupuu et al described the progression of this optical gap in 3 stages:
- Stage 1 represents the mild disruption of the EZ in the foveal region
- Stage 2 represents additional photoreceptor loss
- Stage 3 represents the involvement of even the inner retinal layers[39]
Earliest changes are seen in the parafoveal region and not at the central fovea.[42] OCT appears more sensitive in detecting early lesions compared to fundus autofluorescence.[43]
Cai et al demonstrated that the total area of EZ loss gradually expanded at around 0.31 mm/year.[44] ELM thickening may represent an early sign in pediatric patients. While Burke et al proposed that ELM thickening could signify a Muller cell response,[40] Pang et al suggested that it could represent the migration and retraction of the inner segment of the photoreceptors.[41]
Multiple studies have used enhanced depth imaging (EDI) OCT and swept-source (SS)-OCT to study the choroidal thickness in STGD1 patients. Many studies have revealed that the choroid thickness reduces, affecting mainly the tiny choroidal vessels. Similarly, there is a significant reduction in the choroidal vascularity index. So far, there are 4 patterns of choroidal morphology described. Pattern 1 is characterized by a normal-appearing choroid, pattern 2 by a reduction of the thickness of either the Sattler or Haller layer, pattern 3 by a reduction of the thickness of both Sattler and Haller layers, and pattern 4 by a reduction of both Sattler and Haller layers with choroidal caverns. These patterns positively correlated with degenerative retinal changes on OCT and signs of progression.[45]
Other than thickness, hyperreflective foci in the choroid are also noted. Piri et al demonstrated that the number of hyperreflective foci in the Bruch membrane/RPE complex, choriocapillaris, and Sattler layer correlated with increasing disease severity in terms of central macular thickness, visual acuity, and duration of the disease.[46]
v) OCT-angiography: Study results have demonstrated reduced superficial and deep vessel densities and choriocapillaris vessel density attenuation. OCT-angiography may also help diagnose choroidal neovascular membrane (CNVM), which is a rare and late complication
vi) Fundus autofluorescence: Monitoring the disease progression uses both short-wavelength fundus autofluorescence (SW-FAF) and near infrared-wavelength fundus autofluorescence (NIR-FAF). Flecks are seen on SW-FAF as hyper AF (Figure 2. ), while they appear hypo AF on NIR-FAF. Flecks can be seen on NIR-FAF before they appear on SW-FAF. The atrophic areas show decreased AF on both types of FAF, with the area appearing larger on NIR-FAF than on SW-FAF, which may be due to lipofuscin released by the degenerating photoreceptors, producing a falsely typical image despite the presence of multiple atrophic RPE lesions. Hyper AF lesions on SW-FAF are usually the earliest features and may appear before flecks become visible. The RPE loss expands at a rate of 0.28 to 1.58 mm/year.[44]
Fujinami et al classified the AF appearance on SW-AF images into 3 types. Localized low foveal AF signal surrounded by a homogenous AF background characterizes type 1. Type 2 appears as localized low macular AF surrounded by a homogenous AF background with multiple areas of abnormal signal. Multiple areas of low AF signal at the posterior pole and a heterogenous background FAF are identifiable in type 3.[47] The rate of atrophy enlargement (RAE) in each of the 3 types was estimated to be 0.06, 0.67, and 4.37 mm2/year, respectively.[47]
Kuehlewein and colleagues have divided the lesions into definitely decreased AF (DDAF) and questionably decreased AF (QDAF). Lesions with darkness ≥ 90% of the darkness of the optic nerve head (ONH) are described in DDAF; QDAF has lesions with a darkness level of 50% to 90% of the ONH.[48]
The ProgStart study report number 5 revealed that around 50% of the eyes that did not present with DDAF developed the lesion in less than 5 years (median 4.93 years).[49] Klufas et al described a classification for the ultra-widefield (UWF) FAF.[50] Lesions isolated only to the macular region are in type 1, an atrophic macula, and peripheral flecks identify type 2, while type 3 appears as an atrophic macula with peripheral atrophy.[50]
Quantitative AF (qAF) was first described by Delori et al.[51] This classification is a standardized approach for assessing the number of lipofuscin levels in the retina. The qAF levels in STGD1 are raised, even without fundus abnormalities; therefore, the classification helps establish the diagnosis in borderline cases and monitor the efficacy of the various therapeutic interventions.
vii) Adaptive optics scanning laser ophthalmoscope: Observed are areas with increased spacing between the photoreceptor (both rods and cones). Findings from adaptive optics scanning laser ophthalmoscopy (AOSLO) suggest that photoreceptor degeneration is the primary pathology rather than an event secondary to RPE cell death. Song et al further demonstrated the centrifugal progression of the disease.[52]
viii) Visual field:[53] This test shows a progressive central scotoma. Loss of mid and peripheral VFs is usually associated with more severe disease. Schroeder et al classified the VF severity into 3 groups. Group 1 has a central scotoma within 10°, group 2 has central scotomas within 10° to 35°, and group 3 only has "temporal residues."[54]
ix) Microperimetry: The ProgStar study is a reliable investigation for detecting macular function in studies evaluating the various treatment modalities. The study results showed that measuring visual acuity is not a sensitive outcome for monitoring the disease in various treatment trials. VF testing can miss small scotomas. Patients with STGD1 show varying degrees of reduced sensitivity on microperimetry (MP), depending on the stage of the disease. Macular sensitivity is reduced in the areas with flecks and abnormal AF.
The ProgStar study results also found the mean sensitivity reduces by around 0.68 dB yearly.[55] The "Scotopic Microperimetric Assessment of Rod function in Stargardt Disease" (SMART) study, the ancillary study to ProgStart, demonstrated that the loss of scotopic macular function in the extrafoveal spots was faster than the loss of mesopic macular function.[56]
x) Visual electrophysiology: Lois and colleagues classified STGD1 cases into 3 groups based on their electrophysiological properties.[57]
- Group 1 shows severe abnormalities in pattern electroretinogram (PERG) with normal photopic and scotopic full-field electroretinogram (ffERG).
- Group 2 includes additional abnormalities (loss) in photopic ffERG (abnormal cone function).
- Group 3 shows loss of both photopic and scotopic full-field ERG response (abnormal cone and rod function).[57][58][57]
Fujinami and colleagues noted that patients with initial rod involvement in ERG had clinically significant electrophysiological deterioration with time (mean follow-up of around 10 years).[58] About 22% of group 1 patients showed clinically significant progression with time.[58] Multifocal ERG can detect foveal dysfunction, and the area of dysfunction may be larger than the area of morphological changes.[59] Electrooculogram is affected variably and is more involved in eyes with flecks (69%) than eyes without flecks (42.5%).[60]
xi) Genetic testing: Genetic testing is vital as various changes noted in the ABCA4 gene can cause a variety of phenotypes. Genetic testing also helps in genetic counseling, prognostication, and preconception planning.
Treatment / Management
Currently, no treatment modality is recommended by the FDA to prevent or reverse visual loss in patients with STGD1. Patients should use photoprotection to delay the disease progression and low-vision aids for those with visual field loss. Prescribe patients with proper refractive correction and advise patients to avoid smoking. Intravitreal anti-vascular endothelial growth factor (VEGF) injections are the preferred treatment modality for patients who develop CNVM. However, several ongoing clinical trials are investigating various novel therapeutic modalities for this condition.
Pharmacological Therapy
The investigation of several drugs that can reduce lipofuscin accumulation is currently underway. Reducing lipofuscin can be achieved by either inhibiting the retinoid cycle by inhibiting the key enzymes (emixustat and isotretinoin) or disrupting the transporters like retinol-binding protein 4 (RBP4) to decrease the toxic bisretinoid formation (fenretinide, A1120, BPN-14136, STG-001, LBS-008 or tinlarebant).[61] These drugs include the following:
- Emixustat hydrochloride (ACU-4429): inhibits the enzyme isomerase RPE65 (all-trans: 11-cis retinol isomerase) in a dose-dependent reversible manner. ACU-4429 prevents the isomerization of all-trans-retinyl ester to 11-cis-retinol, thus reducing the production of toxic by-products like A2E. A Phase 1a study demonstrated a dose-dependent reversible suppression (returned to the baseline by day 7) of the enzyme, which manifested as the slow recovery of the rod b-wave amplitude after photobleaching on ERG.[62] A Phase 1b study revealed that the drug was systematically safe but had few reversible ocular adverse effects like chromatopsia, blurred vision, and reduced visual acuity.[63] A phase 2 clinical trial studied the effect of 3 doses (2.5 mg, 5 mg, or 10 mg) of daily emixustat for 1 month. The results showed that 10 mg, 5mg, and 2.5 mg doses resulted in near-complete (mean = 91.86%, median = 96.69%), moderate (mean = 52.2%, median = 68.0%), and no suppression (mean = −3.31%, median= −12.23%) of the rod-derived b-wave amplitude recovery rate on ERG after photobleaching respectively.[64] (A1)
- ALK-001 (deuterated vitamin A): ALK-001 slows the rate of vitamin A dimerization, decelerating the rate of A2E and lipofuscin formation. Results from studies evaluating its efficacy in murine models have shown promising results with no adverse effects on retinal functions.[65] A phase 2 clinical trial evaluated the drug’s pharmacokinetics and 1-year interim safety in 50 subjects aged ≥ 18 suffering from ABCA4-related STGD1 and had a well-delineated area of atrophy. Results of the trial showed that it was well-tolerated, and there were no adverse reactions such as night blindness, impaired dark adaptation, and clinically significant derangement of liver function tests or serum vitamin A levels. An expanded study is underway to recruit new participants ≥ 12 years.
- Fenretinide (retinoid-based RBP4 antagonist): Fenretinide is a synthetic derivative of vitamin A and is a competitive inhibitor of retinol, as it prevents retinol binding to the retinal binding protein (RBP) in the eye. The RBP-fenretinide complex is excreted in the urine, thus reducing the circulating RBP levels; this significantly reduces the growth of geographic atrophy and the onset of CNVM in patients with dry age-related macular degeneration (ARMD). However, there are concerns regarding its long-term safety due to reports of teratogenicity and hemangiosarcomas formation in mice.[66] (A1)
- Vitamin A: Per the current recommendations, avoid vitamin A dietary supplements, as excess vitamin A is considered detrimental. However, these recommendations have a theoretical basis only. To date, there is little evidence to support or exclude the role of vitamin A in deteriorating the disease.[67]
Gene Therapy
The goal is to introduce a functional ABCA4 gene that can produce an adequate amount of the standard, active transporter protein in the photoreceptor cells, thus preventing disease progression. Adeno-associated viruses (AAV) deliver genes to the ocular tissues effectively. However, the AAV vector’s limited packaging capacity (4.7 kb) renders it insufficient to pack the large ABCA4 gene (7 kb). The help of a “dual-vector strategy,” ie, large genes split in half and packaged in 2 separate AAV vectors, can solve this problem.[68]
Recently developed lentivirus-based vectors deliver large genes. One such lentivirus-based vector is delivered subretinally.[69] The gene can be delivered via deoxyribonucleic acid (DNA) nanoparticles and DNA-encapsulating liposomes. An intravitreal or subretinal injection can accomplish delivery.[61](B3)
Stem-cell therapy: This therapy can treat the disease by regenerating the RPE cells.[61]
Genetic counseling: Genetic counseling is an integral part of managing this condition. Genes can help predict the prognosis and progression of the disease (ie, STGD1 is autosomal recessive, STGD3 and STGD4 are autosomal dominant).
Differential Diagnosis
Several inherited macular dystrophies have a clinical appearance similar to STGD1. Distinguishing STGD1 among these similar-appearing conditions is essential for prognostication, systemic associations, and genetic counseling.[70]
Pattern Dystrophy
Marmor and Byers were the first to propose “pattern dystrophy” when describing a spectrum of characteristic fundus patterns (ie, butterfly, reticular pigmentary). Pattern dystrophy (PD) most commonly masquerades as STGD1.[71] PD is an autosomal dominant condition with mild or no visual loss in the second to fifth decade. PD associated with the PRPH2 gene (multifocal pattern dystrophy simulating fundus flavimaculatus) primarily manifests as a macular disease. However, the phenotype may range from macular flecks to diffuse pigmentation in the periphery. Peripapillary sparing is a characterization of PD similar to STGD1. However, PD is associated with a higher incidence of CNVM. FFA helps distinguish the two as there will be window defects in PD, in contrast to a “masked choroid” in STGD1.[72]
Autosomal Dominant Stargardt-like Macular Dystrophies
These dystrophies include variants associated with the ELOVL4 (STGD2 or STGD3) and the PROM1 (STGD4) genes.[70]
Batten Disease/Juvenile Neuronal Ceroid Lipofuscinosis
Defects in the processing of lysosomal storage characterize Batten disease/juvenile neuronal ceroid lipofuscinosis, a rare metabolic disease. These defects lead to apoptosis of both the photoreceptor (retina) and the neuronal (brain) cells. Visual loss and severe color vision deficiency are common, and the life expectancy of patients with this condition is usually 20 to 30 years.
The disease progression is usually rapid. Ocular examination shows vascular attenuation and optic disc pallor. OCT reveals a thinned retinal nerve fiber layer, while dark-adapted full-field ERG shows an absent or electronegative wave. Maintain high suspicion in younger patients diagnosed with severe STGD1 with rapid progression.[73]
Maternally Inherited Diabetes and Deafness or Other Mitochondrial Diseases (Including Mitochondrial Encephalomyopathy, Lactic Acidosis, and Stroke-Like Episodes)
Patients with maternally inherited diabetes and deafness (MIDD) or other mitochondrial diseases present in the third to seventh decade. Ocular examination shows pigmentary changes at the macula, areas of RPE atrophy, and fleck-like deposits.[74] FAF shows hyperautofluorescent spots with corresponding subretinal deposits on OCT. Patients with this condition have distinguishing features such as personal or family history of early-onset diabetes mellitus and hearing loss. Other distinguishing features include the absence of a dark choroid on FFA and the presence of outer retinal tubulations on OCT.[75]
Age-Related Macular Degeneration
This condition can mimic late-onset STGD1, as the flecks may be mistaken for drusens. A careful examination can help distinguish the two, as drusen tend to become confluent toward the center of the macula. Flecks show intense hyper AF, while drusens may be mildly hyperautofluorescent or hypoautofluorescent.[76] Sub-RPE accumulation on OCT characterizes drusens, while STGD1 is associated with hyperreflective thickening of the RPE. Age-related macular degeneration does not show a dark choroid on FFA.
Pentosan Polysulfate Maculopathy
This condition is associated with the intake of pentosan, used in treating interstitial cystitis. The FAF changes may not spare the peripapillary region in pentosan polysulfate maculopathy.[31]
Prognosis
Many patients with this condition progress to legal blindness from losing central vision, although peripheral vision tends to remain preserved. The prognosis is widely variable and is highly dependent on the age of onset of the disease; therefore, patients presenting with early onset have poor outcomes. Visual acuity in such patients declines rapidly, along with the appearance of progressive retinal degeneration—and can even lead to profound chorioretinal atrophy. Thus, patients with this phenotype have a severe disease with a poor prognosis.[34]
Rapid-onset chorioretinopathy (ROC) is a distinct sub-phenotype characterized by rapid disease progression.[7] Patients with late-onset disease maintain good visual acuity, even at an older age. The disease does not impact the patient’s general health or life expectancy.
Complications
The complications associated with STGD1 include the following:
Subretinal fibrosis: Minor ocular trauma can lead to subretinal fibrosis and RPE hypertrophy.[77][78] Grandinetti et al proposed that ocular trauma leads to the release of lipofuscin, which disrupts the RPE cells, leading to subretinal fibrosis and RPE hypertrophy.[77] (Gass et al presented a similar hypothesis.)[78] Rossi et al proposed that even microtrauma can lead to subretinal fibrosis as they found patients developing subretinal fibrosis without a distinct history of ocular trauma or contact sports.[79] Patients with STGD1 are advised to use eye protection while playing competitive sports.[78][79]
Choroidal neovascular membrane: This rare condition is a late complication of STGD1.
Legal blindness: Many patients develop legal blindness with age.
Deterrence and Patient Education
Patient education and counseling are critical for the management of this disease. Counsel patients about their mental state and explain that the disease is progressive and their declining vision may begin to interfere with performing daily activities.[80] Inform patients about low-vision aids that can improve their ability to perform daily activities. Patients should have an ophthalmological screening and genetic counseling, helping them understand the chances of their offspring developing the same disease.[81]
Enhancing Healthcare Team Outcomes
Stargardt disease is one of the common causes of visual loss in young patients. It is an autosomal recessive disease caused by mutations in the ABCA4 gene. Several patients initially seek the opinion of an optometrist for reduced vision. However, due to myriad presentations, the diagnosis may be delayed. As a result, the patients may have multiple investigations like neuroimaging, lumbar punctures, and therapies like occlusion.[2]
Pediatricians and other clinicians are essential in appropriate referral and parent/patient education. Clinicians should know how to perform and interpret various imaging modalities, which will help establish an early diagnosis. Multimodal imaging also helps monitor the disease progression and evaluate the efficacy of the treatment modalities. Several ongoing clinical trials are investigating novel therapies for this condition, as there currently are no approved treatment modalities to prevent or reverse visual loss in patients with this condition. Few patients may have systemic associations; therefore, improving patient outcomes requires a multidisciplinary approach.[2]
Media
(Click Image to Enlarge)
Short-Wavelength Fundus Autofluorescence. Short-wavelength fundus autofluorescence of the right eye showing a hypoautofluorescence in the center of the macula surrounded by multiple flecks of hyperautofluorescence.
Contributed by Sourav Damodaran, MD
(Click Image to Enlarge)

Fundus With Bronze-Beaten Appearance. Fundus photograph of the right eye showing a bronze-beaten appearance at the macula surrounded by multiple flecks.
Contributed by Sourav Damodaran, MD
(Click Image to Enlarge)

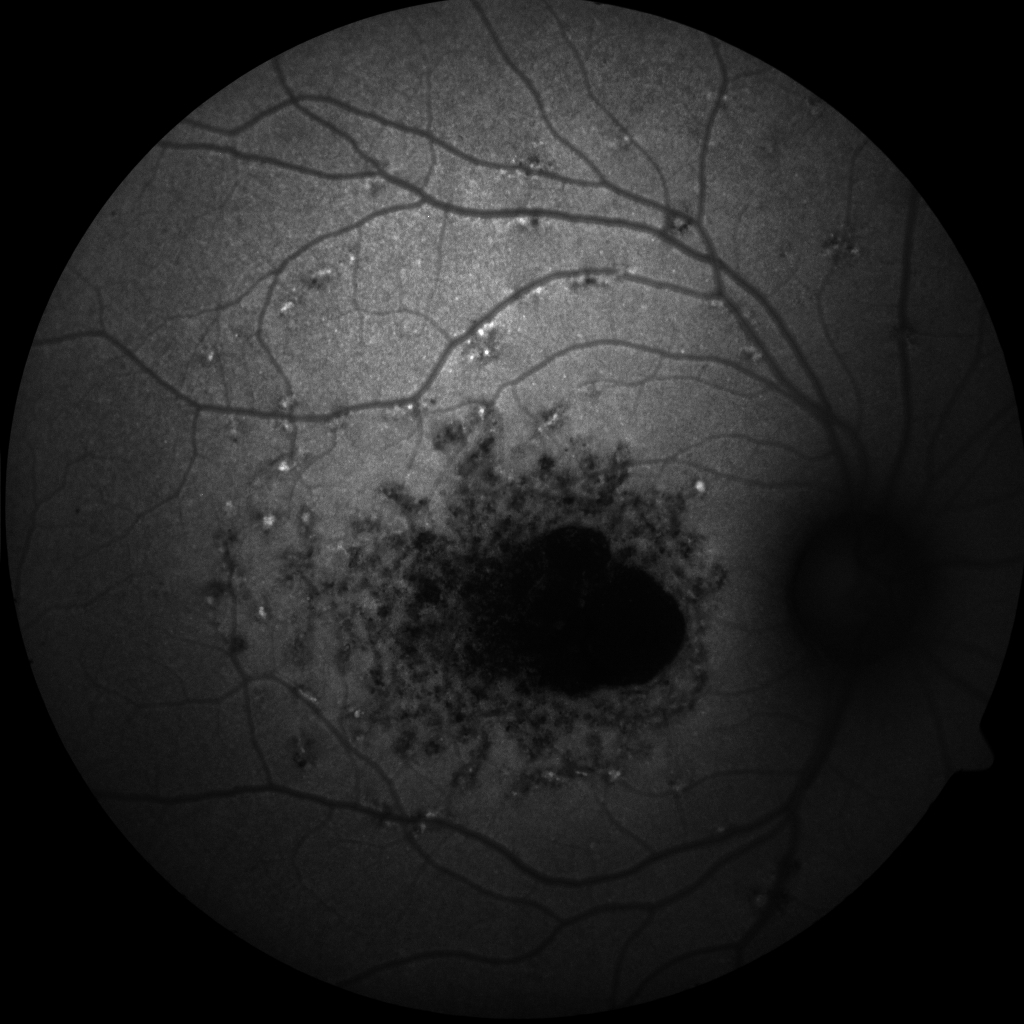
Stargardt Disease. Flecks and macular atrophy in a patient with autosomal recessive Stargardt disease.
Contributed by K Tripathy, MD
(Click Image to Enlarge)
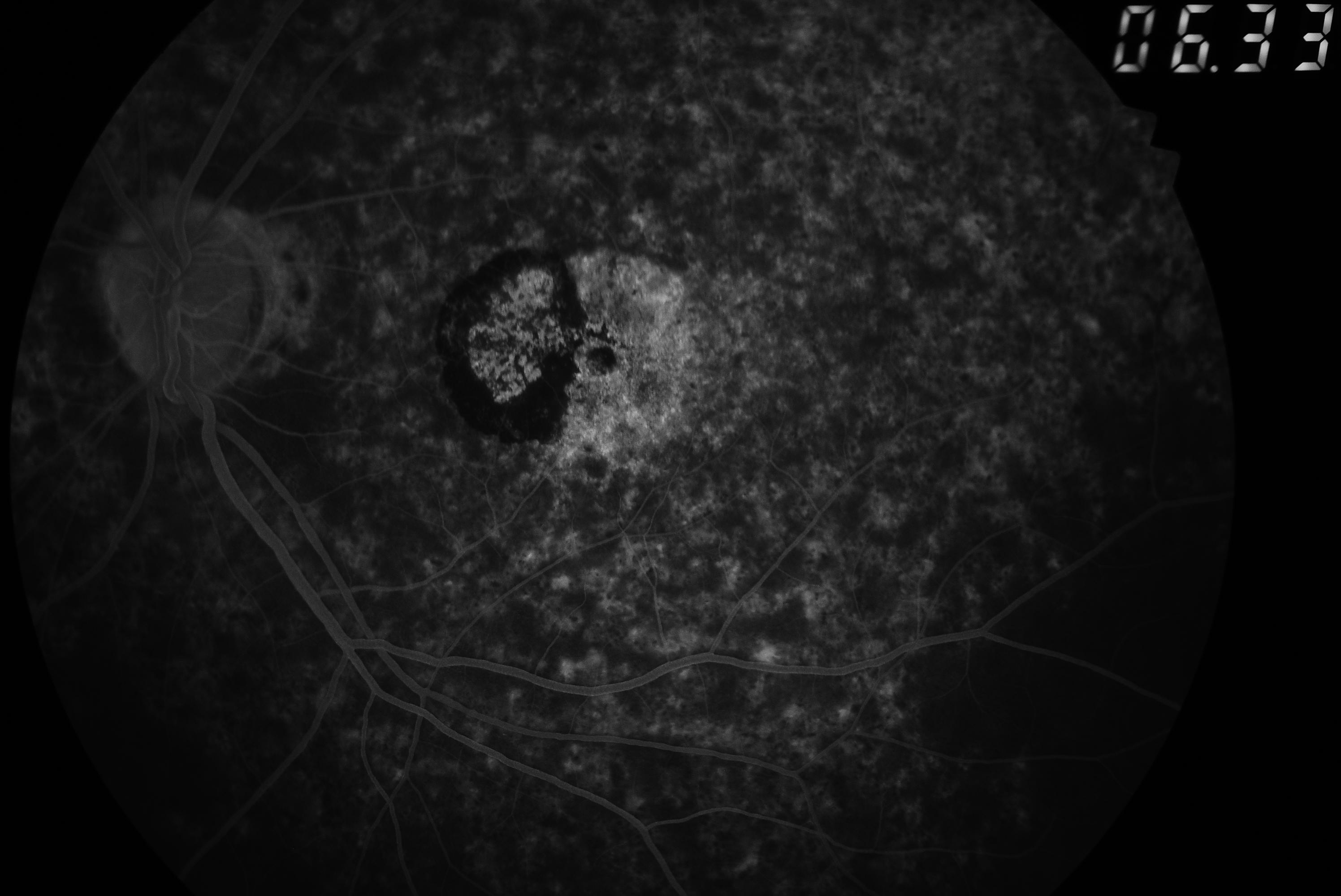
Silent Choroid in Fluorescein Angiogram. Silent choroid is evidenced in the angiogram of this patient with Stargardt disease specifically at the right lower border of the image. The flecks are causing areas of hyper-fluorescence.
Contributed by Koushik Tripathy, MD
(Click Image to Enlarge)
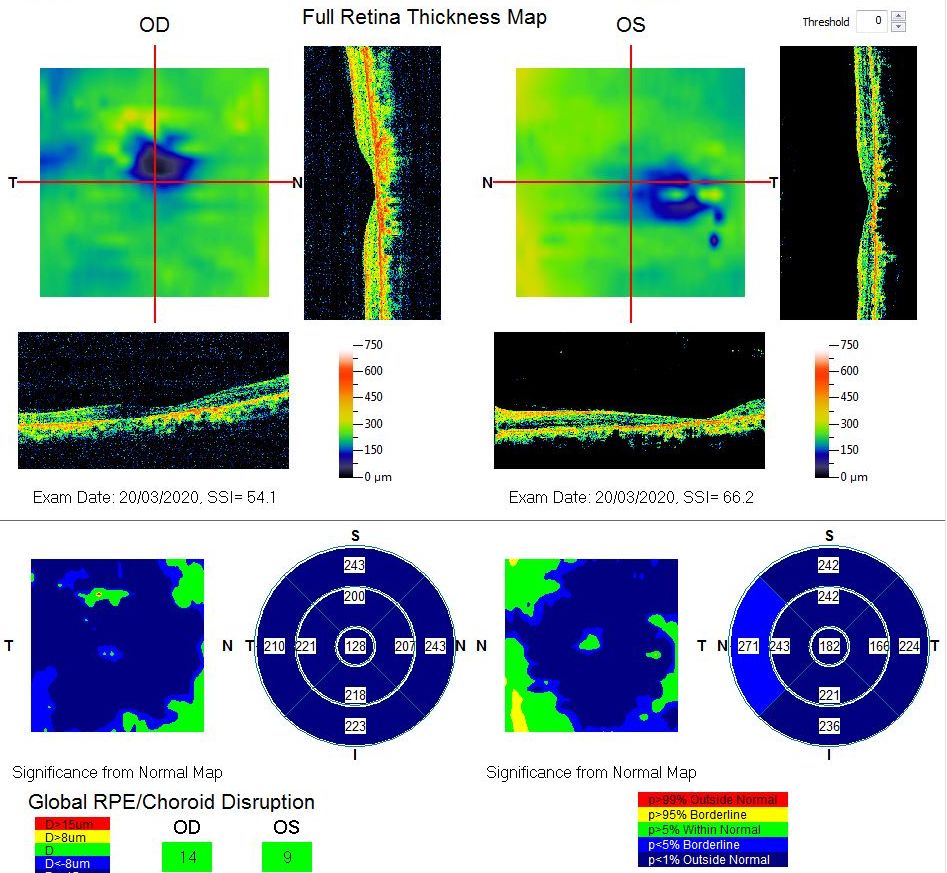
Macular Thickness Map. This map of a patient with Stargardt disease shows foveal thinning. The vertical scan in the left eye is not centered on the fovea (and the calculated central macular thickness is incorrect) due to poor vision, for which fixation may be poor.
Contributed by Koushik Tripathy, MD
(Click Image to Enlarge)
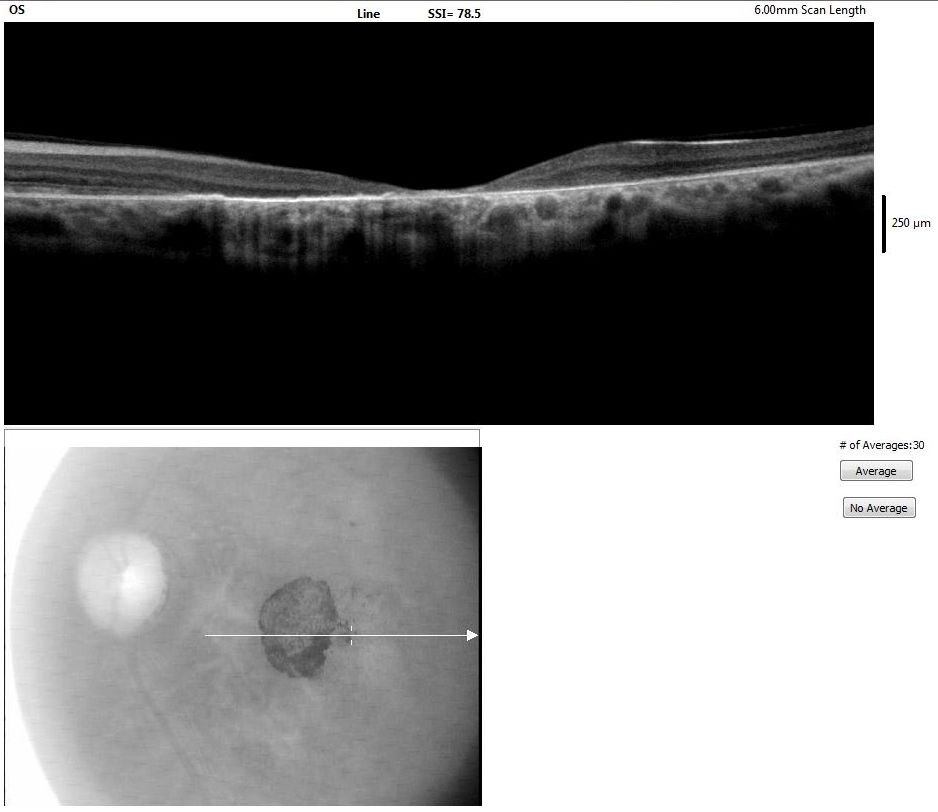
Optical Coherence Tomography of the Macula. Tomography image of the macula in a patient with advanced Stargardt disease showing foveal thinning.
Contributed by K Tripathy, MD
References
Strauss RW, Ho A, Muñoz B, Cideciyan AV, Sahel JA, Sunness JS, Birch DG, Bernstein PS, Michaelides M, Traboulsi EI, Zrenner E, Sadda S, Ervin AM, West S, Scholl HP, Progression of Stargardt Disease Study Group. The Natural History of the Progression of Atrophy Secondary to Stargardt Disease (ProgStar) Studies: Design and Baseline Characteristics: ProgStar Report No. 1. Ophthalmology. 2016 Apr:123(4):817-28. doi: 10.1016/j.ophtha.2015.12.009. Epub 2016 Jan 16 [PubMed PMID: 26786511]
Tanna P, Strauss RW, Fujinami K, Michaelides M. Stargardt disease: clinical features, molecular genetics, animal models and therapeutic options. The British journal of ophthalmology. 2017 Jan:101(1):25-30. doi: 10.1136/bjophthalmol-2016-308823. Epub 2016 Aug 4 [PubMed PMID: 27491360]
Level 3 (low-level) evidenceAl-Khuzaei S, Shah M, Foster CR, Yu J, Broadgate S, Halford S, Downes SM. The role of multimodal imaging and vision function testing in ABCA4-related retinopathies and their relevance to future therapeutic interventions. Therapeutic advances in ophthalmology. 2021 Jan-Dec:13():25158414211056384. doi: 10.1177/25158414211056384. Epub 2021 Dec 19 [PubMed PMID: 34988368]
Level 3 (low-level) evidenceGlazer LC, Dryja TP. Understanding the etiology of Stargardt's disease. Ophthalmology clinics of North America. 2002 Mar:15(1):93-100, viii [PubMed PMID: 12064087]
Level 3 (low-level) evidenceSheffield VC, Stone EM. Genomics and the eye. The New England journal of medicine. 2011 May 19:364(20):1932-42. doi: 10.1056/NEJMra1012354. Epub [PubMed PMID: 21591945]
Al-Khuzaei S, Broadgate S, Foster CR, Shah M, Yu J, Downes SM, Halford S. An Overview of the Genetics of ABCA4 Retinopathies, an Evolving Story. Genes. 2021 Aug 13:12(8):. doi: 10.3390/genes12081241. Epub 2021 Aug 13 [PubMed PMID: 34440414]
Level 3 (low-level) evidenceTanaka K, Lee W, Zernant J, Schuerch K, Ciccone L, Tsang SH, Sparrow JR, Allikmets R. The Rapid-Onset Chorioretinopathy Phenotype of ABCA4 Disease. Ophthalmology. 2018 Jan:125(1):89-99. doi: 10.1016/j.ophtha.2017.07.019. Epub 2017 Sep 22 [PubMed PMID: 28947085]
Lee W, Zernant J, Nagasaki T, Tsang SH, Allikmets R. Deep Scleral Exposure: A Degenerative Outcome of End-Stage Stargardt Disease. American journal of ophthalmology. 2018 Nov:195():16-25. doi: 10.1016/j.ajo.2018.07.018. Epub 2018 Jul 26 [PubMed PMID: 30055151]
Stone EM, Nichols BE, Kimura AE, Weingeist TA, Drack A, Sheffield VC. Clinical features of a Stargardt-like dominant progressive macular dystrophy with genetic linkage to chromosome 6q. Archives of ophthalmology (Chicago, Ill. : 1960). 1994 Jun:112(6):765-72 [PubMed PMID: 8002834]
Kniazeva M, Chiang MF, Morgan B, Anduze AL, Zack DJ, Han M, Zhang K. A new locus for autosomal dominant stargardt-like disease maps to chromosome 4. American journal of human genetics. 1999 May:64(5):1394-9 [PubMed PMID: 10205271]
Zhang K, Bither PP, Park R, Donoso LA, Seidman JG, Seidman CE. A dominant Stargardt's macular dystrophy locus maps to chromosome 13q34. Archives of ophthalmology (Chicago, Ill. : 1960). 1994 Jun:112(6):759-64 [PubMed PMID: 8002833]
Zhang K, Kniazeva M, Han M, Li W, Yu Z, Yang Z, Li Y, Metzker ML, Allikmets R, Zack DJ, Kakuk LE, Lagali PS, Wong PW, MacDonald IM, Sieving PA, Figueroa DJ, Austin CP, Gould RJ, Ayyagari R, Petrukhin K. A 5-bp deletion in ELOVL4 is associated with two related forms of autosomal dominant macular dystrophy. Nature genetics. 2001 Jan:27(1):89-93 [PubMed PMID: 11138005]
Level 3 (low-level) evidenceGriesinger IB, Sieving PA, Ayyagari R. Autosomal dominant macular atrophy at 6q14 excludes CORD7 and MCDR1/PBCRA loci. Investigative ophthalmology & visual science. 2000 Jan:41(1):248-55 [PubMed PMID: 10634627]
Ruia S, Tripathy K. Fluorescein Angiography. StatPearls. 2024 Jan:(): [PubMed PMID: 35015403]
Imani S, Cheng J, Shasaltaneh MD, Wei C, Yang L, Fu S, Zou H, Khan MA, Zhang X, Chen H, Zhang D, Duan C, Lv H, Li Y, Chen R, Fu J. Genetic identification and molecular modeling characterization reveal a novel PROM1 mutation in Stargardt4-like macular dystrophy. Oncotarget. 2018 Jan 2:9(1):122-141. doi: 10.18632/oncotarget.22343. Epub 2017 Nov 9 [PubMed PMID: 29416601]
Lee W, Paavo M, Zernant J, Stong N, Laurente Z, Bearelly S, Nagasaki T, Tsang SH, Goldstein DB, Allikmets R. Modification of the PROM1 disease phenotype by a mutation in ABCA4. Ophthalmic genetics. 2019 Aug:40(4):369-375. doi: 10.1080/13816810.2019.1660382. Epub 2019 Sep 6 [PubMed PMID: 31576780]
Sajovic J, Meglič A, Volk M, Maver A, Jarc-Vidmar M, Hawlina M, Fakin A. Stargardt-like Clinical Characteristics and Disease Course Associated with Variants in the WDR19 Gene. Genes. 2023 Jan 22:14(2):. doi: 10.3390/genes14020291. Epub 2023 Jan 22 [PubMed PMID: 36833218]
Spiteri Cornish K, Ho J, Downes S, Scott NW, Bainbridge J, Lois N. The Epidemiology of Stargardt Disease in the United Kingdom. Ophthalmology. Retina. 2017 Nov-Dec:1(6):508-513. doi: 10.1016/j.oret.2017.03.001. Epub 2017 Apr 21 [PubMed PMID: 31047443]
Runhart EH, Dhooge P, Meester-Smoor M, Pas J, Pott JWR, van Leeuwen R, Kroes HY, Bergen AA, de Jong-Hesse Y, Thiadens AA, van Schooneveld MJ, van Genderen M, Boon C, Klaver C, van den Born LI, Cremers FPM, Hoyng CB. Stargardt disease: monitoring incidence and diagnostic trends in the Netherlands using a nationwide disease registry. Acta ophthalmologica. 2022 Jun:100(4):395-402. doi: 10.1111/aos.14996. Epub 2021 Aug 25 [PubMed PMID: 34431609]
Molday RS, Zhong M, Quazi F. The role of the photoreceptor ABC transporter ABCA4 in lipid transport and Stargardt macular degeneration. Biochimica et biophysica acta. 2009 Jul:1791(7):573-83. doi: 10.1016/j.bbalip.2009.02.004. Epub 2009 Feb 20 [PubMed PMID: 19230850]
Level 3 (low-level) evidenceDelori FC, Dorey CK, Staurenghi G, Arend O, Goger DG, Weiter JJ. In vivo fluorescence of the ocular fundus exhibits retinal pigment epithelium lipofuscin characteristics. Investigative ophthalmology & visual science. 1995 Mar:36(3):718-29 [PubMed PMID: 7890502]
Delori FC, Staurenghi G, Arend O, Dorey CK, Goger DG, Weiter JJ. In vivo measurement of lipofuscin in Stargardt's disease--Fundus flavimaculatus. Investigative ophthalmology & visual science. 1995 Oct:36(11):2327-31 [PubMed PMID: 7558729]
Eldred GE, Lasky MR. Retinal age pigments generated by self-assembling lysosomotropic detergents. Nature. 1993 Feb 25:361(6414):724-6 [PubMed PMID: 8441466]
Westerfeld C, Mukai S. Stargardt's disease and the ABCR gene. Seminars in ophthalmology. 2008 Jan-Feb:23(1):59-65. doi: 10.1080/08820530701745249. Epub [PubMed PMID: 18214793]
Sparrow JR, Marsiglia M, Allikmets R, Tsang S, Lee W, Duncker T, Zernant J. Flecks in Recessive Stargardt Disease: Short-Wavelength Autofluorescence, Near-Infrared Autofluorescence, and Optical Coherence Tomography. Investigative ophthalmology & visual science. 2015 Jul:56(8):5029-39. doi: 10.1167/iovs.15-16763. Epub [PubMed PMID: 26230768]
Bonilha VL, Rayborn ME, Bell BA, Marino MJ, Fishman GA, Hollyfield JG. Retinal Histopathology in Eyes from a Patient with Stargardt disease caused by Compound Heterozygous ABCA4 Mutations. Ophthalmic genetics. 2016 Jun:37(2):150-60. doi: 10.3109/13816810.2014.958861. Epub 2014 Sep 29 [PubMed PMID: 25265374]
Birnbach CD, Järveläinen M, Possin DE, Milam AH. Histopathology and immunocytochemistry of the neurosensory retina in fundus flavimaculatus. Ophthalmology. 1994 Jul:101(7):1211-9 [PubMed PMID: 8035984]
Level 3 (low-level) evidenceCremers FPM, Lee W, Collin RWJ, Allikmets R. Clinical spectrum, genetic complexity and therapeutic approaches for retinal disease caused by ABCA4 mutations. Progress in retinal and eye research. 2020 Nov:79():100861. doi: 10.1016/j.preteyeres.2020.100861. Epub 2020 Apr 9 [PubMed PMID: 32278709]
Tripathy K, Sharma YR, Chawla R, Basu K, Vohra R, Venkatesh P. Triads in Ophthalmology: A Comprehensive Review. Seminars in ophthalmology. 2017:32(2):237-250. doi: 10.3109/08820538.2015.1045150. Epub 2015 Jul 6 [PubMed PMID: 26148300]
Fujinami K, Zernant J, Chana RK, Wright GA, Tsunoda K, Ozawa Y, Tsubota K, Robson AG, Holder GE, Allikmets R, Michaelides M, Moore AT. Clinical and molecular characteristics of childhood-onset Stargardt disease. Ophthalmology. 2015 Feb:122(2):326-34. doi: 10.1016/j.ophtha.2014.08.012. Epub 2014 Oct 12 [PubMed PMID: 25312043]
Level 2 (mid-level) evidenceMargines JB, Tripathy K, Hobbs SD. Pentosan Polysulfate Maculopathy. StatPearls. 2025 Jan:(): [PubMed PMID: 36944010]
Heath Jeffery RC, Chen FK. Stargardt disease: Multimodal imaging: A review. Clinical & experimental ophthalmology. 2021 Jul:49(5):498-515. doi: 10.1111/ceo.13947. Epub 2021 Jun 1 [PubMed PMID: 34013643]
van Huet RA, Bax NM, Westeneng-Van Haaften SC, Muhamad M, Zonneveld-Vrieling MN, Hoefsloot LH, Cremers FP, Boon CJ, Klevering BJ, Hoyng CB. Foveal sparing in Stargardt disease. Investigative ophthalmology & visual science. 2014 Oct 16:55(11):7467-78. doi: 10.1167/iovs.13-13825. Epub 2014 Oct 16 [PubMed PMID: 25324290]
Lambertus S, van Huet RA, Bax NM, Hoefsloot LH, Cremers FP, Boon CJ, Klevering BJ, Hoyng CB. Early-onset stargardt disease: phenotypic and genotypic characteristics. Ophthalmology. 2015 Feb:122(2):335-44. doi: 10.1016/j.ophtha.2014.08.032. Epub 2014 Oct 17 [PubMed PMID: 25444351]
Level 2 (mid-level) evidenceFishman GA. Fundus flavimaculatus. A clinical classification. Archives of ophthalmology (Chicago, Ill. : 1960). 1976 Dec:94(12):2061-7 [PubMed PMID: 999551]
Noble KG, Carr RE. Stargardt's disease and fundus flavimaculatus. Archives of ophthalmology (Chicago, Ill. : 1960). 1979 Jul:97(7):1281-5 [PubMed PMID: 454263]
Jayasundera T, Rhoades W, Branham K, Niziol LM, Musch DC, Heckenlively JR. Peripapillary dark choroid ring as a helpful diagnostic sign in advanced stargardt disease. American journal of ophthalmology. 2010 Apr:149(4):656-660.e2. doi: 10.1016/j.ajo.2009.11.005. Epub 2010 Feb 6 [PubMed PMID: 20138608]
Level 2 (mid-level) evidenceQuerques G, Leveziel N, Benhamou N, Voigt M, Soubrane G, Souied EH. Analysis of retinal flecks in fundus flavimaculatus using optical coherence tomography. The British journal of ophthalmology. 2006 Sep:90(9):1157-62 [PubMed PMID: 16754647]
Nõupuu K, Lee W, Zernant J, Tsang SH, Allikmets R. Structural and genetic assessment of the ABCA4-associated optical gap phenotype. Investigative ophthalmology & visual science. 2014 Oct 9:55(11):7217-26. doi: 10.1167/iovs.14-14674. Epub 2014 Oct 9 [PubMed PMID: 25301883]
Level 2 (mid-level) evidenceBurke TR, Yzer S, Zernant J, Smith RT, Tsang SH, Allikmets R. Abnormality in the external limiting membrane in early Stargardt disease. Ophthalmic genetics. 2013 Mar-Jun:34(1-2):75-7. doi: 10.3109/13816810.2012.707271. Epub 2012 Aug 7 [PubMed PMID: 22871184]
Level 3 (low-level) evidencePang CE, Suqin Y, Sherman J, Freund KB. New insights into Stargardt disease with multimodal imaging. Ophthalmic surgery, lasers & imaging retina. 2015 Feb:46(2):257-61. doi: 10.3928/23258160-20150213-09. Epub [PubMed PMID: 25707054]
Level 3 (low-level) evidenceKhan KN, Kasilian M, Mahroo OAR, Tanna P, Kalitzeos A, Robson AG, Tsunoda K, Iwata T, Moore AT, Fujinami K, Michaelides M. Early Patterns of Macular Degeneration in ABCA4-Associated Retinopathy. Ophthalmology. 2018 May:125(5):735-746. doi: 10.1016/j.ophtha.2017.11.020. Epub 2018 Jan 6 [PubMed PMID: 29310964]
Georgiou M, Kane T, Tanna P, Bouzia Z, Singh N, Kalitzeos A, Strauss RW, Fujinami K, Michaelides M. Prospective Cohort Study of Childhood-Onset Stargardt Disease: Fundus Autofluorescence Imaging, Progression, Comparison with Adult-Onset Disease, and Disease Symmetry. American journal of ophthalmology. 2020 Mar:211():159-175. doi: 10.1016/j.ajo.2019.11.008. Epub 2019 Dec 6 [PubMed PMID: 31812472]
Arrigo A, Grazioli A, Romano F, Aragona E, Bordato A, di Nunzio C, Sperti A, Bandello F, Battaglia Parodi M. Choroidal Patterns in Stargardt Disease: Correlations with Visual Acuity and Disease Progression. Journal of clinical medicine. 2019 Sep 5:8(9):. doi: 10.3390/jcm8091388. Epub 2019 Sep 5 [PubMed PMID: 31491905]
Cai CX, Light JG, Handa JT. Quantifying the Rate of Ellipsoid Zone Loss in Stargardt Disease. American journal of ophthalmology. 2018 Feb:186():1-9. doi: 10.1016/j.ajo.2017.10.032. Epub 2017 Nov 7 [PubMed PMID: 29126757]
Piri N, Nesmith BL, Schaal S. Choroidal hyperreflective foci in Stargardt disease shown by spectral-domain optical coherence tomography imaging: correlation with disease severity. JAMA ophthalmology. 2015 Apr:133(4):398-405. doi: 10.1001/jamaophthalmol.2014.5604. Epub [PubMed PMID: 25590640]
Level 2 (mid-level) evidenceFujinami K, Lois N, Mukherjee R, McBain VA, Tsunoda K, Tsubota K, Stone EM, Fitzke FW, Bunce C, Moore AT, Webster AR, Michaelides M. A longitudinal study of Stargardt disease: quantitative assessment of fundus autofluorescence, progression, and genotype correlations. Investigative ophthalmology & visual science. 2013 Dec 17:54(13):8181-90. doi: 10.1167/iovs.13-12104. Epub 2013 Dec 17 [PubMed PMID: 24265018]
Kuehlewein L, Hariri AH, Ho A, Dustin L, Wolfson Y, Strauss RW, Scholl HP, Sadda SR. COMPARISON OF MANUAL AND SEMIAUTOMATED FUNDUS AUTOFLUORESCENCE ANALYSIS OF MACULAR ATROPHY IN STARGARDT DISEASE PHENOTYPE. Retina (Philadelphia, Pa.). 2016 Jun:36(6):1216-21. doi: 10.1097/IAE.0000000000000870. Epub [PubMed PMID: 26583307]
Strauss RW, Muñoz B, Ho A, Jha A, Michaelides M, Mohand-Said S, Cideciyan AV, Birch D, Hariri AH, Nittala MG, Sadda S, Scholl HPN, ProgStar Study Group. Incidence of Atrophic Lesions in Stargardt Disease in the Progression of Atrophy Secondary to Stargardt Disease (ProgStar) Study: Report No. 5. JAMA ophthalmology. 2017 Jul 1:135(7):687-695. doi: 10.1001/jamaophthalmol.2017.1121. Epub [PubMed PMID: 28542697]
Klufas MA, Tsui I, Sadda SR, Hosseini H, Schwartz SD. ULTRAWIDEFIELD AUTOFLUORESENCE IN ABCA4 STARGARDT DISEASE. Retina (Philadelphia, Pa.). 2018 Feb:38(2):403-415. doi: 10.1097/IAE.0000000000001567. Epub [PubMed PMID: 28248825]
Delori F, Greenberg JP, Woods RL, Fischer J, Duncker T, Sparrow J, Smith RT. Quantitative measurements of autofluorescence with the scanning laser ophthalmoscope. Investigative ophthalmology & visual science. 2011 Dec 9:52(13):9379-90. doi: 10.1167/iovs.11-8319. Epub 2011 Dec 9 [PubMed PMID: 22016060]
Song H, Rossi EA, Latchney L, Bessette A, Stone E, Hunter JJ, Williams DR, Chung M. Cone and rod loss in Stargardt disease revealed by adaptive optics scanning light ophthalmoscopy. JAMA ophthalmology. 2015 Oct:133(10):1198-203. doi: 10.1001/jamaophthalmol.2015.2443. Epub [PubMed PMID: 26247787]
Ruia S, Tripathy K. Humphrey Visual Field. StatPearls. 2025 Jan:(): [PubMed PMID: 36256759]
Schroeder M, Kjellström U. Full-field ERG as a predictor of the natural course of ABCA4-associated retinal degenerations. Molecular vision. 2018:24():1-16 [PubMed PMID: 29386879]
Schönbach EM, Wolfson Y, Strauss RW, Ibrahim MA, Kong X, Muñoz B, Birch DG, Cideciyan AV, Hahn GA, Nittala M, Sunness JS, Sadda SR, West SK, Scholl HPN, ProgStar Study Group. Macular Sensitivity Measured With Microperimetry in Stargardt Disease in the Progression of Atrophy Secondary to Stargardt Disease (ProgStar) Study: Report No. 7. JAMA ophthalmology. 2017 Jul 1:135(7):696-703. doi: 10.1001/jamaophthalmol.2017.1162. Epub [PubMed PMID: 28542693]
Kong X, Ibrahim-Ahmed M, Bittencourt MG, Strauss RW, Birch DG, Cideciyan AV, Ervin AM, Ho A, Sunness JS, Audo IS, Michaelides M, Zrenner E, Sadda S, Ip MS, West S, Scholl HPN, SMART Study Group. Longitudinal Changes in Scotopic and Mesopic Macular Function as Assessed with Microperimetry in Patients With Stargardt Disease: SMART Study Report No. 2. American journal of ophthalmology. 2022 Apr:236():32-44. doi: 10.1016/j.ajo.2021.10.014. Epub 2021 Oct 23 [PubMed PMID: 34695402]
Lois N, Holder GE, Bunce C, Fitzke FW, Bird AC. Phenotypic subtypes of Stargardt macular dystrophy-fundus flavimaculatus. Archives of ophthalmology (Chicago, Ill. : 1960). 2001 Mar:119(3):359-69 [PubMed PMID: 11231769]
Level 2 (mid-level) evidenceFujinami K, Lois N, Davidson AE, Mackay DS, Hogg CR, Stone EM, Tsunoda K, Tsubota K, Bunce C, Robson AG, Moore AT, Webster AR, Holder GE, Michaelides M. A longitudinal study of stargardt disease: clinical and electrophysiologic assessment, progression, and genotype correlations. American journal of ophthalmology. 2013 Jun:155(6):1075-1088.e13. doi: 10.1016/j.ajo.2013.01.018. Epub 2013 Mar 15 [PubMed PMID: 23499370]
Level 2 (mid-level) evidenceKretschmann U, Seeliger MW, Ruether K, Usui T, Apfelstedt-Sylla E, Zrenner E. Multifocal electroretinography in patients with Stargardt's macular dystrophy. The British journal of ophthalmology. 1998 Mar:82(3):267-75 [PubMed PMID: 9602623]
Level 2 (mid-level) evidenceStavrou P, Good PA, Misson GP, Kritzinger EE. Electrophysiological findings in Stargardt's-fundus flavimaculatus disease. Eye (London, England). 1998:12 ( Pt 6)():953-8 [PubMed PMID: 10325994]
Level 2 (mid-level) evidenceHuang D, Heath Jeffery RC, Aung-Htut MT, McLenachan S, Fletcher S, Wilton SD, Chen FK. Stargardt disease and progress in therapeutic strategies. Ophthalmic genetics. 2022 Feb:43(1):1-26. doi: 10.1080/13816810.2021.1966053. Epub 2021 Aug 29 [PubMed PMID: 34455905]
Kubota R, Boman NL, David R, Mallikaarjun S, Patil S, Birch D. Safety and effect on rod function of ACU-4429, a novel small-molecule visual cycle modulator. Retina (Philadelphia, Pa.). 2012 Jan:32(1):183-8. doi: 10.1097/IAE.0b013e318217369e. Epub [PubMed PMID: 21519291]
Level 1 (high-level) evidenceKubota R, Al-Fayoumi S, Mallikaarjun S, Patil S, Bavik C, Chandler JW. Phase 1, dose-ranging study of emixustat hydrochloride (ACU-4429), a novel visual cycle modulator, in healthy volunteers. Retina (Philadelphia, Pa.). 2014 Mar:34(3):603-9. doi: 10.1097/01.iae.0000434565.80060.f8. Epub [PubMed PMID: 24056528]
Level 1 (high-level) evidenceKubota R, Birch DG, Gregory JK, Koester JM. Randomised study evaluating the pharmacodynamics of emixustat hydrochloride in subjects with macular atrophy secondary to Stargardt disease. The British journal of ophthalmology. 2022 Mar:106(3):403-408. doi: 10.1136/bjophthalmol-2020-317712. Epub 2020 Nov 19 [PubMed PMID: 33214244]
Level 1 (high-level) evidenceCharbel Issa P, Barnard AR, Herrmann P, Washington I, MacLaren RE. Rescue of the Stargardt phenotype in Abca4 knockout mice through inhibition of vitamin A dimerization. Proceedings of the National Academy of Sciences of the United States of America. 2015 Jul 7:112(27):8415-20. doi: 10.1073/pnas.1506960112. Epub 2015 Jun 23 [PubMed PMID: 26106163]
Mata NL, Lichter JB, Vogel R, Han Y, Bui TV, Singerman LJ. Investigation of oral fenretinide for treatment of geographic atrophy in age-related macular degeneration. Retina (Philadelphia, Pa.). 2013 Mar:33(3):498-507. doi: 10.1097/IAE.0b013e318265801d. Epub [PubMed PMID: 23023528]
Level 1 (high-level) evidenceFederspiel CA, Bertelsen M, Kessel L. Vitamin A in Stargardt disease-an evidence-based update. Ophthalmic genetics. 2018 Oct:39(5):555-559. doi: 10.1080/13816810.2018.1488174. Epub 2018 Jun 25 [PubMed PMID: 29939824]
Trapani I, Toriello E, de Simone S, Colella P, Iodice C, Polishchuk EV, Sommella A, Colecchi L, Rossi S, Simonelli F, Giunti M, Bacci ML, Polishchuk RS, Auricchio A. Improved dual AAV vectors with reduced expression of truncated proteins are safe and effective in the retina of a mouse model of Stargardt disease. Human molecular genetics. 2015 Dec 1:24(23):6811-25. doi: 10.1093/hmg/ddv386. Epub 2015 Sep 29 [PubMed PMID: 26420842]
Binley K, Widdowson P, Loader J, Kelleher M, Iqball S, Ferrige G, de Belin J, Carlucci M, Angell-Manning D, Hurst F, Ellis S, Miskin J, Fernandes A, Wong P, Allikmets R, Bergstrom C, Aaberg T, Yan J, Kong J, Gouras P, Prefontaine A, Vezina M, Bussieres M, Naylor S, Mitrophanous KA. Transduction of photoreceptors with equine infectious anemia virus lentiviral vectors: safety and biodistribution of StarGen for Stargardt disease. Investigative ophthalmology & visual science. 2013 Jun 12:54(6):4061-71. doi: 10.1167/iovs.13-11871. Epub 2013 Jun 12 [PubMed PMID: 23620430]
Level 3 (low-level) evidenceRicca AM, Han IC, Sohn EH. Stargardt disease masquerades. Current opinion in ophthalmology. 2021 May 1:32(3):214-224. doi: 10.1097/ICU.0000000000000750. Epub [PubMed PMID: 33653979]
Level 3 (low-level) evidenceMarmor MF, Byers B. Pattern dystrophy of the pigment epithelium. American journal of ophthalmology. 1977 Jul:84(1):32-44 [PubMed PMID: 900215]
Level 3 (low-level) evidenceFrancis PJ, Schultz DW, Gregory AM, Schain MB, Barra R, Majewski J, Ott J, Acott T, Weleber RG, Klein ML. Genetic and phenotypic heterogeneity in pattern dystrophy. The British journal of ophthalmology. 2005 Sep:89(9):1115-9 [PubMed PMID: 16113362]
Kuper WFE, Talsma HE, van Schooneveld MJ, Pott JWR, Huijgen BCH, de Wit GC, van Hasselt PM, van Genderen MM. Recognizing differentiating clinical signs of CLN3 disease (Batten disease) at presentation. Acta ophthalmologica. 2021 Jun:99(4):397-404. doi: 10.1111/aos.14630. Epub 2020 Oct 18 [PubMed PMID: 33073538]
Tripathy K, Sarma B, Mazumdar S. Outer retinal tubulation and inner retinal pseudocysts in a patient with maternally inherited diabetes and deafness evaluated with optical coherence tomography angiogram. Indian journal of ophthalmology. 2020 Jan:68(1):250-253. doi: 10.4103/ijo.IJO_577_19. Epub [PubMed PMID: 31856543]
Raja MS, Goldsmith C, Burton BJ. Outer retinal tubulations in maternally inherited diabetes and deafness (MIDD)-associated macular dystrophy. Graefe's archive for clinical and experimental ophthalmology = Albrecht von Graefes Archiv fur klinische und experimentelle Ophthalmologie. 2013 Sep:251(9):2265-7. doi: 10.1007/s00417-012-2217-z. Epub 2013 Jan 12 [PubMed PMID: 23314478]
Level 3 (low-level) evidenceWesteneng-van Haaften SC, Boon CJ, Cremers FP, Hoefsloot LH, den Hollander AI, Hoyng CB. Clinical and genetic characteristics of late-onset Stargardt's disease. Ophthalmology. 2012 Jun:119(6):1199-210. doi: 10.1016/j.ophtha.2012.01.005. Epub 2012 Mar 24 [PubMed PMID: 22449572]
Level 2 (mid-level) evidenceGrandinetti AA, Portella E, Arana J, Iskorostenski NT. Subretinal fibrosis in Stargardt's disease: case report. Arquivos brasileiros de oftalmologia. 2011 Nov-Dec:74(6):449-51 [PubMed PMID: 22331122]
Level 3 (low-level) evidenceGass JD, Hummer J. Focal retinal pigment epithelial dysplasia associated with fundus flavimaculatus. Retina (Philadelphia, Pa.). 1999:19(4):297-301 [PubMed PMID: 10458294]
Level 3 (low-level) evidenceRossi S, Testa F, Attanasio M, Orrico A, de Benedictis A, Corte MD, Simonelli F. Subretinal Fibrosis in Stargardt's Disease with Fundus Flavimaculatus and ABCA4 Gene Mutation. Case reports in ophthalmology. 2012 Sep:3(3):410-7. doi: 10.1159/000345415. Epub 2012 Dec 20 [PubMed PMID: 23341817]
Level 3 (low-level) evidencePaterick TE, Patel N, Tajik AJ, Chandrasekaran K. Improving health outcomes through patient education and partnerships with patients. Proceedings (Baylor University. Medical Center). 2017 Jan:30(1):112-113 [PubMed PMID: 28152110]
Lee SM, Cho JC. Low vision devices for children. Community eye health. 2007 Jun:20(62):28-9 [PubMed PMID: 17612694]