Introduction
A pheochromocytoma is a rare tumor originating from chromaffin cells in the adrenal medulla. Pheotochromocytomas clinical manifestations result from excessive catecholamine secretion. Catecholamines are a group of hormones and neurotransmitters crucial for regulating homeostasis and managing the body's response to stress. These chemicals are primarily produced by the adrenal gland and nerve tissue, including the brain. The principal catecholamines are dopamine, norepinephrine, and epinephrine.
Pheochromocytoma is a neoplasm that can be either benign or malignant and is often associated with familial syndromes such as neurofibromatosis type 1 (NF1), multiple endocrine neoplasia type 2 (MEN2), and von Hippel-Lindau (VHL) disease. In addition, sporadic cases are also significant, as they are among the most commonly overlooked causes of secondary hypertension (see Image. Gross Specimen of a Giant Pheochromocytoma).
The clinical manifestations of these tumors are primarily due to the excessive secretion of catecholamines. Tumors that arise from extra-adrenal chromaffin cells are known as paragangliomas, and both types are often studied together as neuroendocrine tumors due to their similar characteristics. Pheochromocytomas account for 80% to 85% of these tumors, while sympathetic paragangliomas account for 15% to 20%.[1][2] Although most pheochromocytomas are benign, a small percentage can be malignant.[3]
In the past, pheochromocytomas were primarily identified during evaluations for secondary hypertension. However, they are now increasingly found as incidental findings on abdominal imaging conducted for other conditions or through surveillance screening in individuals with known genetic disorders.[2]
Pheochromocytomas generally exhibit a predominant type of catecholamine production. Around half primarily secrete epinephrine with varying levels of norepinephrine. Others, including sympathetic paragangliomas, mainly produce norepinephrine with dopamine as a by-product.[2] Dopamine production is considered an independent predictor of malignancy, likely due to its role in promoting angiogenesis.[4]
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
Pheochromocytomas can arise sporadically or have a familial origin. Recent studies suggest that up to 35% of cases may be linked to germline mutations.[5] Familial syndromes known to be associated with pheochromocytomas include VHL disease, MEN2, and NF1.
Over half of pheochromocytomas occur sporadically, without any known connection to inherited disorders, and their underlying cause often remains unknown. However, individuals with a family history of pheochromocytoma or paraganglioma have an increased risk of developing the condition.
Pheochromocytomas can develop due to the factors mentioned below.
- Genetics: Inherited mutations in various genes, including RET, VHL, or NF1, can lead to pheochromocytoma.
- Sporadic: Approximately two-thirds of pheochromocytomas occur spontaneously, without a known family history. However, genetic factors may still be involved in these instances.
- Additional factors: Other contributors to pheochromocytoma (or its discovery) include intense physical activity, trauma, emotional stress, childbirth, anesthesia, and surgery.
Epidemiology
Similar to many secondary causes of hypertension, pheochromocytomas are often underdiagnosed. An autopsy study revealed undiagnosed pheochromocytomas in 0.05% of individuals.[6] In a single-center study of 4180 patients in Brooklyn, pheochromocytomas were identified in 0.2% of those with hypertension, resulting in an average annual incidence rate of 0.5 cases per 100,000 person-years.[7]
Pathophysiology
Catecholamines are produced in chromaffin cells, beginning with the rate-limiting conversion of tyrosine to dihydroxyphenylalanine (DOPA) by tyrosine hydroxylase. DOPA is then converted into dopamine by the action of DOPA decarboxylase, which is further converted into norepinephrine by dopamine β-hydroxylase. Finally, norepinephrine is methylated by phenylethanolamine-N-methyltransferase to produce epinephrine. These catecholamines are stored in vesicles and released into the blood circulation, with cardiac manifestations mediated through adrenoreceptors.[8]
Pheochromocytomas release catecholamines in different patterns, classified as paroxysmal, continuous, or mixed. Norepinephrine is typically released continuously, leading to persistent hypertension, while epinephrine is released in a paroxysmal manner, which can cause tachyarrhythmias.[9][10] The alpha-1 and alpha-2 (α-1 and α-2) adrenoreceptors and beta-1 and beta-2 (β-1 and β-2) adrenoreceptors bind epinephrine and norepinephrine with varying affinities. Epinephrine binds to β-1 and β-2 receptors with similar affinity, while norepinephrine is approximately 10-fold more selective for the β-1 receptor.[11] The α-1 receptors exhibit greater selectivity for norepinephrine than for epinephrine,[12] and dopamine has an affinity for α-2 receptors. Please see StatPearls' companion resource, "Adrenergic Drugs," for more information. Glucocorticoids and thyroid hormones can influence these adrenoceptors by either increasing their numbers or affecting their affinity.[13][14][15]
The heart contains β-1 adrenoceptors, and their stimulation activates adenylate cyclase via the guanine triphosphate protein-coupled receptor. This activation converts adenosine triphosphate (ATP) to cyclic adenosine monophosphate (cAMP). The increase in cAMP activates hyperpolarization-activated cyclic nucleotide-gated channels and protein kinase A. This signaling cascade may increase ionotropy in SA nodal cells and dromotropy in AV nodal cells.[16]
β-2 adrenoceptors are found in peripheral blood vessels and can induce vasodilation when activated by epinephrine and norepinephrine. In contrast, α-adrenoceptors located in vascular smooth muscle cells can cause hypertension through vasoconstriction when activated by norepinephrine and epinephrine. Additionally, α-2 adrenoceptors at synaptic nerve terminals inhibit the release of norepinephrine.
Histopathology
Histopathological evaluation of pheochromocytomas typically reveals a zellballen pattern characterized by nests of chromaffin cells. These cells show strong positivity for chromogranin, synaptophysin, CD56, and focal positivity for S100.[17][18] The presence of chromaffin cells in the extra-adrenal tissue is the only pathognomonic characteristic of the malignant form of the entity.[17]
History and Physical
In pheochromocytoma, episodes of hypertension are often paroxysmal and have been associated with symptoms such as headaches, tachycardia, and sweating. However, patients may also present with sustained resistant hypertension. The presence of hypertension that is resistant to multiple antihypertensive therapies should raise suspicion for pheochromocytoma. Severe hypertension episodes may be triggered by dopamine receptor antagonists, non-selective β-blockers, tricyclic antidepressants, corticosteroids, sympathomimetics, and neuromuscular agents. Symptoms can also arise during surgery or anesthesia induction.[1]
The condition is often diagnosed through screening tests after establishing a familial diagnosis in familial syndromes associated with pheochromocytoma. Many of these patients do not present with hypertension at the time of diagnosis. Giant lesions may paradoxically present solely as abdominal masses with minimal characteristic symptoms of pheochromocytoma. This occurs due to tumoral necrosis, a higher proportion of interstitial tissue relative to chromaffin cells, and encapsulation by connective tissues (see Image. A Giant Pheochromocytoma).[17]
Familial Pheochromocytomas
Pheochromocytomas are associated with several hereditary syndromes, including VHL disease, MEN2, and NF1. All these syndromes follow an autosomal dominant pattern of inheritance.
von Hippel-Lindau disease: The phenotype of VHL disease typically includes hemangioblastomas of the brain and spine, retinal angiomas, clear cell renal cell carcinomas, pheochromocytomas, paragangliomas, pancreatic neuroendocrine tumors, endolymphatic sac tumors of the middle ear, and cystadenomas of the pancreas, epididymis, and broad ligament. The condition is usually caused by mutations in the VHL tumor suppressor gene.[19] Patients with VHL type 1 have a lower risk of developing pheochromocytomas compared to those with VHL type 2.[20] Additionally, individuals with VHL disease tend to exhibit higher levels of the norepinephrine metabolite normetanephrine than those with MEN2.[21]
Multiple endocrine neoplasia type 2: The typical phenotype of MEN2 includes medullary thyroid cancer, pheochromocytomas, primary hyperparathyroidism, mucocutaneous neuromas, skeletal deformities, intestinal ganglioneuromas, and cutaneous lichen amyloidosis. This syndrome is caused by mutations in the RET proto-oncogene and is inherited in an autosomal dominant manner.[22] Patients with MEN2 often exhibit elevated levels of metanephrines, particularly the epinephrine metabolite, and tend to be more symptomatic, with a higher incidence of hypertension compared to those with VHL disease.[21]
Neurofibromatosis type 1: NF1 is caused by mutations in the NF1 tumor suppressor gene and is inherited in an autosomal dominant manner.[23] Diagnosis is typically made clinically, with characteristic features including café-au-lait macules, axillary freckling, Lisch nodules, optic gliomas, osseous lesions, and neurofibromas.[24] Pheochromocytomas occur in a small proportion of NF1 patients, with studies reporting their presence in 0.1% to 5.7% of patients with NF1.[25]
Subclinical Presentation
Pheochromocytomas can present with varied clinical presentations depending on their secretory patterns. Dopamine-secreting tumors and those with minimal hormone secretion may lead to subclinical or mild disease. Dopamine-secreting tumors often simultaneously release norepinephrine, and the counteractive effects of these hormones on blood vessels can reduce the development of overt clinical signs and symptoms.[26]
The recent increase in genetic screening of family members has led to increased detection of patients with subclinical disease. Tumors with succinate dehydrogenase complex iron-sulfur subunit B (SDHB) mutations often exhibit an undifferentiated catecholamine biosynthetic phenotype with low catecholamine concentrations. As a result, these tumors can present subclinically until they grow large enough to produce noticeable signs and symptoms.[27]
Catecholamine secretory patterns can produce varying clinical presentations. Epinephrine-secreting tumors often cause paroxysmal hypertensive crises but may also have a silent course. In contrast, norepinephrine-secreting tumors typically result in essential hypertension. Chronically elevated levels of norepinephrine can lead to downregulation of adrenoreceptors, which may contribute to a milder clinical presentation.[28][29]
Patients with subclinical disease are also at significant cardiovascular risk, with case reports describing sudden hypertensive crises leading to sudden death.[30][31] Mechanical factors such as abdominal palpation, sexual intercourse, coughing, sneezing, defecation, pain, extreme emotions, and exposure to cold can trigger hypertensive crises in otherwise asymptomatic catecholamine-secreting tumors. Annual screening with plasma or urinary metanephrines is recommended for genetic carriers to facilitate early detection of disease.
Another important aspect of subclinical presentation involves incidentalomas discovered through imaging, most commonly on computed tomography (CT). These findings are prevalent in 6% of autopsy studies and 4% of CT scans.[32][33] The adrenal glands develop nodules with advancing age and are reported to have a 7% prevalence in individuals aged 70 or older. More than 4% were reported to be pheochromocytomas in a large series of incidentalomas.[34] Screening with plasma or urinary metanephrines is recommended surgery involving adrenal incidentalomas to ensure appropriate management.
In summary, examination findings may include:
- Hypertension
- Tachycardia
- Anxiety
- Diaphoresis
- Subcutaneous neurofibromas
- Café-au-lait macules
- Thyroid mass
- Axillary freckling
- Iridic Lisch nodules
- Retinal angiomas
- Abdominal mass
Evaluation
Laboratory Studies
In the appropriate clinical scenario, diagnosis of pheochromocytoma can be established by biochemical confirmation of hypersecretion of metanephrines and catecholamines. According to the latest Endocrine Society Clinical Practice Guidelines, the initial biochemical evaluation should include either plasma-fractionated metanephrines or urinary-fractionated metanephrines.[1]
Catecholamines are rapidly inactivated by the enzyme catechol-O-methyltransferase, producing metanephrine and normetanephrine, which are subsequently conjugated with sulfate. These metabolites have a longer half-life and are excreted in the urine, making them more suitable for measurement than catecholamines. Elevated plasma metanephrines are considered more specific for diagnosing pheochromocytoma than urinary metanephrines, with higher levels correlating with larger tumor sizes. The European Society Clinical Practice Guidelines recommend liquid chromatography with mass spectrometry or electrochemical detection for optimal accuracy. Reported sensitivities for urinary metanephrines range from 86% to 97%, with specificities between 69% and 95%.[35]
Key Considerations in Biochemical Testing for Pheochromocytoma
- Although pheochromocytoma is a rare condition that causes catecholamine excess, the low pretest probability of the disease means that test results must be interpreted with caution.[1]
- Plasma-free metanephrines are the most reliable test for confirming or ruling out pheochromocytoma.
- Given the frequency of testing and the low incidence of true pheochromocytoma, false positives significantly outnumber true positives—a pattern also observed with paraganglioma.[36][37]
- Additional laboratory findings may include hyperglycemia, hypercalcemia, and erythrocytosis.
Several studies have shown that plasma metanephrine concentrations tend to be higher when measured in the seated position compared to the supine position, leading to reduced specificity and an increase in false positive results.[38][39][40] Guidelines recommend drawing blood with the patient in the supine position after they have been fully recumbent for at least 30 minutes before sampling.[1] When logistical constraints make this unfeasible, positive results from seated samples should be confirmed with repeat testing in the supine position. Reference intervals specific to the patient’s position during sampling must be applied.
Imaging Studies
Following biochemical confirmation, the next suggested evaluation step is to locate the tumor using imaging studies. Per guidelines, a CT of the abdomen and pelvis is the recommended initial imaging test to locate the tumor. Current guidelines suggest an initial CT scan of the abdomen and pelvis. Pheochromocytomas on CT typically present as low-density lesions relative to soft tissue. However, lesions larger than 3 cm can display variable appearances, potentially mimicking other adrenal tumors.
Magnetic resonance imaging (MRI) serves as a viable alternative, especially when avoiding radiation or contrast is preferred, although it offers lower spatial resolution than CT. Pheochromocytomas typically appear as hyperintense lesions on T2-weighted imaging. For metastatic disease, fluorodeoxyglucose F18 (FDG) positron emission tomography (PET) is recommended. A 123I-metaiodobenzylguanidine (MIBG) scan is an alternate option, particularly when radiotherapy with 123I-MIBG is being considered.[1]
Genetic Testing
Given that up to 35% of pheochromocytoma cases may be related to germline disease-causing mutations, genetic testing should be considered for all patients diagnosed with pheochromocytoma.[5] Bilateral tumors and those that present at younger ages are more frequently associated with hereditary syndromes.
Once pheochromocytoma is diagnosed, a thorough clinical evaluation and family history can help identify characteristic symptoms and signs of hereditary syndromes associated with the condition. In such cases, targeted genetic testing for VHL, MEN2, and NF1 syndromes is recommended. After identifying a pathogenic mutation, genetic screening for first-degree relatives should also be considered.
Typical sporadic cases of pheochromocytoma are unilateral and associated with an adverse family history, as well as an absence of syndromic symptoms and signs. Individuals with sporadic cases should be clinically monitored for the development of characteristic signs of hereditary syndromes. Genetic testing should be considered in sporadic cases where clinical suspicion arises. A study from Germany found that characteristic mutations were detected in 24% of patients with apparently sporadic pheochromocytoma.[41]
Several genes associated with the development of pheochromocytomas have been identified and categorized into 3 clusters based on the mechanism of tumorigenesis:
- Pseudohypoxia pathway (cluster 1): This cluster has been associated with mutations in the EGLN1, EGLN2, DLST, FH, IDH3B, MDH2, SDHA, SDHAF2, SDHB, SDHC, SDHD, VHL, EPAS1, IDH1, and IDH2 genes.
- Kinase-signaling pathway (cluster 2): This has been associated with mutations in the NF1, MAX, MERTK, MET, MYCN, RET, and TMEM127 genes.
- Somatic cluster (cluster 3): This has been associated with alterations in the Wnt signaling pathway.[42]
Treatment / Management
The definitive treatment for pheochromocytoma is surgical resection, with preoperative α- and β-adrenergic blockade being crucial.[43]
Unilateral Pheochromocytoma
Most sporadic tumors are unilateral. According to the Endocrine Society Clinical Practice Guidelines, adrenalectomy via a minimally invasive approach is the preferred treatment for most unilateral adrenal pheochromocytomas. Open adrenalectomy may be considered for larger tumors or in cases where the tumor is considered invasive.[1](A1)
Bilateral Pheochromocytoma
Bilateral neoplasms are most commonly associated with hereditary cases, particularly MEN2 and VHL disease.[44] In these cases, bilateral total adrenalectomy necessitates lifelong steroid replacement, which can lead to long-term adverse effects.[45] Alternatively, bilateral partial or cortical-sparing adrenalectomy can be performed via any surgical approach. This option has demonstrated comparable survival rates despite tumor recurrence and better preservation of adrenocortical function, resulting in a reduced need for lifelong glucocorticoid replacement therapy.[45][46](B2)
Preoperative Preparation
No randomized studies exist to guide medical optimization approaches before surgery. However, it is well-recognized that initiating medical therapy with α- and β-blockade in the preoperative period can significantly reduce the risk of uncontrolled hypertension, hypertensive crises, tachycardia, and volume expansion during the perioperative period.[43]
According to the Endocrine Society Clinical Practice Guidelines, the first-line drugs for preoperative preparation are α-adrenergic receptor blockers.[1] A typical regimen involves initiating phenoxybenzamine 7 to 14 days before surgery, starting at a dosage of 10 mg orally twice daily and carefully titrating up to a maximum of 1 mg/kg/d. Doxazosin, an α-1 selective agent, is an acceptable alternative due to its favorable adverse effect profile.(A1)
At least 3 to 4 days after starting α-blockade, β-blockade with propranolol, atenolol, or metoprolol should be initiated to manage tachycardia. These agents should not be started without prior α-blockade, as doing so may precipitate a hypertensive crisis due to unopposed α-receptor stimulation.[1] Calcium channel blockers, such as amlodipine and nifedipine, can serve as alternative or add-on agents to effectively control hypertension in the preoperative period.[47](A1)
Volume contraction related to catecholamine excess in patients with pheochromocytoma can be managed by starting a high-sodium diet a few days after initiating α-blockade. This approach can also help reduce the risk of hypotension following surgery.[9](B3)
Perioperative Concerns
The key elements of perioperative management are 4-fold. First, prevention of catecholamine surge is achieved through minimal handling of the lesion and avoiding spillage of tumor contents, particularly in cystic lesions. Second, early intraoperative control of the adrenal vein is crucial. Lastly, effective management of sudden hypotension, characterized by a decrease in peripheral vascular resistance following surgical removal of the lesion, is essential.[17](B3)
Nonsurgical Management
Nonsurgical management of pheochromocytomas may involve radiotherapy, chemotherapy, and palliative care. According to the 2017 National Comprehensive Cancer Network guidelines, treatment options for malignant paraganglioma and pheochromocytoma include cytoreductive surgery when feasible, systemic chemotherapy (commonly consisting of cyclophosphamide, vincristine, and dacarbazine or temozolomide), or 131I-MIBG.
Percutaneous ablation has emerged as a minimally invasive local treatment option for pheochromocytoma.[48][49] Small retrospective studies have indicated that external beam radiation therapy may serve as a useful local treatment modality for some patients with advanced or unresectable malignant pheochromocytoma.[50][51](A1)
Embolization in Pheochromocytoma Treatment
Embolization involves blocking the artery supplying blood to a tumor. Spontaneous remission of hypercatecholaminergic crises associated with pheochromocytomas, particularly those with extensive necrosis, has been linked to the exhaustion of catecholamine secretion before surgical tumor resection.[52] In situations where emergent surgery and medical control using α-1 blockade pose a high risk, treatment options may include mechanical circulatory support with a cardiopulmonary device or intra-aortic balloon pump or the removal of catecholamines through continuous hemodiafiltration.[53] (B3)
Alternatively, emergent transcatheter arterial embolization is being considered a safer modality for the urgent treatment of critical blood pressure fluctuations associated with pheochromocytoma-related hypercatecholaminergic crises. This approach can quickly stabilize blood pressure, allowing for future elective surgical tumor resection in a more controlled setting.[54](B3)
Palliative Treatment in Advanced Metastatic Pheochromocytoma
The palliative treatment approach for advanced metastatic pheochromocytoma focuses on relieving symptoms and improving quality of life. This may involve surgical intervention to remove as much of the tumor as possible or radiation therapy to target areas affected by the cancer. Additionally, medications such as α- and β-blockers can be used to manage disease-related symptoms effectively.
Differential Diagnosis
The differential diagnosis of pheochromocytoma includes various causes of primary and secondary hypertension, including:
- Hyperthyroidism
- Anxiety disorder
- Renal artery stenosis
- Hyperaldosteronism
- Migraine headache
- Preeclampsia
- Cardiomyopathy
- Postural tachycardia syndrome
- Drug-induced hypertension
- Cushing syndrome
- Carcinoid syndrome
- Acrodynia
Surgical Oncology
Metastatic Pheochromocytomas
Approximately 10% of pheochromocytomas are classified as malignant, characterized by the presence of metastasis. Currently, no definitive histological or biochemical features exist that can reliably distinguish malignant pheochromocytomas from benign ones, except for the identification of chromaffin cells in extra-adrenal tissue.[17][55] The Pheochromocytoma of the Adrenal gland Scaled Score (PASS) has been applied to differentiate benign lesions from their malignant counterparts.[17][18]
Surgery
Resection of both primary and metastatic disease should be considered when technically feasible, although it is unlikely to result in a complete cure. The surgical approach must be individualized based on the location of the metastases and may involve either open or minimally invasive techniques. Larger tumors are typically treated using an open approach, as this method is associated with a lower risk of tumor rupture and may facilitate the simultaneous removal of metastases.[55][56]
Debulking both primary and metastatic lesions may alleviate symptoms associated with catecholamine surges.[55][56] However, the reported 5-year survival rate is only 50%.[17]
Pertinent Studies and Ongoing Trials
A phase II trial of temozolomide and olaparib is currently underway. This trial aims to assess whether combining olaparib (a PARP inhibitor) with temozolomide (a chemotherapeutic agent) is more effective for treating metastatic or unresectable neuroendocrine tumors compared to using temozolomide alone. (https://clinicaltrials.gov/study/NCT04394858)
The mechanism of olaparib is the inhibition of PARP proteins, which are involved in the repair of DNA damage in tumor cells. By blocking this repair mechanism, olaparib may hinder the ability of tumor cells to recover from DNA damage, potentially leading to their death or slowed growth. Temozolomide is a chemotherapy drug that kills tumor cells or prevents their division and spread by inducing DNA damage.
The trial aims to determine whether the synergistic effect of combining these two drugs can enhance treatment effectiveness for neuroendocrine cancers that are challenging to treat with existing methods. Other phase II trials of the vascular endothelial growth factor (VEGF) inhibitors lenvatinib and dovitinib are ongoing for patients with pheochromocytoma or paraganglioma.
Treatment Planning
The treatment for pheochromocytoma or paraganglioma that has metastasized to nearby organs or lymph nodes typically involves surgical resection to completely remove the tumor. This may include excising not only the primary tumor but also affected adjacent organs, such as the kidney or liver, as well as sections of major blood vessels and lymph nodes that have been invaded by cancer.
Toxicity and Adverse Effect Management
Phenoxybenzamine is a medication used to manage and treat paroxysmal hypertension and sweating associated with pheochromocytoma.
Adverse Effects
Adverse effects of phenoxybenzamine include:
- Inhibited ejaculation
- Orthostatic hypotension
- Tachycardia (reflex tachycardia)
- Miosis
- Drowsiness
- Fatigue
- Gastrointestinal irritation
Toxicity
An overdose of phenoxybenzamine can result in blockade of the sympathetic nervous system and circulating epinephrine, leading to symptoms such as postural hypotension (especially upon standing), tachycardia, dizziness, fainting, lethargy, vomiting, and shock. According to the US Food and Drug Administration (FDA) drug label, case reports have associated long-term use of phenoxybenzamine with the development of carcinoma, indicating that prolonged therapy is not advised.
Medical Oncology
External Beam Radiation Therapy
External beam radiation therapy can be effective for controlling primary and metastatic disease across various sites. Data from smaller studies suggest that these treatments can provide symptom relief and help in disease debulking.[51][50]
Systemic Chemotherapy
Systemic chemotherapy may be considered for patients with extensive metastatic or unresectable disease. Reported treatment regimens for metastatic pheochromocytoma have included combinations of cyclophosphamide, doxorubicin, dacarbazine, and vincristine.[57][58] A meta-analysis indicates that approximately 37% of patients respond to chemotherapy, although complete responses are rare.[59][55] Nonetheless, chemotherapy can reduce tumor size and improve blood pressure control.[57][60]
Radionuclide Therapy
MIBG: This treatment can only be considered for tumors capable of taking up MIBG and is used in cases of unresectable disease or high tumor burden. Due to its structural similarity to norepinephrine, MIBG scintigraphy helps localize catecholamine-secreting tissues.[61] Treatment with iobenguane I-131 can lead to symptomatic relief in many patients and a reduction in tumor size in a considerable proportion of patients, which is 24% to 45% of cases.[62][63][64][65]
Novel Therapies
Recent advances in treating metastatic pheochromocytomas involve the use of agents that inhibit angiogenesis and proliferative signaling within tumor cells. These processes are driven by the interaction of growth factors, such as VEGF and platelet-derived growth factors (PDGF), with tyrosine kinase receptors, making them key therapeutic targets.[55]
Sunitinib: Sunitinib inhibits multiple receptors, including VEGF1, VEGF2, VEGF3, PDGF-α, PDGF-β, c-kit, fms-related tyrosine kinase 3, and the RET proto-oncogene, targeting angiogenesis and tumor cell proliferation.[55][66] Sunitinib has been evaluated in small studies, with disease control rates—comprising stable disease and partial responses—ranging between 57% and 83%.[67][68] Median progression-free survival has been reported between 4 and 13 months.[67][68] As sunitinib can induce hypertension, antihypertensive therapy may require adjustment to maintain adequate blood pressure control during treatment.
Axitinib: This medication is an inhibitor of VEGF receptor 2 (VEGFR2), which is especially important in metastatic pheochromocytoma. These tumors often present a pseudohypoxic environment that stimulates VEGF synthesis, promoting angiogenesis.[55] In phase 2 clinical trials, a partial response was observed in 36% of patients. Dosage adjustments are often necessary, as this medication can significantly worsen blood pressure control in a significant proportion of treated patients.[55]
Other Investigational Targeted Therapies
Investigational therapies include cabozantinib (a VEGFR2 and c-MET receptor inhibitor) and hypoxia-inducible factor 2α (HIF2A) inhibitors.
Prognosis
The risk of pheochromocytoma recurrence exists in both sporadic and familial cases. A study of 192 patients with pheochromocytomas and paragangliomas found that recurrence was more common in familial cases, as well as in right adrenal and extra-adrenal tumors.[69]
A recent meta-analysis estimates that the recurrence rate after curative surgery is low, at approximately 3%, with a mean follow-up of 77 months.[70] The Endocrine Society Clinical Practice Guidelines recommend lifelong annual biochemical testing to monitor for recurrent or metastatic disease.[1]
Complications
Complications may include:
- Myocardial infarction
- Cardiogenic shock
- Cerebrovascular accident
- Renal failure
- Pulmonary edema
- Acute respiratory distress syndrome
- Cardiac arrhythmias
- Lactic acidosis
- Hypertensive retinopathy
- Hypertensive encephalopathy
- Seizures (in children)
- Polydipsia (in children)
- Polyuria (in children)
- Cerebral vasculitis
- Ischemic enterocolitis
- Renal infarction
- Anxiety
- Depression
Postoperative and Rehabilitation Care
The most common complication following tumor removal is hypotension, which may require fluid therapy and vasopressor support for several hours. However, advances in surgical and anesthetic techniques have reduced the risk of severe complications and mortality associated with the procedure in high-volume centers.
Deterrence and Patient Education
Patient education is essential in managing pheochromocytomas. Patients should understand the episodic nature of the symptoms associated with the condition and be informed that episodes of severe hypertension can be triggered by medications such as dopamine receptor antagonists, nonselective β-blockers, tricyclic antidepressants, corticosteroids, sympathomimetics, and neuromuscular agents. They should also be aware that such episodes may occur during surgery or anesthesia induction.
Common episodic symptoms include headaches, sweating, heart racing, shortness of breath, chest pain, and anxiety.
All patients diagnosed with pheochromocytoma, particularly those with a known family history of VHL syndrome, MEN2, or NF1, should be advised about the importance of genetic screening for their relatives. Several blood, urine, and imaging tests are available to aid in the diagnosis of pheochromocytoma. Treatment options include medications to manage blood pressure and other symptoms, as well as surgical removal of the tumor, which may be considered based on various factors.
Pearls and Other Issues
Pheochromocytomas and Pregnancy
Pheochromocytomas present a rare yet anxiety-provoking condition in pregnant patients, with reported prevalence rates ranging from 1 in 15,000 to 1 in 54,000 pregnancies.[71][72] The clinical course is often unpredictable, as increased catecholamine secretion into maternal circulation can lead to severe hypertension, uteroplacental ischemia, arrhythmias, and even death. Due to the rarity of this condition, there are currently no established guidelines for management in this setting.
The authors provided valuable evidence to assist clinicians in a large international multicenter retrospective cohort study.[73] They found that unrecognized and suboptimally treated pheochromocytoma and paraganglioma (PPGL) were associated with maternal or fetal death in 33 out of 230 pregnancies (14%). Patients with undiagnosed or untreated PPGLs faced a significantly increased risk of maternal and fetal complications, with an odds ratio of 27.0 (95% CI 3.5–3473.1) compared to those with PPGLs identified before pregnancy. This highlights the importance of initiating adrenoceptor blockade following the diagnosis of the tumor.
The authors reported that severe maternal complications occurred in 7% of pregnancies associated with untreated PPGL, with a mortality rate of 1%. Overall, fetal mortality was reported at 9%, a rate significantly lower than those found in previously published literature. Abdominal and pelvic PPGLs were associated with a higher incidence of adverse events, likely due to compression by the uterus or increased catecholamine release. Tumor size was not associated with a higher rate of complications at delivery, and the type of delivery did not correlate with adverse outcomes.
Antepartum surgery did not show improved outcomes, with adverse events occurring in 8% of such pregnancies. If surgery is necessary, the second trimester is considered safer than the first. Metastatic PPGL was not associated with adverse outcomes, likely due to closer monitoring and treatment. Additionally, the authors reported lower adverse outcomes in syndromic PPGL, which was thought to be secondary to earlier diagnosis and treatment.
In conclusion, undiagnosed and untreated PPGL was associated with a significantly higher risk of maternal and fetal complications compared to the excellent outcomes observed in patients treated with α-adrenergic blockade agents, such as phenoxybenzamine and doxazosin, during pregnancy. The transperitoneal approach is preferred for laparoscopic resection, as the prone positioning required for the extraperitoneal approach is contraindicated during pregnancy.[74]
Enhancing Healthcare Team Outcomes
Patients with pheochromocytoma are at risk for numerous complications, making early identification and management essential for reducing morbidity and mortality. A collaborative approach among healthcare professionals is crucial to ensure patient-centered care and improve overall outcomes. Endocrinologists, endocrine surgeons, emergency medicine physicians, critical care physicians, advanced practitioners, nurses, pharmacists, and other healthcare providers involved in the care of these patients must possess the necessary clinical skills and knowledge for accurate diagnosis and management. This expertise includes recognizing the diverse clinical presentations and understanding the intricacies of diagnostic testing and laboratory interpretation.
A strategic approach is equally essential, incorporating evidence-based strategies to optimize treatment plans and minimize adverse effects. Ethical considerations should guide decision-making, ensuring informed consent and respecting patient autonomy in treatment choices. Each healthcare professional must understand their responsibilities and contribute their unique expertise to the patient's care plan, thereby fostering a multidisciplinary approach. Effective interprofessional communication is vital, facilitating seamless information exchange and collaborative decision-making among healthcare team members.
Care coordination is crucial in ensuring that the patient's journey from diagnosis to treatment and follow-up is effectively managed, minimizing errors and enhancing patient safety. By embracing principles of skill, strategy, ethics, responsibility, interprofessional communication, and care coordination, healthcare professionals can provide patient-centered care, ultimately improving outcomes and enhancing team performance in the management of pheochromocytomas.
Media
(Click Image to Enlarge)

A Giant Pheochromocytoma.
Contributed by S Munakomi, MD
(Click Image to Enlarge)
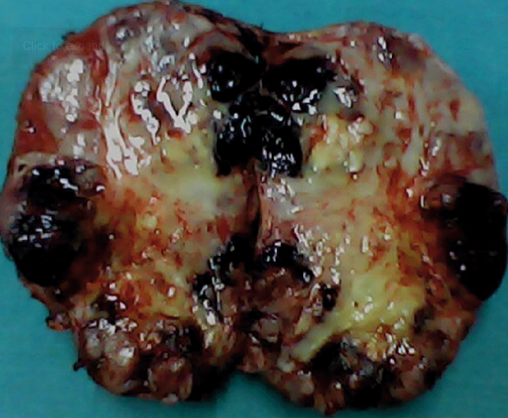
Gross Specimen of a Giant Pheochromocytoma.
Contributed by S Munakomi, MD
References
Lenders JW, Duh QY, Eisenhofer G, Gimenez-Roqueplo AP, Grebe SK, Murad MH, Naruse M, Pacak K, Young WF Jr, Endocrine Society. Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. The Journal of clinical endocrinology and metabolism. 2014 Jun:99(6):1915-42. doi: 10.1210/jc.2014-1498. Epub [PubMed PMID: 24893135]
Level 1 (high-level) evidenceLenders JWM, Kerstens MN, Amar L, Prejbisz A, Robledo M, Taieb D, Pacak K, Crona J, Zelinka T, Mannelli M, Deutschbein T, Timmers HJLM, Castinetti F, Dralle H, Widimský J, Gimenez-Roqueplo AP, Eisenhofer G. Genetics, diagnosis, management and future directions of research of phaeochromocytoma and paraganglioma: a position statement and consensus of the Working Group on Endocrine Hypertension of the European Society of Hypertension. Journal of hypertension. 2020 Aug:38(8):1443-1456. doi: 10.1097/HJH.0000000000002438. Epub [PubMed PMID: 32412940]
Level 3 (low-level) evidencePlouin PF, Chatellier G, Fofol I, Corvol P. Tumor recurrence and hypertension persistence after successful pheochromocytoma operation. Hypertension (Dallas, Tex. : 1979). 1997 May:29(5):1133-9 [PubMed PMID: 9149678]
Osinga TE, Links TP, Dullaart RPF, Pacak K, van der Horst-Schrivers ANA, Kerstens MN, Kema IP. Emerging role of dopamine in neovascularization of pheochromocytoma and paraganglioma. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2017 Jun:31(6):2226-2240. doi: 10.1096/fj.201601131R. Epub 2017 Mar 6 [PubMed PMID: 28264974]
Neumann HP, Young WF Jr, Krauss T, Bayley JP, Schiavi F, Opocher G, Boedeker CC, Tirosh A, Castinetti F, Ruf J, Beltsevich D, Walz M, Groeben HT, von Dobschuetz E, Gimm O, Wohllk N, Pfeifer M, Lourenço DM Jr, Peczkowska M, Patocs A, Ngeow J, Makay Ö, Shah NS, Tischler A, Leijon H, Pennelli G, Villar Gómez de Las Heras K, Links TP, Bausch B, Eng C. 65 YEARS OF THE DOUBLE HELIX: Genetics informs precision practice in the diagnosis and management of pheochromocytoma. Endocrine-related cancer. 2018 Aug:25(8):T201-T219. doi: 10.1530/ERC-18-0085. Epub 2018 May 24 [PubMed PMID: 29794110]
Lo CY, Lam KY, Wat MS, Lam KS. Adrenal pheochromocytoma remains a frequently overlooked diagnosis. American journal of surgery. 2000 Mar:179(3):212-5 [PubMed PMID: 10827323]
Level 2 (mid-level) evidenceAriton M, Juan CS, AvRuskin TW. Pheochromocytoma: clinical observations from a Brooklyn tertiary hospital. Endocrine practice : official journal of the American College of Endocrinology and the American Association of Clinical Endocrinologists. 2000 May-Jun:6(3):249-52 [PubMed PMID: 11421540]
Mariani-Costantini R, Shen Y, Cheng L. Biochemical Diagnosis of Pheochromocytoma and Paraganglioma. Paraganglioma: A Multidisciplinary Approach. 2019 Jul 2:(): [PubMed PMID: 31294941]
Pacak K. Preoperative management of the pheochromocytoma patient. The Journal of clinical endocrinology and metabolism. 2007 Nov:92(11):4069-79 [PubMed PMID: 17989126]
Level 3 (low-level) evidenceGupta G, Pacak K, AACE Adrenal Scientific Committee. PRECISION MEDICINE: AN UPDATE ON GENOTYPE/BIOCHEMICAL PHENOTYPE RELATIONSHIPS IN PHEOCHROMOCYTOMA/PARAGANGLIOMA PATIENTS. Endocrine practice : official journal of the American College of Endocrinology and the American Association of Clinical Endocrinologists. 2017 Jun:23(6):690-704. doi: 10.4158/EP161718.RA. Epub 2017 Mar 23 [PubMed PMID: 28332883]
Xu X, Kaindl J, Clark MJ, Hübner H, Hirata K, Sunahara RK, Gmeiner P, Kobilka BK, Liu X. Binding pathway determines norepinephrine selectivity for the human β(1)AR over β(2)AR. Cell research. 2021 May:31(5):569-579. doi: 10.1038/s41422-020-00424-2. Epub 2020 Oct 22 [PubMed PMID: 33093660]
Perez DM. α(1)-Adrenergic Receptors in Neurotransmission, Synaptic Plasticity, and Cognition. Frontiers in pharmacology. 2020:11():581098. doi: 10.3389/fphar.2020.581098. Epub 2020 Sep 29 [PubMed PMID: 33117176]
Sundaresan PR, Banerjee SP. Differential regulation of beta-adrenergic receptor-coupled adenylate cyclase by thyroid hormones in rat liver and heart: possible role of corticosteroids. Hormone research. 1987:27(2):109-18 [PubMed PMID: 2820855]
Level 3 (low-level) evidenceMotulsky HJ, Insel PA. Adrenergic receptors in man: direct identification, physiologic regulation, and clinical alterations. The New England journal of medicine. 1982 Jul 1:307(1):18-29 [PubMed PMID: 6123082]
Level 3 (low-level) evidenceCiaraldi TP, Marinetti GV. Hormone action at the membrane level. VIII. Adrenergic receptors in rat heart and adipocytes and their modulation by thyroxine. Biochimica et biophysica acta. 1978 Jul 3:541(3):334-46 [PubMed PMID: 149563]
Level 3 (low-level) evidenceNazari MA, Rosenblum JS, Haigney MC, Rosing DR, Pacak K. Pathophysiology and Acute Management of Tachyarrhythmias in Pheochromocytoma: JACC Review Topic of the Week. Journal of the American College of Cardiology. 2020 Jul 28:76(4):451-464. doi: 10.1016/j.jacc.2020.04.080. Epub [PubMed PMID: 32703516]
Munakomi S, Rajbanshi S, Adhikary PS. Case Report: A giant but silent adrenal pheochromocytoma - a rare entity. F1000Research. 2016:5():290 [PubMed PMID: 27785358]
Level 3 (low-level) evidenceCajipe KM, Gonzalez G, Kaushik D. Giant cystic pheochromocytoma. BMJ case reports. 2017 Nov 8:2017():. pii: bcr-2017-222264. doi: 10.1136/bcr-2017-222264. Epub 2017 Nov 8 [PubMed PMID: 29122903]
Level 3 (low-level) evidenceLonser RR, Glenn GM, Walther M, Chew EY, Libutti SK, Linehan WM, Oldfield EH. von Hippel-Lindau disease. Lancet (London, England). 2003 Jun 14:361(9374):2059-67 [PubMed PMID: 12814730]
Zbar B, Kishida T, Chen F, Schmidt L, Maher ER, Richards FM, Crossey PA, Webster AR, Affara NA, Ferguson-Smith MA, Brauch H, Glavac D, Neumann HP, Tisherman S, Mulvihill JJ, Gross DJ, Shuin T, Whaley J, Seizinger B, Kley N, Olschwang S, Boisson C, Richard S, Lips CH, Lerman M. Germline mutations in the Von Hippel-Lindau disease (VHL) gene in families from North America, Europe, and Japan. Human mutation. 1996:8(4):348-57 [PubMed PMID: 8956040]
Eisenhofer G, Walther MM, Huynh TT, Li ST, Bornstein SR, Vortmeyer A, Mannelli M, Goldstein DS, Linehan WM, Lenders JW, Pacak K. Pheochromocytomas in von Hippel-Lindau syndrome and multiple endocrine neoplasia type 2 display distinct biochemical and clinical phenotypes. The Journal of clinical endocrinology and metabolism. 2001 May:86(5):1999-2008 [PubMed PMID: 11344198]
Frank-Raue K, Rondot S, Raue F. Molecular genetics and phenomics of RET mutations: Impact on prognosis of MTC. Molecular and cellular endocrinology. 2010 Jun 30:322(1-2):2-7. doi: 10.1016/j.mce.2010.01.012. Epub 2010 Jan 18 [PubMed PMID: 20083156]
Gutmann DH, Ferner RE, Listernick RH, Korf BR, Wolters PL, Johnson KJ. Neurofibromatosis type 1. Nature reviews. Disease primers. 2017 Feb 23:3():17004. doi: 10.1038/nrdp.2017.4. Epub 2017 Feb 23 [PubMed PMID: 28230061]
DeBella K, Szudek J, Friedman JM. Use of the national institutes of health criteria for diagnosis of neurofibromatosis 1 in children. Pediatrics. 2000 Mar:105(3 Pt 1):608-14 [PubMed PMID: 10699117]
Walther MM, Herring J, Enquist E, Keiser HR, Linehan WM. von Recklinghausen's disease and pheochromocytomas. The Journal of urology. 1999 Nov:162(5):1582-6 [PubMed PMID: 10524872]
Mannelli M, Lenders JW, Pacak K, Parenti G, Eisenhofer G. Subclinical phaeochromocytoma. Best practice & research. Clinical endocrinology & metabolism. 2012 Aug:26(4):507-15. doi: 10.1016/j.beem.2011.10.008. Epub 2012 May 22 [PubMed PMID: 22863392]
Timmers HJ, Kozupa A, Eisenhofer G, Raygada M, Adams KT, Solis D, Lenders JW, Pacak K. Clinical presentations, biochemical phenotypes, and genotype-phenotype correlations in patients with succinate dehydrogenase subunit B-associated pheochromocytomas and paragangliomas. The Journal of clinical endocrinology and metabolism. 2007 Mar:92(3):779-86 [PubMed PMID: 17200167]
Level 2 (mid-level) evidenceTsujimoto G, Manger WM, Hoffman BB. Desensitization of beta-adrenergic receptors by pheochromocytoma. Endocrinology. 1984 Apr:114(4):1272-8 [PubMed PMID: 6323140]
Level 3 (low-level) evidenceStreeten DH, Anderson GH Jr. Mechanisms of orthostatic hypotension and tachycardia in patients with pheochromocytoma. American journal of hypertension. 1996 Aug:9(8):760-9 [PubMed PMID: 8862222]
Song G, Joe BN, Yeh BM, Meng MV, Westphalen AC, Coakley FV. Risk of catecholamine crisis in patients undergoing resection of unsuspected pheochromocytoma. International braz j urol : official journal of the Brazilian Society of Urology. 2011 Jan-Feb:37(1):35-40;discussion 40-1 [PubMed PMID: 21385478]
Level 3 (low-level) evidenceShen SJ, Cheng HM, Chiu AW, Chou CW, Chen JY. Perioperative hypertensive crisis in clinically silent pheochromocytomas: report of four cases. Chang Gung medical journal. 2005 Jan:28(1):44-50 [PubMed PMID: 15804148]
Level 3 (low-level) evidenceKloos RT, Gross MD, Francis IR, Korobkin M, Shapiro B. Incidentally discovered adrenal masses. Endocrine reviews. 1995 Aug:16(4):460-84 [PubMed PMID: 8521790]
Glazer HS, Weyman PJ, Sagel SS, Levitt RG, McClennan BL. Nonfunctioning adrenal masses: incidental discovery on computed tomography. AJR. American journal of roentgenology. 1982 Jul:139(1):81-5 [PubMed PMID: 6979870]
Mantero F, Terzolo M, Arnaldi G, Osella G, Masini AM, Alì A, Giovagnetti M, Opocher G, Angeli A. A survey on adrenal incidentaloma in Italy. Study Group on Adrenal Tumors of the Italian Society of Endocrinology. The Journal of clinical endocrinology and metabolism. 2000 Feb:85(2):637-44 [PubMed PMID: 10690869]
Level 2 (mid-level) evidenceFarrugia FA, Charalampopoulos A. Pheochromocytoma. Endocrine regulations. 2019 Jul 1:53(3):191-212. doi: 10.2478/enr-2019-0020. Epub [PubMed PMID: 31517632]
Schürfeld R, Pamporaki C, Peitzsch M, Rayes N, Sabri O, Rohm S, Biemann R, Sandner B, Tönjes A, Eisenhofer G. False-positive results for pheochromocytoma associated with norepinephrine reuptake blockade. Endocrine-related cancer. 2024 Jan 1:31(1):. doi: 10.1530/ERC-23-0063. Epub 2023 Dec 1 [PubMed PMID: 37955319]
Kline GA, Boyd J, Leung AA, Tang A, Sadrzadeh HM. Very high rate of false positive biochemical results when screening for pheochromocytoma in a large, undifferentiated population with variable indications for testing. Clinical biochemistry. 2020 Mar:77():26-31. doi: 10.1016/j.clinbiochem.2020.01.005. Epub 2020 Jan 21 [PubMed PMID: 31978379]
Lenders JW, Willemsen JJ, Eisenhofer G, Ross HA, Pacak K, Timmers HJ, Sweep CG. Is supine rest necessary before blood sampling for plasma metanephrines? Clinical chemistry. 2007 Feb:53(2):352-4 [PubMed PMID: 17200132]
Level 2 (mid-level) evidencede Jong WH, Eisenhofer G, Post WJ, Muskiet FA, de Vries EG, Kema IP. Dietary influences on plasma and urinary metanephrines: implications for diagnosis of catecholamine-producing tumors. The Journal of clinical endocrinology and metabolism. 2009 Aug:94(8):2841-9. doi: 10.1210/jc.2009-0303. Epub 2009 Jun 30 [PubMed PMID: 19567530]
Deutschbein T, Unger N, Jaeger A, Broecker-Preuss M, Mann K, Petersenn S. Influence of various confounding variables and storage conditions on metanephrine and normetanephrine levels in plasma. Clinical endocrinology. 2010 Aug:73(2):153-60. doi: 10.1111/j.1365-2265.2009.03761.x. Epub 2009 Dec 18 [PubMed PMID: 20039892]
Level 2 (mid-level) evidenceNeumann HP, Bausch B, McWhinney SR, Bender BU, Gimm O, Franke G, Schipper J, Klisch J, Altehoefer C, Zerres K, Januszewicz A, Eng C, Smith WM, Munk R, Manz T, Glaesker S, Apel TW, Treier M, Reineke M, Walz MK, Hoang-Vu C, Brauckhoff M, Klein-Franke A, Klose P, Schmidt H, Maier-Woelfle M, Peçzkowska M, Szmigielski C, Eng C, Freiburg-Warsaw-Columbus Pheochromocytoma Study Group. Germ-line mutations in nonsyndromic pheochromocytoma. The New England journal of medicine. 2002 May 9:346(19):1459-66 [PubMed PMID: 12000816]
Level 2 (mid-level) evidenceSarkadi B, Saskoi E, Butz H, Patocs A. Genetics of Pheochromocytomas and Paragangliomas Determine the Therapeutical Approach. International journal of molecular sciences. 2022 Jan 27:23(3):. doi: 10.3390/ijms23031450. Epub 2022 Jan 27 [PubMed PMID: 35163370]
Friedman LR, Ramamoorthy B, Nilubol N. Progress in surgical approaches and outcomes of patients with pheochromocytoma and paraganglioma. Best practice & research. Clinical endocrinology & metabolism. 2024 Sep 21:():101954. doi: 10.1016/j.beem.2024.101954. Epub 2024 Sep 21 [PubMed PMID: 39366823]
Lee JE, Curley SA, Gagel RF, Evans DB, Hickey RC. Cortical-sparing adrenalectomy for patients with bilateral pheochromocytoma. Surgery. 1996 Dec:120(6):1064-70; discussion 1070-1 [PubMed PMID: 8957496]
Level 2 (mid-level) evidenceNeumann HP, Reincke M, Bender BU, Elsner R, Janetschek G. Preserved adrenocortical function after laparoscopic bilateral adrenal sparing surgery for hereditary pheochromocytoma. The Journal of clinical endocrinology and metabolism. 1999 Aug:84(8):2608-10 [PubMed PMID: 10443647]
Neumann HPH, Tsoy U, Bancos I, Amodru V, Walz MK, Tirosh A, Kaur RJ, McKenzie T, Qi X, Bandgar T, Petrov R, Yukina MY, Roslyakova A, van der Horst-Schrivers ANA, Berends AMA, Hoff AO, Castroneves LA, Ferrara AM, Rizzati S, Mian C, Dvorakova S, Hasse-Lazar K, Kvachenyuk A, Peczkowska M, Loli P, Erenler F, Krauss T, Almeida MQ, Liu L, Zhu F, Recasens M, Wohllk N, Corssmit EPM, Shafigullina Z, Calissendorff J, Grozinsky-Glasberg S, Kunavisarut T, Schalin-Jäntti C, Castinetti F, Vlcek P, Beltsevich D, Egorov VI, Schiavi F, Links TP, Lechan RM, Bausch B, Young WF Jr, Eng C, International Bilateral-Pheochromocytoma-Registry Group. Comparison of Pheochromocytoma-Specific Morbidity and Mortality Among Adults With Bilateral Pheochromocytomas Undergoing Total Adrenalectomy vs Cortical-Sparing Adrenalectomy. JAMA network open. 2019 Aug 2:2(8):e198898. doi: 10.1001/jamanetworkopen.2019.8898. Epub 2019 Aug 2 [PubMed PMID: 31397861]
Level 2 (mid-level) evidenceUlchaker JC, Goldfarb DA, Bravo EL, Novick AC. Successful outcomes in pheochromocytoma surgery in the modern era. The Journal of urology. 1999 Mar:161(3):764-7 [PubMed PMID: 10022680]
McBride JF, Atwell TD, Charboneau WJ, Young WF Jr, Wass TC, Callstrom MR. Minimally invasive treatment of metastatic pheochromocytoma and paraganglioma: efficacy and safety of radiofrequency ablation and cryoablation therapy. Journal of vascular and interventional radiology : JVIR. 2011 Sep:22(9):1263-70. doi: 10.1016/j.jvir.2011.06.016. Epub [PubMed PMID: 21856504]
Shah MH, Goldner WS, Benson AB, Bergsland E, Blaszkowsky LS, Brock P, Chan J, Das S, Dickson PV, Fanta P, Giordano T, Halfdanarson TR, Halperin D, He J, Heaney A, Heslin MJ, Kandeel F, Kardan A, Khan SA, Kuvshinoff BW, Lieu C, Miller K, Pillarisetty VG, Reidy D, Salgado SA, Shaheen S, Soares HP, Soulen MC, Strosberg JR, Sussman CR, Trikalinos NA, Uboha NA, Vijayvergia N, Wong T, Lynn B, Hochstetler C. Neuroendocrine and Adrenal Tumors, Version 2.2021, NCCN Clinical Practice Guidelines in Oncology. Journal of the National Comprehensive Cancer Network : JNCCN. 2021 Jul 28:19(7):839-868. doi: 10.6004/jnccn.2021.0032. Epub 2021 Jul 28 [PubMed PMID: 34340212]
Level 1 (high-level) evidenceFishbein L, Bonner L, Torigian DA, Nathanson KL, Cohen DL, Pryma D, Cengel KA. External beam radiation therapy (EBRT) for patients with malignant pheochromocytoma and non-head and -neck paraganglioma: combination with 131I-MIBG. Hormone and metabolic research = Hormon- und Stoffwechselforschung = Hormones et metabolisme. 2012 May:44(5):405-10. doi: 10.1055/s-0032-1308992. Epub 2012 May 7 [PubMed PMID: 22566196]
Level 2 (mid-level) evidenceBreen W, Bancos I, Young WF Jr, Bible KC, Laack NN, Foote RL, Hallemeier CL. External beam radiation therapy for advanced/unresectable malignant paraganglioma and pheochromocytoma. Advances in radiation oncology. 2018 Jan-Mar:3(1):25-29. doi: 10.1016/j.adro.2017.11.002. Epub 2017 Nov 22 [PubMed PMID: 29556576]
Level 3 (low-level) evidenceOhara N, Uemura Y, Mezaki N, Kimura K, Kaneko M, Kuwano H, Ebe K, Fujita T, Komeyama T, Usuda H, Yamazaki Y, Maekawa T, Sasano H, Kaneko K, Kamoi K. Histopathological analysis of spontaneous large necrosis of adrenal pheochromocytoma manifested as acute attacks of alternating hypertension and hypotension: a case report. Journal of medical case reports. 2016 Oct 12:10(1):279 [PubMed PMID: 27729064]
Level 3 (low-level) evidenceKoizumi G, Saiki R, Kurokawa I, Mikura K, Iida T, Murai N, Kaji M, Hashizume M, Kigawa Y, Endo K, Iizaka T, Otsuka F, Isobe T, Norose T, Ohike N, Sasaki J, Hayashi M, Sasaki H, Nagasaka S. Continuous Hemodiafiltration for Pheochromocytoma Crisis with a Positive Outcome. Internal medicine (Tokyo, Japan). 2019 Nov 1:58(21):3113-3119. doi: 10.2169/internalmedicine.2991-19. Epub 2019 Jul 10 [PubMed PMID: 31292390]
Kariyasu T, Machida H, Nishina Y, Tambo M, Miyagawa S, Rakue T, Sumitani Y, Yasuda K, Shibahara J, Yokoyama K. Emergent transcatheter arterial embolization to control critical blood pressure fluctuation associated with hypercatecholaminemic crisis in a patient with an unruptured retroperitoneal paraganglioma. Radiology case reports. 2021 Aug:16(8):2065-2071. doi: 10.1016/j.radcr.2021.05.018. Epub 2021 Jun 8 [PubMed PMID: 34158896]
Level 3 (low-level) evidenceJimenez C. Treatment for Patients With Malignant Pheochromocytomas and Paragangliomas: A Perspective From the Hallmarks of Cancer. Frontiers in endocrinology. 2018:9():277. doi: 10.3389/fendo.2018.00277. Epub 2018 May 28 [PubMed PMID: 29892268]
Level 3 (low-level) evidenceRoman-Gonzalez A, Zhou S, Ayala-Ramirez M, Shen C, Waguespack SG, Habra MA, Karam JA, Perrier N, Wood CG, Jimenez C. Impact of Surgical Resection of the Primary Tumor on Overall Survival in Patients With Metastatic Pheochromocytoma or Sympathetic Paraganglioma. Annals of surgery. 2018 Jul:268(1):172-178. doi: 10.1097/SLA.0000000000002195. Epub [PubMed PMID: 28257320]
Ayala-Ramirez M, Feng L, Habra MA, Rich T, Dickson PV, Perrier N, Phan A, Waguespack S, Patel S, Jimenez C. Clinical benefits of systemic chemotherapy for patients with metastatic pheochromocytomas or sympathetic extra-adrenal paragangliomas: insights from the largest single-institutional experience. Cancer. 2012 Jun 1:118(11):2804-12. doi: 10.1002/cncr.26577. Epub 2011 Oct 17 [PubMed PMID: 22006217]
Tanabe A, Naruse M, Nomura K, Tsuiki M, Tsumagari A, Ichihara A. Combination chemotherapy with cyclophosphamide, vincristine, and dacarbazine in patients with malignant pheochromocytoma and paraganglioma. Hormones & cancer. 2013 Apr:4(2):103-10. doi: 10.1007/s12672-013-0133-2. Epub 2013 Jan 30 [PubMed PMID: 23361939]
Niemeijer ND, Alblas G, van Hulsteijn LT, Dekkers OM, Corssmit EP. Chemotherapy with cyclophosphamide, vincristine and dacarbazine for malignant paraganglioma and pheochromocytoma: systematic review and meta-analysis. Clinical endocrinology. 2014 Nov:81(5):642-51. doi: 10.1111/cen.12542. Epub 2014 Jul 30 [PubMed PMID: 25041164]
Level 1 (high-level) evidenceHuang H, Abraham J, Hung E, Averbuch S, Merino M, Steinberg SM, Pacak K, Fojo T. Treatment of malignant pheochromocytoma/paraganglioma with cyclophosphamide, vincristine, and dacarbazine: recommendation from a 22-year follow-up of 18 patients. Cancer. 2008 Oct 15:113(8):2020-8. doi: 10.1002/cncr.23812. Epub [PubMed PMID: 18780317]
Level 3 (low-level) evidencevan der Harst E, de Herder WW, Bruining HA, Bonjer HJ, de Krijger RR, Lamberts SW, van de Meiracker AH, Boomsma F, Stijnen T, Krenning EP, Bosman FT, Kwekkeboom DJ. [(123)I]metaiodobenzylguanidine and [(111)In]octreotide uptake in begnign and malignant pheochromocytomas. The Journal of clinical endocrinology and metabolism. 2001 Feb:86(2):685-93 [PubMed PMID: 11158032]
Level 2 (mid-level) evidenceLoh KC, Fitzgerald PA, Matthay KK, Yeo PP, Price DC. The treatment of malignant pheochromocytoma with iodine-131 metaiodobenzylguanidine (131I-MIBG): a comprehensive review of 116 reported patients. Journal of endocrinological investigation. 1997 Dec:20(11):648-58 [PubMed PMID: 9492103]
Mukherjee JJ, Kaltsas GA, Islam N, Plowman PN, Foley R, Hikmat J, Britton KE, Jenkins PJ, Chew SL, Monson JP, Besser GM, Grossman AB. Treatment of metastatic carcinoid tumours, phaeochromocytoma, paraganglioma and medullary carcinoma of the thyroid with (131)I-meta-iodobenzylguanidine [(131)I-mIBG]. Clinical endocrinology. 2001 Jul:55(1):47-60 [PubMed PMID: 11453952]
Level 2 (mid-level) evidenceTroncone L, Rufini V. Nuclear medicine therapy of pheochromocytoma and paraganglioma. The quarterly journal of nuclear medicine : official publication of the Italian Association of Nuclear Medicine (AIMN) [and] the International Association of Radiopharmacology (IAR). 1999 Dec:43(4):344-55 [PubMed PMID: 10731785]
Sisson JC. Radiopharmaceutical treatment of pheochromocytomas. Annals of the New York Academy of Sciences. 2002 Sep:970():54-60 [PubMed PMID: 12381541]
Hao Z, Sadek I. Sunitinib: the antiangiogenic effects and beyond. OncoTargets and therapy. 2016:9():5495-505. doi: 10.2147/OTT.S112242. Epub 2016 Sep 8 [PubMed PMID: 27660467]
O'Kane GM, Ezzat S, Joshua AM, Bourdeau I, Leibowitz-Amit R, Olney HJ, Krzyzanowska M, Reuther D, Chin S, Wang L, Brooks K, Hansen AR, Asa SL, Knox JJ. A phase 2 trial of sunitinib in patients with progressive paraganglioma or pheochromocytoma: the SNIPP trial. British journal of cancer. 2019 Jun:120(12):1113-1119. doi: 10.1038/s41416-019-0474-x. Epub 2019 May 20 [PubMed PMID: 31105270]
Ayala-Ramirez M, Chougnet CN, Habra MA, Palmer JL, Leboulleux S, Cabanillas ME, Caramella C, Anderson P, Al Ghuzlan A, Waguespack SG, Deandreis D, Baudin E, Jimenez C. Treatment with sunitinib for patients with progressive metastatic pheochromocytomas and sympathetic paragangliomas. The Journal of clinical endocrinology and metabolism. 2012 Nov:97(11):4040-50. doi: 10.1210/jc.2012-2356. Epub 2012 Sep 10 [PubMed PMID: 22965939]
Level 2 (mid-level) evidenceAmar L, Servais A, Gimenez-Roqueplo AP, Zinzindohoue F, Chatellier G, Plouin PF. Year of diagnosis, features at presentation, and risk of recurrence in patients with pheochromocytoma or secreting paraganglioma. The Journal of clinical endocrinology and metabolism. 2005 Apr:90(4):2110-6 [PubMed PMID: 15644401]
Holscher I, van den Berg TJ, Dreijerink KMA, Engelsman AF, Nieveen van Dijkum EJM. Recurrence Rate of Sporadic Pheochromocytomas After Curative Adrenalectomy: A Systematic Review and Meta-analysis. The Journal of clinical endocrinology and metabolism. 2021 Jan 23:106(2):588-597. doi: 10.1210/clinem/dgaa794. Epub [PubMed PMID: 33125073]
Level 1 (high-level) evidenceHarrington JL, Farley DR, van Heerden JA, Ramin KD. Adrenal tumors and pregnancy. World journal of surgery. 1999 Feb:23(2):182-6 [PubMed PMID: 9880429]
Level 3 (low-level) evidenceLenders JWM, Langton K, Langenhuijsen JF, Eisenhofer G. Pheochromocytoma and Pregnancy. Endocrinology and metabolism clinics of North America. 2019 Sep:48(3):605-617. doi: 10.1016/j.ecl.2019.05.006. Epub 2019 Jun 13 [PubMed PMID: 31345526]
Bancos I, Atkinson E, Eng C, Young WF Jr, Neumann HPH, International Pheochromocytoma and Pregnancy Study Group. Maternal and fetal outcomes in phaeochromocytoma and pregnancy: a multicentre retrospective cohort study and systematic review of literature. The lancet. Diabetes & endocrinology. 2021 Jan:9(1):13-21. doi: 10.1016/S2213-8587(20)30363-6. Epub 2020 Nov 26 [PubMed PMID: 33248478]
Level 2 (mid-level) evidencevan der Weerd K, van Noord C, Loeve M, Knapen MFCM, Visser W, de Herder WW, Franssen G, van der Marel CD, Feelders RA. ENDOCRINOLOGY IN PREGNANCY: Pheochromocytoma in pregnancy: case series and review of literature. European journal of endocrinology. 2017 Aug:177(2):R49-R58. doi: 10.1530/EJE-16-0920. Epub 2017 Apr 5 [PubMed PMID: 28381449]
Level 2 (mid-level) evidence