Introduction
Parkinson-plus syndrome (PPS), also called atypical parkinsonism, refers to a group of neurodegenerative movement disorders that resemble idiopathic Parkinson's disease (PD) with certain distinguishing clinical and pathophysiological features.[1] As in classical Parkinson disease, the primary symptoms of parkinsonism present include apraxia, rigidity, bradykinesia, tremor, and postural instability. Distinctive features of PPS include early-onset dementia, hallucinations, dysautonomia, gaze palsy, myoclonus, pyramidal tract signs, and alien limb phenomenon.[2]
Unlike Parkinson disease, these conditions have a limited response to levodopa with a poor overall prognosis.[3] The most common PPS conditions are Lewy body dementia (LBD), multiple system atrophy (MSA), corticobasal degeneration (CBD), and progressive supranuclear palsy (PSP).[2][4]
Less prevalent disorders are frontotemporal dementia, Pick disease, pallidonigral degeneration, parkinsonian-dementia complex of Guam, Wilson disease, and a rigid variant of Huntington disease.[5][6] This review will discuss the most common PPS conditions, focusing on epidemiology, etiology, pathophysiology, evaluation, and treatment options.
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
The vast majority of Parkinson-plus syndrome cases occur sporadically, and the precise etiology remains unknown. A few examples of familial cases suggest that genetics may play a role in the pathogenesis of PPS.[7] Environmental factors additionally contribute to the evolution of these diseases.[8]
Epidemiology
Lewy Body Dementia
LBD is one of the most common causes of neurodegenerative dementia, accounting for up to 30 percent of all dementia cases. The only forms of dementia more common are Alzheimer disease (AD) and vascular dementia. The prevalence is estimated to be 5 percent of the general population, and the incidence is approximately 0.1 percent annually.[9][10]
Multiple System Atrophy
The approximate prevalence of MSA ranges from 2 to 5 cases per 100,000, and the incidence is 3 per 100,000. The mean age of onset is estimated to be 54 years. Men and women are affected equally.[11]
Corticobasal Degeneration
The approximate annual incidence of CBD ranges between 0.62 to 0.92 per 100,000, and the prevalence ranges from 5 to 7 per 100,000.[12] The disease typically presents at a mean age of 61 to 64 years. There is a suggested predominance of females.
Progressive Supranuclear Palsy
The approximate prevalence of PSP is 3 to 7 per 100,000. The approximate incidence is 1.1 per 100,000 person-years.[12]
Pathophysiology
The pathophysiology of PPS can be divided into two groups, alpha-synucleinopathies and tauopathies. These groups allude to the type of protein (alpha-synuclein and tau proteins) which folds into a toxic and cell-damaging form.[1][2] It is proposed that the misfolded, toxic proteins cause cell death and induce improper folding of other proteins as the disease spreads throughout the brain.[2]
In tauopathies such as PSP and CBD, cytoskeleton proteins become abnormally phosphorylated, leading to the development of tau inclusions in neurons and glial cells.[4] A similar process occurs in the alpha-synucleinopathies PD, LBD, and MSA.[6] The specific inclusions formed by alpha-synuclein are referred to as Lewy bodies in PD and LBD.[6] It is unknown why tau and alpha-synuclein proteins originally misfold, but it is postulated that it is due to sporadic mutations and environmental factors.[1][2]
The specific PPS condition is related to the type of neurons targeted and the location of these toxic proteins in the brain.
Lewy Body Dementia (LBD)
In LBD, alpha-synuclein dysfunction and associated Lewy body deposits contribute to the impairment of particularly susceptible dopaminergic and cholinergic neurons.[13] Neurodegeneration begins in the brainstem and progresses to the olfactory bulb, substantia nigra, limbic areas, and the neocortex.[14][13]
Multiple System Atrophy (MSA)
MSA is suggested to be due to the prion-like spreading of alpha-synuclein proteins primarily, as well as glial cytoplasmic inclusions of tau and ubiquitin proteins.[15] Locations of pathology include the substantia nigra, Purkinje cells of the cerebellum, caudate nucleus, putamen, locus ceruleus, pontine nuclei, and inferior olivary nucleus.[16][17]
Progressive Supranuclear Palsy (PSP)
PSP involves tau cytoplasmic inclusions within neurons, referred to as neurofibrillary tangles.[14] These inclusions lead to neuronal loss, gliosis, and damage to astrocytes and oligodendrocytes. Areas targeted include the basal ganglia (substantia nigra, subthalamic nucleus, internal globus pallidus), oculomotor complex, cerebral cortex periaqueductal gray matter, superior colliculi, basis pontis, and dentate nucleus.[18]
Corticobasal Degeneration (CBD)
CBD is associated with tau astrocytic plaques and inclusions in the glia. These tau proteins lead to severe gliosis, neuronal loss, and ballooned achromatic neurons.[19] In CBD, there is prominent asymmetric frontoparietal atrophy.[19]
History and Physical
Parkinson-plus syndrome presents with parkinsonism as well as a variety of other clinical features that can be elicited by obtaining a focused history and physical along with close neurological monitoring. As in classical PD, there is apraxia, rigidity, bradykinesia, tremor, and postural instability.[1]
Depending on the specific PPS, other possible clinical features include dementia, hallucinations, dysautonomia, symmetrical onset, myoclonus, pyramidal tract signs, supranuclear gaze palsy, and alien limb phenomenon.[5] PPS is also notable for its rapid progression and reduced response to classical PD medications. As the disease progresses, patients experience difficulty with ambulation and activities of daily living, as well as an overall decline in function.[4][5]
Lewy Body Dementia
LBD consists of dementia characterized by deficits in attention, cognition, and visuospatial function. Other notable features include visual hallucinations, rapid eye movement sleep behavior disorder (RBD), repeated falls, autonomic dysfunction, antipsychotic drug sensitivity, delusions, and depression. LBD consists of two clinical entities: dementia with Lewy bodies and PD dementia. Dementia with Lewy bodies is classified as dementia occurring first or within one year of the movement disorder. PD dementia occurs in a patient first receiving the diagnosis of PD and then developing dementia symptoms after one year or more of the diagnosis.[20]
Multiple System Atrophy
The cardinal features of MSA are akinetic-rigid parkinsonism, cerebellar ataxia, autonomic failure, urogenital dysfunction, and pyramidal signs. There are two clinical subtypes of MSA based on motor presentation: MSA with predominant parkinsonism (MSA-P) and MSA with predominant cerebellar ataxia (MSA-C).[5]
The predominant motor feature may also change as the disease progresses. MSA-P is associated with bradykinesia, postural instability, rigidity, and a jerky postural and action tremor. MSA-C is characterized by gait and limb ataxia, dysarthria, and eye movement disturbances. Dysautonomia in both motor subtypes is standard and includes early-onset erectile dysfunction, urinary frequency, incontinence, and late-appearing orthostatic hypotension.[21] Cognitive function in MSA is usually well preserved.
Corticobasal Degeneration
CBD presents as an asymmetric movement disorder, with symptoms affecting one limb initially. Akinesia features include extreme rigidity, focal myoclonus, dystonia, alien limb phenomenon, and apraxia.[5] Cognitive impairment is common and may be the presenting feature. The cognitive decline includes aphasia, executive dysfunction, visuospatial deficits, and behavioral change. Episodic memory is often preserved.[22]
Progressive Supranuclear Palsy
PSP presents a variety of unique phenotypes. The most common and classic form of PSP is known as Richardson syndrome (PSP-RS). PSP-RS initially presents with gait dysfunction that leads to frequent falls. Supranuclear palsy is the hallmark of PSP, as patients cannot move the eyes vertically, focus on objects, or control eyelid movements. Ophthalmoparesis usually appears within four years but may take up to 10 years to develop.[23]
PSP patients are described as having axial rigidity and standing with an extended posture as opposed to the flexed posture in PD. Patients tend to pivot quickly and often fall backward. Other common symptoms include dysphagia, dysarthria, rigidity, frontal cognitive dysfunction, and sleep abnormalities.[24]
Phenotypes of PSP include PSP with predominant oculomotor dysfunction (PSP-OM), postural instability (PSP-PI), frontal presentation (PSP-F), speech/language disorder (PSP-SL), corticobasal syndrome (PSP-CBS), and progressive gait freezing (PSP-PGF).
Evaluation
Although Parkinson-plus syndrome is a clinical diagnosis, a full medical workup consists of detailed laboratory testing and imaging studies to help rule out other common diagnoses. Unfortunately, in most PPS conditions, laboratory and imaging findings are non-diagnostic.[1][2][4]
Lewy Dody Dementia
The evaluation of LBD begins as an analysis of dementia. This consists of a cognitive assessment with a Mini-Mental State Examination (MMSE) or Montreal Cognitive Assessment (MoCA). Laboratory testing includes a basic workup with a chemistry panel, complete blood count (CBC), thyroid function test, and vitamin B12 levels. Neuroimaging is performed with computed tomography (CT) or magnetic resonance imaging (MRI). Imaging findings in LBD include generalized brain atrophy and white matter lesions, pronounced cortical atrophy, degeneration of the putamen, and reduced perfusion, most marked in the occipital areas.[20]
Indicative biomarkers that contribute to the diagnosis of LBD include reduced dopamine transporter uptake in the basal ganglia evident on positron emission tomography (PET) or single-photon emission computerized tomography (SPECT), low uptake of 123-iodine-metaiodobenzylguanidine (MIBG) myocardial scintigraphy, and confirmation of REM sleep without atonia on polysomnography. Supportive biomarkers for diagnosis include relative preservation of medial temporal lobe structures on CT or MRI, low uptake on PET perfusion scans with reduced occipital activity, and posterior slow-wave activity on electroencephalogram (EEG) with periodic fluctuations in the pre-alpha and theta range.[20]
Multiple System Atrophy
As in other PPS diseases, laboratory and imaging studies are non-specific, and diagnosis is based on clinical features. Neuroimaging is used to rule out other diseases but may also show atrophy of the putamen, pons, or middle cerebellar peduncle, along with signal changes in the putamen. A definite diagnosis of MSA is based on post-mortem neurodegenerative changes in the striatonigral or olivopontocerebellar structures, with pathology showing alpha-synuclein inclusions in glial cells.[25] Other useful tests include autonomic function testing and bladder function analysis with sphincter electromyography (EMG).[26][27]
Corticobasal Degeneration
No established biological markers exist for CBD. Routine blood, urine, and cerebrospinal fluid (CSF) analyses are all non-diagnostic, while neuroimaging studies are non-specific. Workup includes laboratory tests such as CBC, electrolytes, vitamin B12, thyroid function tests, syphilis testing (to investigate for causes of cognitive deficits), and copper and ceruloplasmin levels to rule out Wilson's disease. Workup includes erythrocyte sedimentation rate (ESR), antinuclear antibody (ANA), and liver function tests to investigate rheumatologic disease; CSF testing for biomarkers of neurodegeneration can be performed, such as analysis for 14-3-3 protein, neuron-specific enolase, and CSF real-time quaking-induced conversion (CSF RT-QuIC) to test for prionopathies such as Creutzfeldt-Jakob disease (CJD).
Evaluation for AD can be performed by testing for Amyloid beta-42, phosphorylated tau, and total tau. Regarding neuroimaging, CBD has been shown to have more asymmetric atrophy of the frontal and parietal lobes.[28] Cortical atrophy is observed mainly in the central sulci, supplementary motor area, and superior frontal gyrus. PET and SPECT imaging demonstrate asymmetric activity in the subcortical basal ganglia and cortical frontal-parietal regions.[28]
Progressive Supranuclear Palsy
In PSP, a medical workup is based upon eliminating other diagnoses. Although not diagnostic, neuroimaging is notable for the hummingbird sign (or penguin silhouette sign) on MRI, a name derived from predominant midbrain atrophy with preserved pons resembling a hummingbird or penguin in silhouette. PET studies have shown hypometabolism in the frontal cortex, putamen, caudate, thalamus, and brainstem. Fluorodopa (F-dopa) PET study demonstrates a lower influx of F-dopa in the caudate and putamen compared to PD.
Furthermore, sleep studies are often abnormal in PSP patients with lower total sleep time, loss of REM sleep, and increased awakenings. Histologic findings in PSP show prominent involvement in the basal ganglia (specifically the internal globus pallidus, subthalamic nucleus, and substantia nigra), oculomotor complex, superior colliculi, dentate nucleus, periaqueductal gray matter, and basis pontis.
Treatment / Management
In general, there are no disease-modifying treatments for PPS conditions. Treatment is focused on symptom management and improving quality of life. A minority of patients may show some relief of parkinsonism symptoms with levodopa. Alternative agents with limited benefit for parkinsonism include monoamine oxidase type B (MAO-B) inhibitor selegiline, the dopamine-releasing agent amantadine, and dopamine receptor agonists.
Managing tremors includes using propranolol, clonazepam, topiramate, and gabapentin. Anticholinergics and baclofen may be helpful. Physical therapy helps prevent falls, reduces contractures, and promotes mobility. Physical therapists can also help with movement problems through cardiovascular, strengthening, flexibility exercises, gait training, and general fitness programs.
Occupational therapy can assist in promoting independence for activities of daily living. Speech therapy is important in evaluating dysphagia, while dieticians can assist in proper nutrition planning. Palliative and safety measures are important for end-of-life care and limiting stress for the patient and caregivers.[5]
Music or expressive arts therapists may provide meaningful activities that can reduce anxiety and improve well-being. There are also various other treatments and therapies which are specific for individual PPS diseases.
Lewy Body Dementia
In LBD, cholinesterase inhibitors or memantine can be used to treat cognitive or behavioral symptoms. Antipsychotic medications are used when behavioral symptoms are severe. Disabling parkinsonism can be treated with levodopa, although effects are limited.[20] REM sleep behavior disorder is treated with melatonin or clonazepam. Mental health counselors can help LBD patients manage difficult emotions and behaviors.
Multiple System Atrophy
Botulinum toxin injections may alleviate focal dystonias in MSA. Chronic orthostatic hypotension resulting from autonomic dysfunction can be improved with fludrocortisone acetate or midodrine. REM sleep behavior disorder can be treated with melatonin or clonazepam if necessary.[29](B3)
Corticobasal Degeneration
In CBD, dystonia can be treated with botulinum toxin to improve function in the affected limb.[29] Clonazepam is the agent of choice for treating myoclonus. Acetylcholinesterase inhibitors such as donepezil or rivastigmine can be attempted to treat cognitive deficits.[30](B3)
Progressive Supranuclear Palsy
There are a variety of therapeutic options to address the ophthalmological symptoms of PSP. Patients with limited extraocular movements can use mirror-prism lenses to read and perform activities of daily living with more independence. Eyelid opening apraxia and blepharospasm can be relieved with eyelid crutches and botulinum toxin.[29] Artificial tears should be used for treating a decreased blink rate, and dark glasses can reduce photophobia.(B3)
Differential Diagnosis
Each Parkinson-plus syndrome disease and classical PD must be considered when forming a differential diagnosis. Other similar diagnoses include AD, vascular dementia, normal pressure hydrocephalus, CJD, and certain psychiatric diseases.[1][2] Extrapyramidal side effects of medications can also be a cause of parkinsonism.[4]
Prognosis
Compared to idiopathic PD, PPS conditions progress more rapidly and culminate in death earlier.[1][2][4]
Lewy Body Disease
In LBD, symptoms progress rapidly, with an estimated survival of 1.8 to 9.5 years. Studies show that survival for LBD is shorter compared to AD. Cognitive decline is progressive, and the median time to severe dementia is approximately five years. Parkinsonism and non-motor symptoms, including autonomic dysfunction and neuropsychiatric deficits, worsen over time.[20] Behavioral symptoms such as hallucinations, delusions, depression, and anxiety may require nursing home admission.
Multiple System Atrophy
Compared to idiopathic PD, the disease progression of MSA is much quicker. From symptom onset, autonomic dysfunction begins in 2.5 years, wheelchair confinement occurs in 3.5 to 5 years, and patients are bedridden for 5 to 8 years. Following diagnosis, death occurs after 6 to 10 years in most patients.[29]
Corticobasal Degeneration
CBD progresses rapidly to death, with the most common cause of mortality being complications from immobility or dysphagia (pneumonia and sepsis).[31] From the onset of symptoms, the median survival time is approximately 6 to 8 years but ranges from 2 to 13 years.[32]
Progressive Supranuclear Palsy
The progression of PSP is rapid, and patients usually become dependent on care within three to four years from symptom onset. Death occurs approximately six to nine years after the diagnosis.[18] Shorter survival is seen more in the PSP-RS phenotype compared to the other variants.[29]
Complications
There are various complications in PPS. These are often related to aspects of dementia and parkinsonism. Complications of parkinsonism include immobility and falls. Dysphagia can result in aspiration pneumonia and sepsis in many of these diseases. Urinary tract abnormalities may lead to complications like urinary tract infections.[6]
Frequently, medications present many unwanted side effects. Furthermore, autonomic dysfunction can present with cardiac complications.[7] Often overlooked, behavioral symptoms such as anxiety and depression lead to suicidal ideation.[8]
Deterrence and Patient Education
Patient and caregiver education in the context of Parkinson-plus syndrome is crucial and can result in improved disease management, better functional outcomes, and higher quality of life.[2] Patients and families should be aware that PPS conditions progress rapidly, but there are many treatments and therapies that can provide benefits.[2]
The various healthcare professionals should provide education to help manage the disease. Patients and families must acknowledge the symptoms of PPS, including cognitive decline, motor deficits, behavioral changes, hallucinations, and autonomic dysfunction.[1] There are many complications of these conditions. Therefore, a focus on preventing and deterring further comorbidities is crucial.[4]
Enhancing Healthcare Team Outcomes
In managing PPS, an interdisciplinary approach is essential. It should involve healthcare professionals from primary care, neurology, radiology, psychiatry, occupational therapy, physical therapy, speech pathology, nutrition, neuropsychology, social work, palliative care, and specialty-trained nursing.[1][2]
Collaboration among interprofessional healthcare team members can enhance disease management and improve functional outcomes for patients. Because PPS conditions advance rapidly and result in profound functional impairments, healthcare professionals must coordinate care promptly to limit disease progression and alleviate debilitating symptoms. Medications require strict management to maximize therapeutic benefits and limit harmful side effects. Therapies and support services provided early in the disease course can improve patient function and limit the development of comorbidities.[2] This includes contributions from clinicians, nurses, pharmacists, and therapists, all engaged in open communication and contributing from their areas of expertise to achieve the best possible patient outcomes. [Level 5]
Since the prognosis is poor and most patients end up in long-term facilities where the quality of life might not be optimal, caregiver education is vital to improve care for patients while they are at home.[29] Home care nurses can play a significant role in regularly monitoring patients for disease progression or developing new health issues that require further specialist attention.[6]
Media
(Click Image to Enlarge)
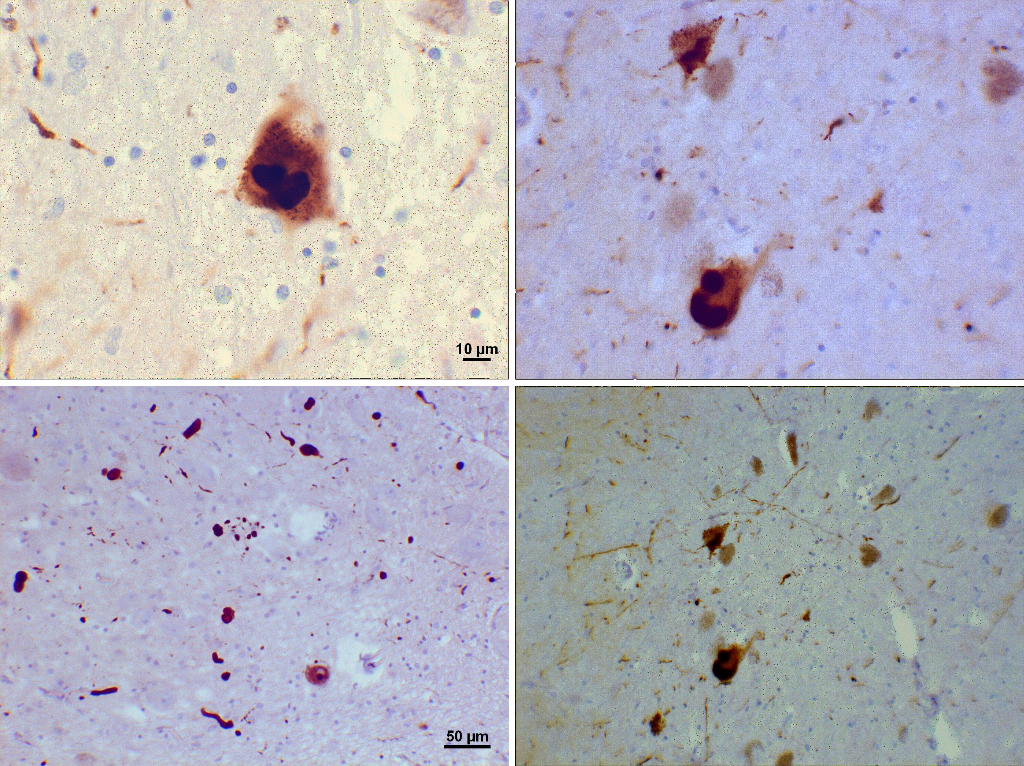
Alpha-synuclein deposits, Lewy bodies, Pathology, Histology, Substantia Nigra Used with Permission from Suraj Rajan at Creative Commons. No alterations were made to this image. License: https://creativecommons.org/licenses/by-sa/3.0/legalcode
(Click Image to Enlarge)

Dementia with Lewy Bodies Contributed by Djekidel Mehdi MD
References
Fabre N, Brefel-Courbon C, Rascol O. [Parkinson "plus"]. La Revue du praticien. 1997 May 15:47(10):1077-82 [PubMed PMID: 9208670]
Mark MH. Lumping and splitting the Parkinson Plus syndromes: dementia with Lewy bodies, multiple system atrophy, progressive supranuclear palsy, and cortical-basal ganglionic degeneration. Neurologic clinics. 2001 Aug:19(3):607-27, vi [PubMed PMID: 11532646]
Miyasaki JM. Treatment of Advanced Parkinson Disease and Related Disorders. Continuum (Minneapolis, Minn.). 2016 Aug:22(4 Movement Disorders):1104-16. doi: 10.1212/CON.0000000000000347. Epub [PubMed PMID: 27495200]
Jankovic J. Parkinsonism-plus syndromes. Movement disorders : official journal of the Movement Disorder Society. 1989:4 Suppl 1():S95-119 [PubMed PMID: 2657411]
Sjöström AC, Holmberg B, Strang P. Parkinson-plus patients--an unknown group with severe symptoms. The Journal of neuroscience nursing : journal of the American Association of Neuroscience Nurses. 2002 Dec:34(6):314-9 [PubMed PMID: 12506814]
Tysnes OB, Vilming ST. [Atypical parkinsonism]. Tidsskrift for den Norske laegeforening : tidsskrift for praktisk medicin, ny raekke. 2008 Sep 25:128(18):2077-80 [PubMed PMID: 18846125]
Srivanitchapoom P, Pitakpatapee Y, Suengtaworn A. Parkinsonian syndromes: A review. Neurology India. 2018 Mar-Apr:66(Supplement):S15-S25. doi: 10.4103/0028-3886.226459. Epub [PubMed PMID: 29503324]
Stacy M, Jankovic J. Differential diagnosis of Parkinson's disease and the parkinsonism plus syndromes. Neurologic clinics. 1992 May:10(2):341-59 [PubMed PMID: 1584178]
Mueller C, Soysal P, Rongve A, Isik AT, Thompson T, Maggi S, Smith L, Basso C, Stewart R, Ballard C, O'Brien JT, Aarsland D, Stubbs B, Veronese N. Survival time and differences between dementia with Lewy bodies and Alzheimer's disease following diagnosis: A meta-analysis of longitudinal studies. Ageing research reviews. 2019 Mar:50():72-80. doi: 10.1016/j.arr.2019.01.005. Epub 2019 Jan 6 [PubMed PMID: 30625375]
Level 1 (high-level) evidenceHanson JC, Lippa CF. Lewy body dementia. International review of neurobiology. 2009:84():215-28. doi: 10.1016/S0074-7742(09)00411-5. Epub [PubMed PMID: 19501720]
Vanacore N. Epidemiological evidence on multiple system atrophy. Journal of neural transmission (Vienna, Austria : 1996). 2005 Dec:112(12):1605-12 [PubMed PMID: 16284906]
Level 3 (low-level) evidenceConstantinides VC, Paraskevas GP, Paraskevas PG, Stefanis L, Kapaki E. Corticobasal degeneration and corticobasal syndrome: A review. Clinical parkinsonism & related disorders. 2019:1():66-71. doi: 10.1016/j.prdoa.2019.08.005. Epub 2019 Aug 30 [PubMed PMID: 34316603]
Yousaf T, Dervenoulas G, Valkimadi PE, Politis M. Neuroimaging in Lewy body dementia. Journal of neurology. 2019 Jan:266(1):1-26. doi: 10.1007/s00415-018-8892-x. Epub 2018 May 14 [PubMed PMID: 29761296]
Mitra K,Gangopadhaya PK,Das SK, Parkinsonism plus syndrome--a review. Neurology India. 2003 Jun; [PubMed PMID: 14570999]
Yang F, Li WJ, Huang XS. Alpha-synuclein levels in patients with multiple system atrophy: a meta-analysis. The International journal of neuroscience. 2018 May:128(5):477-486. doi: 10.1080/00207454.2017.1394851. Epub 2018 Jan 3 [PubMed PMID: 29053035]
Level 1 (high-level) evidenceWenning GK, Braune S. Multiple system atrophy: pathophysiology and management. CNS drugs. 2001:15(11):839-52 [PubMed PMID: 11700149]
Level 3 (low-level) evidenceKoga S, Dickson DW. Recent advances in neuropathology, biomarkers and therapeutic approach of multiple system atrophy. Journal of neurology, neurosurgery, and psychiatry. 2018 Feb:89(2):175-184. doi: 10.1136/jnnp-2017-315813. Epub 2017 Aug 31 [PubMed PMID: 28860330]
Level 3 (low-level) evidenceCoughlin DG, Litvan I. Progressive supranuclear palsy: Advances in diagnosis and management. Parkinsonism & related disorders. 2020 Apr:73():105-116. doi: 10.1016/j.parkreldis.2020.04.014. Epub 2020 May 25 [PubMed PMID: 32487421]
Level 3 (low-level) evidenceWakabayashi K, Takahashi H. [Neuropathology of corticobasal degeneration]. No to shinkei = Brain and nerve. 1996 Jun:48(6):521-32 [PubMed PMID: 8703555]
Armstrong MJ. Lewy Body Dementias. Continuum (Minneapolis, Minn.). 2019 Feb:25(1):128-146. doi: 10.1212/CON.0000000000000685. Epub [PubMed PMID: 30707190]
Damon-Perrière N, Tison F, Meissner WG. [Multiple system atrophy]. Psychologie & neuropsychiatrie du vieillissement. 2010 Sep:8(3):179-91. doi: 10.1684/pnv.2010.0212. Epub [PubMed PMID: 20739256]
Level 2 (mid-level) evidenceArmstrong RA. Visual signs and symptoms of corticobasal degeneration. Clinical & experimental optometry. 2016 Nov:99(6):498-506. doi: 10.1111/cxo.12429. Epub 2016 Aug 23 [PubMed PMID: 27553583]
Armstrong MJ. Progressive Supranuclear Palsy: an Update. Current neurology and neuroscience reports. 2018 Feb 17:18(3):12. doi: 10.1007/s11910-018-0819-5. Epub 2018 Feb 17 [PubMed PMID: 29455271]
Ali F, Josephs K. The diagnosis of progressive supranuclear palsy: current opinions and challenges. Expert review of neurotherapeutics. 2018 Jul:18(7):603-616. doi: 10.1080/14737175.2018.1489241. Epub 2018 Jun 28 [PubMed PMID: 29902389]
Level 3 (low-level) evidencePalma JA, Norcliffe-Kaufmann L, Kaufmann H. Diagnosis of multiple system atrophy. Autonomic neuroscience : basic & clinical. 2018 May:211():15-25. doi: 10.1016/j.autneu.2017.10.007. Epub 2017 Oct 23 [PubMed PMID: 29111419]
Ahmed Z, Asi YT, Sailer A, Lees AJ, Houlden H, Revesz T, Holton JL. The neuropathology, pathophysiology and genetics of multiple system atrophy. Neuropathology and applied neurobiology. 2012 Feb:38(1):4-24. doi: 10.1111/j.1365-2990.2011.01234.x. Epub [PubMed PMID: 22074330]
Level 3 (low-level) evidenceDel Campo N, Phillips O, Ory-Magne F, Brefel-Courbon C, Galitzky M, Thalamas C, Narr KL, Joshi S, Singh MK, Péran P, Pavy-LeTraon A, Rascol O. Broad white matter impairment in multiple system atrophy. Human brain mapping. 2021 Feb 1:42(2):357-366. doi: 10.1002/hbm.25227. Epub 2020 Oct 16 [PubMed PMID: 33064319]
Svenningsson P. Corticobasal degeneration: advances in clinicopathology and biomarkers. Current opinion in neurology. 2019 Aug:32(4):597-603. doi: 10.1097/WCO.0000000000000707. Epub [PubMed PMID: 31145128]
Level 3 (low-level) evidenceHawley JS, Robottom BJ, Weiner WJ. Multiple system atrophy. Reviews in neurological diseases. 2010 Spring-Summer:7(2-3):e45-55 [PubMed PMID: 20944523]
Level 3 (low-level) evidenceArmstrong MJ. Diagnosis and treatment of corticobasal degeneration. Current treatment options in neurology. 2014 Mar:16(3):282. doi: 10.1007/s11940-013-0282-1. Epub [PubMed PMID: 24469408]
Grijalvo-Perez AM, Litvan I. Corticobasal degeneration. Seminars in neurology. 2014 Apr:34(2):160-73. doi: 10.1055/s-0034-1381734. Epub 2014 Jun 25 [PubMed PMID: 24963675]
Saranza GM, Whitwell JL, Kovacs GG, Lang AE. Corticobasal degeneration. International review of neurobiology. 2019:149():87-136. doi: 10.1016/bs.irn.2019.10.014. Epub 2019 Nov 21 [PubMed PMID: 31779825]